
Abstract
Tumour hypoxia represents a significant challenge to the curability of human tumours leading to treatment resistance and enhanced tumour progression. Tumour hypoxia can be detected by non-invasive and invasive techniques but the inter-relationships between these remains largely undefined. 18F-MISO and Cu-ATSM-PET, and BOLD-MRI are the lead contenders for human application based on their non-invasive nature, ease of use and robustness, measurement of hypoxia status, validity, ability to demonstrate heterogeneity and general availability, these techniques are the primary focus of this review. We discuss where developments are required for hypoxia imaging to become clinically useful and explore potential new uses for hypoxia imaging techniques including biological conformal radiotherapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The suspicion that tumour hypoxia increased resistance to radiotherapy was first considered in the 1930’s but it was not until 1955 that Tomlinson and Gray showed that chronic hypoxia occurred in human bronchial carcinomas with necrosis occurring approximately 150 μm from blood vessels [1], which is a little larger than the currently known diffusion distance of soluble oxygen in tissues (approximately 70 μm). Decades of research in radiation therapy then followed, much of which focused on attempts to circumvent hypoxia-mediated radio-resistance but these efforts were only moderately successful. Over the last decade, it has become evident that hypoxia changes the patterns of gene expression in several ways that alters the malignant potential of tumours, leading to more aggressive survival traits. As a result, hypoxic cancers are difficult to treat, particularly by radiation and photodynamic therapy [2], but also by cytotoxic chemotherapy. Attempts at circumventing the cure-limiting impact of hypoxia have included the use of hyperbaric oxygen and radiation sensitizer drugs but these have, in general, not proved widely advantageous. However, attempts to take advantage of the presence of tumour hypoxia, such as hypoxia-specific cytotoxins, are more promising. As hypoxia-directed therapies enter into clinical trials, it has become important to non-invasively assess for the presence of hypoxia and to be able to follow how it is modulated by new therapies. Hypoxia imaging may help select the most appropriate population that would benefit from novel hypoxia-directed therapies. In this review we describe the causes for and the effects of tumour hypoxia, as well as summarise the lead contenders for human tumour imaging. We also assess where developments are required for them to become clinically useful imaging tests and explore potential new uses for hypoxia imaging techniques including biologically-directed conformal radiotherapy.
Overview of tumour hypoxia & its importance
For the majority of solid tumours hypoxia develops because of the inability of the vascular system to supply the growing tumour mass with adequate amounts of oxygen. Consequently, both low oxygen tensions and nutrient deprivation contribute to impaired tumour growth such that growth beyond 2 mm requires tumour neovascularisation. The major factors that play roles in the development of tumour cell hypoxia are the known abnormalities in structure and functioning of tumour microvessels [3], the increased diffusion distances between blood vessels (many of which may not even carry oxygenated red blood cells), the expanding tumour cell mass competing for oxygen and the reduced oxygen carrying capacity of blood due to disease- or treatment related anaemia. Thus, there are three distinct types of tumour hypoxia [4]: (1) Perfusion related (acute) hypoxia that results from inadequate blood flow in tumours that is generally the consequence of recognised structural and functional abnormalities of the tumour neovasculature. Such acute hypoxia is often transient, caused by temporary occlusions and temporary rises in interstitial pressure and can affect all cells right up to the vessel wall; (2) Diffusion related (chronic) hypoxia is caused by increased oxygen diffusion distances due to tumour expansion and affects cells greater than 70–100 μm from the nearest capillary, depending on where tumour cells lie in relation to the arterial or venous end of a capillary; (3) Anaemic hypoxia, which relates to reduced O2-carrying capacity of the blood and may be tumour associated or treatment related.
The presence of hypoxia within human tumours before starting treatment has been observed in a variety of tumour types including squamous cell carcinomas, gliomas, adenocarcinomas (breast & pancreas) and in sarcomas. For example, in the normal cervix the pO2 is a median of 42 mmHg compared to a median of 10 mmHg in squamous carcinomas, and for cervix cancer the oxygenation status is independent of size, stage, histopathological type, and grade of malignancy [5]. Oxygen probes, that is, electrodes implanted directly into tumours to measure oxygen concentration by a polarographic technique [6–8] have shown (1) heterogeneity within and between the same tumour types of oxygen concentration and, (2) that hypoxia contributes to poor prognosis; pO2 < 10 mmHg results in poor local tumour control, disease-free survival and overall survival in squamous carcinomas of the head and neck and of the cervix [9, 10].
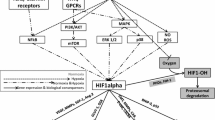
A large body of clinical evidence suggests that the hypoxia-mediated aggressive behavior of cancer cells and their resistance to therapy is orchestrated by the heterodimeric transcription factor, hypoxia inducible factor-1 alpha (HIF-1α), via a number of molecular events required for the adaptation of tumour cells to hypoxia (including unregulated glycolysis, angiogenesis and mutant p53) [11]. It is also important to realise that in some tumours including uterine lieomyomas, HIF expression is not always correlated with the presence of hypoxia suggesting that other factors including genetic events also contribute to activation of HIF, the most significant one of which is the loss of function of the Von Hippel-Lindau (VHL) tumour suppressor protein which results in constitutive activation of the HIF pathway. HIF-1 controls the expression of a variety of genes, the protein products of which play crucial roles in the acute and chronic adaptation of tumour cells to oxygen deficiency, including enhanced erythropoiesis & glycolysis, promotion of cell survival, inhibition of apoptosis, inhibition of cell differentiation, and angiogenesis. Thus, adaptive changes in the proteome and genome of neoplastic cells result in the emergence of more aggressive clones which are of cells that are more able to overcome nutrient deprivation or escape their hostile environments. Selection pressures by hypoxia and clonal expansion of the more aggressive cell types can result in exacerbations of regional hypoxia, further promoting the development of cell phenotypes that are treatment resistant (Fig. 1). Given the central role of HIF-1 in hypoxia mediated aggressive behaviour of cancer cells and their resistance to therapy, HIF-1 has become a target for the development of anti-cancer drugs [12].

Stylised diagram showing how hypoxia leads to therapy resistance and the development of an aggressive tumour cell phenotype. Figure adapted from [4]
There is debate about whether there is a critical intratumoural pO2 below which detrimental changes begin to occur that is common across cell types. This occurs because experiments performed in cell cultures may not be applicable to in-vivo environments and some of the literature variation can be attributed to the tumour cell type chosen for experiments and the demands of host tissues. With these caveats in mind, the critical pO2 tensions below which cellular functions progressively cease or anticancer treatments are impaired are approximately as follows [13]: Effectiveness of immunotherapy becomes impaired (30–35 mmHg); Photodynamic therapy (15–35 mmHg); Cell death on exposure to radiation (25–30 mmHg); Binding of hypoxia immunohistochemical markers (10–20 mmHg); Proteome changes (1–15 mmHg) and Genome changes (0.2–1 mmHg). The differences in these numbers are smaller than the similarities so that, from a practical perspective, for solid tissue tumors in vivo, a value of between 5–15 mmHg is a good number to remember because of its impact on therapy. This number is in contrast to ischaemic hypoxia in the myocardium or stroke where detrimental effects are experienced at higher O2 [14]. In all these instances the critical oxygen level in tissues reflects the drive to match delivery with metabolic demand.
As noted above, the presence of tumour hypoxia appears to impair the effectiveness of radiotherapy and radiosensitivity is progressively limited as tumour pO2 levels fall. Hypoxia-induced radioresistance is multifactorial with the presence of oxygen mediating DNA damage through the formation of oxygen free radicals which occurs after the interaction of radiation with intracellular water. The ratio of doses administered under well-oxygenated to hypoxic conditions needed to achieve the same biological effect (i.e., cell kill) is called the oxygen enhancement ratio (OER). For sparsely ionising radiations such as x- and gamma rays, the OER at therapeutic doses is between 2.5–3.5 [15]. That is, well oxygenated cells are about three times more sensitive to x- and gamma radiation than the same cells when they are hypoxic. Half maximal sensitivity to x- and gamma rays occurs at oxygen tensions of approximately 2–5 mmHg; above pO2 values of approximately 10–15 mmHg near maximal oxygen effects are seen. However, it should be recognised that sensitivity of cells to radiation is dependent on the phase of the cell cycle, with cells in the G1 phase having a lower OER (i.e., more radiosensitive) than cells in S-phase. As noted above, the oxygen effect is not the only mechanism for radioresistance in hypoxic tumour cells. Evidence is accumulating that the hypoxia-mediated proteomic and genomic changes may also contribute to radioresistance by increasing the levels of heat shock proteins (heat shock proteins (HSPs), are induced in response to environmental stresses like heat, cold and oxygen deprivation [16]) or by increasing the number of tumour cells that can resist apoptosis by mutating p53 (the slowing of cell division is dependent on a protein brake known as p53; the disruption of the functioning of this protein is associated with approximately 50–55% of human cancers).
Clinical imaging of hypoxia
As tumour hypoxia is an important biological characteristic and there is no good or easy clinical way to predict its presence, it has been suggested that imaging may be a good way of non-invasively selecting cancer patients who would benefit from treatments that overcome, circumvent or take advantage of the presence of hypoxia. Since tumour hypoxia is a key mechanism that leads to radioresistance, it has been repeatedly suggested that a hypoxia mapping technique could be integrated with conformal radiotherapy techniques to improve target delineation and dose delivery; this is discussed in more detail below. Imaging could also be used to document whether or not and the extent to which reoxygenation of tumours occurs during radiotherapy. Key requirements of any method that evaluates tumour hypoxia include non-invasive assessments that allow serial changes during treatment to be monitored and evaluation of heterogeneity between and within tumours.
There are a number of ways in which tissue oxygenation status can be assessed in vivo (both invasive and non-invasive) or in vitro using material from biopsy. Non-imaging methods of assessing for the presence of hypoxia in tissues include histological appearance, immunohistochemical staining for intrinsic markers of hypoxia (e.g., carbonic anhydrase IX (CA-IX) and hypoxia inducible factor-1 (HIF-1)) and for the binding of externally administered nitroimidazoles [17, 18].
From an imaging perspective, an ideal test would: (1) distinguish normoxia / hypoxia /anoxia/necrosis, (2) distinguish between perfusion-related (acute) and diffusion-related (chronic) hypoxia if possible, (3) reflect cellular in preference to vascular pO2, (4) be applicable to any tumour site with complete loco-regional evaluation, (5) be simple to perform, non-toxic and allow repeated measurements, and (6) be sensitive at pO2 levels relevant to tumour therapy. Therefore, the challenge for hypoxia imaging is to measure low levels of tissue pO2 on a spatial scale similar to the O2-diffusion distance (70–100 μm); a much smaller dimension than can be achieved with human imaging techniques. Currently available MRI and PET methods were compared at a National Institute of Health/National Cancer Institute of the USA sponsored workshop in April 2004 and it was noted that only a few techniques have potential for in vivo assessment in humans particularly for repeated, sequential measurements. 18F-MISO and 60/64Cu-ATSM PET, and BOLD-MRI are the lead contenders for human application based on their non-invasive nature, ease of use and robustness, measurement of hypoxia status, validity, ability to demonstrate heterogeneity and general availability (Table 1).
18F-MISO
[18F]Fluoromisonidazole, 3-fluoro-1-(2′-nitro-1′-imidazolyl)-2-propanol or (18F-MISO), is the prototype hypoxia imaging agent whose uptake is homogeneous in most normal tissues, reflecting its high partition coefficient that nears unity, and whose delivery to tumours is not limited by perfusion [19]. The initial distribution of 18F-MISO is flow dependent, as with any freely diffusible tracer, but local oxygen tension is the major determinant of its retention above normal background in tissues after 2 hours (Fig. 2). 18F-MISO accumulates in tissues by binding to intracellular macromolecules when pO2 < 10 mmHg. Retention within tissues is dependent on nitroreductase activity (that is, on reduction status of a NO2 group on the imidazole ring) (Fig. 3) and accumulation in hypoxic tissues over a range of blood flows has been noted, including within the intestinal lumen where it is retained in anaerobes!

The 18FDG-PET image (bottom left panel) shows increase uptake in both the oropharyngeal tumour (arrow) and in the left neck nodal metastasis (asterix). The 18F-MISO images (bottom right panel) were acquired in a dynamic mode and representative images after 1 minute, 30 minutes and 240 minutes are shown together with time-activity curves from the two regions of interest indicated in the FDG-PET image. The early distribution (1 minute) shows hyperperfusion in the region of the primary tumour and metastasis because of the high partition coefficient of 18F-MISO. After 2 hours, only the left neck nodal metastasis is shown to be hypoxic

The structure of [18F]-fluoromisonidazole, 18F-MISO, and its mechanism of retention in hypoxic tissues. The partition coefficient of 18F-MISO is near unity so the molecule diffuses freely into all cells. Once 18F-MISO is in an environment where electron transport is occurring (viable tissues), the –NO2 substituent (which has a high electron affinity) takes on an electron to form the radical anion reduction product. If O2 is also present, that electron is rapidly transferred to oxygen and 18F-MISO changes back to its original structure and can leave the cell. However, if a second electron from cellular metabolism reacts with the nitroimidazole to form the 2-electron reduction product, the molecule reacts non-discriminately with peptides and RNA within the cell and becomes trapped. Thus, retention of FMISO is inversely related to the intracellular partial pressure of O2 as shown in the lower left panel [45]. This mechanism is confirmed by the autoradiograph of a tumour spheroid (bottom right panel) with a radius of approximately 0.5 mm that shows no retention in the necrotic core or in the well-oxygenated outer sphere but intense uptake (white spots) in a donut like ring where cells are hypoxic
Hypoxia can be imaged with 18F-MISO PET in a procedure that is well-tolerated by the patients. Imaging requires 20–30 min and starts anywhere from 75 to 150 min after injection, making it similar to the bone scan with which most cancer patients are familiar. Useful and well-validated images can be achieved with a modest dose of radiation, typically 250 MBq. No arterial sampling or metabolite analysis is required and synthesis is achieved through relatively simple modifications of nucleophilic displacement / deprotection synthesis boxes such as are used for fluoro-deoxyglucose (18F-FDG). In the USA, F-MISO has investigational new drug (IND) authorisation from the Food and Drug Administration (FDA) as an investigational product for use in humans. Unlike Eppendorf pO2 histography, 18F-MISO is only sensitive to the presence of hypoxia in viable cells; 18F-MISO is not retained in necrosis because the electron transport chain that reduces the nitroimidazole to a bioreductive alkylating agent is no longer active (Fig. 3). Limitations of 18F-MISO PET include the modest signal-to-noise ratio of raw 18F-MISO PET images but if a venous blood sample is acquired during the mid-course of the imaging procedure and used to calculate a Tumour:Blood (T/B) ratio image, then normoxic uptake (T/B < 1) can be electronically subtracted to increase image contrast. Several studies in a range of hypoxic tumours, stroke and hypoxic myocardium [14] have shown that a T/B of >1.2 reliably identifies the presence of hypoxia. The presence of high normal liver uptake impairs complete assessment of liver lesions and urinary excretion interferes with imaging near the bladder.
18F-MISO PET is able to monitor the changing hypoxia status of lung tumours during radiotherapy [20]. Studies in sarcoma [21] and head and neck cancer [22–24] have demonstrated a correlation of 18F-FMISO uptake with poor outcome to radiation and chemotherapy.
Cu-ATSM
Cu-diacetyl-bis(N4-methylthiosemicarbazone) (Cu-ATSM) holds exceptional promise as an agent for delineating the extent of hypoxia within tumours with PET. Numerous pre-clinical studies have evaluated and validated its use for imaging of hypoxia in tumours and other tissues [25–32]. The mechanism of retention of the reagent in hypoxic tissues is largely attributed to the low oxygen tensions and the subsequent altered redox environment of hypoxic tumours (increased NADH levels) (Fig. 4). Clinical studies, well-tolerated by patients, involved 60Cu-ATSM imaging sessions of about 60 minutes with analysis of 30–60 minute summed-images. This time frame not only yields excellent data with good image quality (Fig. 4) in a very short time frame which opens up the opportunity with the shorter-lived 60Cu to perform multiple imaging sessions. A number of radioactive copper isotopes with longer half lives are available, e.g. 64Cu (t1/2 = 12.74 h) [33], enabling wide geographic distribution and the United States FDA recently approved an IND application for the study of 64Cu-ATSM for the imaging of hypoxia in human tumours.

Retention mechanism of Cu(II)ATSM in hypoxic tissues. (a) Cu(II)ATSM is bioreduced (Cu(II) to Cu(I)) once entering the cell. The reduced intermediate species (likely to be [Cu-ATSM]-) is trapped within the cell because of its charge. This transient complex can then go through one of two competing pathways: reoxidation to the uncharged Cu(II) species (which can escape by diffusion), or proton-induced dissociation (which releases copper to be irreversibly sequestered by intracellular proteins). [Cu-ATSM]- favours the reoxidation route because it is easily oxidised but chemically more resistant to protonation. Copper from Cu(II) ATSM is trapped reversibly as [Cu-ATSM]- (if oxygen is absent), with the possibility of irreversible trapping by dissociation over a longer period. Cu(II)ATSM is thus hypoxia-selective. (b) High quality axial 60Cu-ATSM-PET image through the mid-upper thorax demonstrates heterogeneously increased uptake (arrow) within a known lung cancer in the aorto-pulmonary window and left suprahilar region. PET images representing summed data were obtained from 30 to 60 minutes after injection of 60Cu-ATSM
In human studies of lung [34] and cervix cervical cancers [35], encouraging evidence has emerged that 60Cu-ATSM can act as a prognostic indicator for response to therapy. In the prospective study of 14 humans with non-small cell lung cancer, a semi-quantitative analysis of the 60Cu-ATSM muscle-to-tumour ratio was able to discriminate those likely to respond to therapy from non-responders [34]. A similar study in 14 women with cervical cancer demonstrated a similar predictive value in the tumour response to therapy [35].
BOLD-MRI
Blood oxygenation level dependent (BOLD) and intrinsic susceptibility weighted MRI are interchangeable terms. As in any MR image, tissue contrast in BOLD images is affected by intrinsic tissue properties including spin-lattice and spin-spin relaxations. Additionally, BOLD MRI contrast is affected by blood flow and paramagnetic deoxyhaemoglobin within red blood cells (oxyhaemoglobin is not paramagnetic). Deoxyhaemoglobin increases the MR transverse relaxation rate (R2*) of water in blood and surrounding tissues thus BOLD-MRI is sensitive to pO2 within, and in tissues adjacent to perfused vessels [36]. Static tissue components include iron content (e.g. myoglobin found in muscle) and presence of fibrosis or ligamentous structures (e.g. in benign prostatic hyperplasia and the suspensory ligaments of the breasts) also affect the appearances of intrinsic susceptibility weighted images. In order to decouple the effects of flow from deoxyhaemoglobin and static components it is necessary to measure the T2* relaxation rate (R2* = 1/ T2*) which can be done by using a multi-echo GRE sequence (Fig. 5). Decoupling of flow from static effects on R2* images occurs because the flow component can be thought of as affecting individual T2* images of a multi-gradient echo sequence equally. It is important to remember that, although synthetic R2* images are free of the contribution of blood flow (that is, they mainly reflect deoxyhaemoglobin content and static tissue components), improving blood flow and vascular functioning will also increase tissue oxygenation, which can be seen by changes in R2* images.

Data acquisition and quantification of BOLD-MRI in the prostate gland. Gradient recalled-echo MR images acquired at 1.5 T with a fixed repetition time and flip angle (TR ~100 msec; alpha 40 degrees) with lengthening echo-time (TE) are acquired through the prostate gland (bottom row of images). These images show increased susceptibility (T2*) effects with increasing TE. The rate of signal intensity decay in the dorsal aspect of the prostate is dependent on intrinsic T2* relaxation (local structure), deoxyhaemoglobin concentration [dHb] and local blood flow. Synthetic R2* (=1/T2*) images are created by plotting the natural logarithm of the signal intensity against the TE (top left panel). R2* map (top right panel) reflects on the structure of tissues and local [dHb] but inflow effects are minimised; however, R2* maps retain sensitivity to pO2 changes caused by alterations in blood flow
The observations made in the previous paragraph imply two intuitive inferences. (1) BOLD-MRI images are more likely to reflect on acute (perfusion-related) tissue hypoxia which, as stated above, occurs because of transient occlusions of vessels, simply because hypoxic areas extend to the level of the blood vessels. In contradistinction, chronic hypoxia is less likely to be reflected by BOLD-MRI because the red blood cells in vessels are too distant from the area of hypoxia. (2) For BOLD-MRI to be able to inform on tissue oxygenation status, it is important for red blood cells to be delivered to the tissue in question. Human and xenograft studies have shown that tumour perfusion varies widely and that red blood cell perfusion is not simply related to the absence/presence of vessels; plenty of tumour vessels maybe present but perfusion by red blood cells may not occur [37]. This observation probably explains in part why no direct correlations between baseline R2* and tissue pO2 have been observed (that is, R2* does NOT measure tissue pO2). So it is necessary to know or to determine the distribution of blood volume in tissue in order to be able to correctly interpret R2* images in order to infer oxygenation status. Thus, if a tissue is perfused but has a high baseline R2* in one area/region compared to another area/region in the same tissue (i.e. the statistic components are the same), then one can infer that the high R2* region is relatively more hypoxic; this hypothesis is supported by recent preclinical and clinical data [38, 39].
As stated above, the use of BOLD-MRI for assessment of tissue hypoxia is predicated on the assumption that the oxygenation of haemoglobin is proportional to blood arterial pO2 which is in equilibrium with oxygenation of surrounding tissues. Many studies have shown that changes in R2* in response to vasomodulation with Carbogen (95% CO2:5% O2) inhalation, for example, are temporally correlated with changes in tissue pO2. Tumours differ in their responses to carbogen inhalation with only 50–60% of human tumours showing changes in R2* [40, 41]. The reasons for these limited and heterogeneous responses are complex but undoubtedly include the fact that tumours have adapted to widely different perfusion and that, even when vessels are present, red blood cell transport along these vessels may not be effective as demonstrated by Robinson et al. [37]. Thus, hypoxic tumours with high blood volume (due to high microvessel density coupled with large vessels) will not only have raised baseline R2* values but are more likely to respond to Carbogen. This will be reflected by large changes in R2*; and it is these hypoxic tumours that show positive radiosensitisation with Carbogen. On the other hand, hypoxic tumours with low blood volume (due to lower microvessel density, or due to small vessels) will have lower baseline R2* values and are thus less likely to respond to Carbogen. In this situation, there will be negligible changes in R2* and such hypoxic tumours do not show radiosensitisation with Carbogen [42]. Readers should also note that the BOLD response to Carbogen is also dependent on the ability of the underlying maturity of the vasculature with mature vessels able to respond actively to vasoconstrictory and vasodilatory stimuli [43].
The primary advantages of BOLD-MRI are that there is no need to administer exogenous radioactive contrast material and images at high temporal and with high spatial resolution can be obtained and repeated as needed. It is possible to decouple the effects of flow and deoxyhaemoglobin which are seen in native BOLD images and so to demonstrate changes in oxygenation independent of changes in blood flow. Major limitations of BOLD-MRI include the fact that they do not measure tissue pO2 directly (either in blood or tissues because of a non-linear relationship of R2* and tissue pO2), the images obtained have low signal to noise ratio and clinical studies with Carbogen vasomodulation are technically challenging (approximately 25–35% of patient examinations fail due to respiratory distress caused by an increased respiratory drive induced by Carbogen [40, 41]. BOLD-MRI appears most sensitive to oxygen levels adjacent to perfused vessels (that is, perfusion related or acute hypoxia) and BOLD-MRI sensitivity to more distant diffusion related or chronic hypoxia is an unknown.
Hypoxia guided radiotherapy
Recently introduced, technological improvements in radiotherapy delivery systems, including intensity-modulated radiotherapy (IMRT), have provided a means for shaping the dose distribution not only to the geometry of target volumes, and also to the differences in radiobiology across tumours [44]. Thus, it is now possible to define an additional “target within the target” as 3D pixel maps of the prescribed dose incorporating biological information derived from functional images; sometimes called dose painting by numbers [45, 46]. This allows treatment to the desired dose with escalation based on biologically relevant data, such as hypoxia which was discussed above, that is mechanistically related to therapeutic outcome. As an example of this, the spatial distribution of 60/64Cu-ATSM uptake on PET was successfully fused with CT radiotherapy planning images to show a proof of concept. This theoretical treatment planning would deliver higher doses of radiation via intensity modulated radiotherapy techniques (IMRT) to the most hypoxic regions of head and neck tumours [47].
In the case of hypoxia, it is important to remember that if the tumour stays hypoxic throughout the course of a treatment, more (escalating) radiation alone may not be sufficient to overcome the hypoxia-induced resistance. Hence, from a therapeutic perspective it is additionally important to know when and if reoxygenation occurs after initial radiotherapy (either by a reduction of oxygen consumption by the arrest of proliferation or by death of well oxygenated cells [15] (Fig. 6). However, data correlating treatment success with hypoxia and reoxygenation changes occurring during treatment are sparse; the extent and rapidity of reoxygenation is impossible to predict for individual tumours. The complexity of the processes leading to reoxygenation suggests the need for repeated imaging during the initial phase of treatment to determine the best time for dose adaptation. Koh et al. used 18F-MISO PET imaging to detect reoxygenation of some lung tumours after just a few treatments, whereas other patients exhibited no reduction in hypoxia even over the full course of radiotherapy [20]. Thus, dose escalation to target hypoxic areas in all patients at the beginning of the treatment could be a wasted effort because reoxygenation would change its distribution. Dose escalation is possibly best undertaken towards the end of treatment, at a time when hypoxic radioresistant cells are predominate.

The re-oxygenation phenomenon. Tumours contain mixtures of aerated and hypoxic cells. Radiation is effective at eliminating well oxygenated cells because they are radiosensitive. Reoxygenation cause the preradiation pattern to return which can be eliminated by further radiation fractions but a progressive decrease in the tumour mass occurs after a series of fractions. Figure adapted from [15]
Recently, Thorwarth et al. presented a study where dynamic (mapping of perfusion) and static (mapping of hypoxia) 18F-MISO scans were obtained in 15 patients with head-and-neck cancers who were subsequently treated with chemoradiation [48]. Their data suggested that subsequent treatment failure was related to both pretherapy hypoxia and concurrent poor perfusion. This result hints that reoxygenation did not occur as a consequence of the deficient vasculature of tumours [49]. Thus, it may be necessary not only to target hypoxic regions but also areas of poor blood flow. Dose escalation maps can be derived from dynamic 18F-MISO PET scans for targeting poor perfusion, while functional planning target volumes could be obtained from late static scans (that reflect hypoxia); both concepts lend themselves equally well to IMRT (Fig. 7). As a caution, simulated treatment plans also suggest that some patients may require such high radiation doses that will lead to a high risk of late complications in the vicinity of the tumour bed, such as necrosis, rupture of blood vessels or excessive fibrosis.

Dose escalation map superimposed on CT with Planning Target Volume (red). Isodose lines show conformality to hypoxic lymph node with a maximum dose increase by 20%. This is the same patient illustrated in Fig. 2
Challenges for hypoxia imaging techniques
As hypoxia imaging techniques move from academic research environments to routine clinical usage, it becomes important to recognise the unique challenges of clinical translation. For example, it is important that patient examination times are short to improve patient compliance particularly for repeated examinations. Thus, the need for doing dynamic scanning followed by several static scans over a prolonged period of time with PET techniques could prove a disincentive for patients. However, as noted above, not all patients undergoing hypoxia studies by PET need dynamic imaging although it may be useful in selected patients where the aim is to demonstrate both perfusion and hypoxia (for example to demonstrate reperfusion-reoxygenation). In-contradistinction, the interpretation of BOLD-MRI does require that the distribution of blood flow/volume is known and this can be done in clinical studies using dynamic contrast enhanced MRI (DCE-MRI) [50]. Whatever the chosen technique for clinical translation, there needs to be standardisation of imaging procedures and analysis methods in order to allow techniques to become more completely validated, for use in clinical trials. Amongst other issues that require addressing when clinical trials are being designed include the need for quantification, test-retest variability and data collection in body parts where there is a large degree of physiological movement such as the lungs and liver.
A practical question often asked is whether it is necessary to quantify imaging data to answer important clinical questions. Subjective assessments work well enough in the clinic; however it is important to realise that subjective criteria cannot be applied simply from one centre to another particularly, when different equipment and imaging routines and human observers are used. Quantification techniques aim to minimise errors that can result from the use of different equipment and imaging protocols. Quantification techniques also enable the derivation of parameters that are based on some understanding of physiological processes and so can provide insights into tumour biology, for example the simple T/B and T/M ratios described for the two PET procedures. Quantification techniques are preferred when serial imaging studies are anticipated, for example when evaluating response to novel anticancer therapeutics.
The reproducibility of the imaging technique should also be known in order to estimate the sample size required to evaluate therapy efficacy. Variation between measurements of the same quantity on the same individual can be caused either by measurement error or by physiological changes between measurements. Whilst it is possible (in theory) to reduce measurement error, physiological variation is inherent, and can cause difficulty in attempts to characterise disease or to monitor the effects of therapy. An estimate of measurement error enables us to decide whether a change in observation represents a real change. Data addressing the precision and measurement variability of hypoxia imaging techniques are urgently needed and should be an integral part of any prospective study that evaluates functional response to therapy to allow assessments of individual patients and group changes.
It is intuitive that analysis and presentation of imaging data needs to take into account the heterogeneity of tumour hypoxia. The presence of motion can invalidate functional parameter estimates particularly for pixel-by-pixel analyses and this is especially true for high spatial resolution techniques such as BOLD-MRI. Motion is averaged in PET imaging because imaging times are long. In that case, pixel-analyses of the data and the issues of heterogeneity assessment can become less meaningful. The first step in heterogeneity analysis includes ROI definition which should be performed independent on the hypoxia imaging being assessed. For BOLD-MRI this could be done by anatomic MRI images and for PET studies could include the CT component of CT-PET studies although some groups have used ROIs defined in 18FDG-PET images. However, ROIs defined on 18FDG-PET images are know to be prone to error as far as tumour boundaries are concerned and are highly dependent on the level of threshold chosen. Whole tumour ROIs yield outputs with good signal-to-noise ratio, but lack spatial resolution and are prone to partial volume averaging errors and thus are unable to evaluate tumour heterogeneity. Pixel mapping has the advantages of an improved appreciation of heterogeneity of hypoxia and the risk of missing important diagnostic information and of creating ROIs that contain more than one tissue type is reduced. An important advantage of pixel mapping is being able to spatially map tumour characteristics such as hypoxia, glucose metabolism and blood flow and to be able to probe the spatial correlations between different kinetic parameters, providing unique insights into tumour structure, function and response to treatment.
Conclusions
To summarise, tumour hypoxia is common and its effects represents a significant challenge to the curability of human tumours, leading to treatment resistance and enhanced tumour progression. Tumour hypoxia can be detected by non-invasive and invasive techniques but the inter-relationship between these techniques needs to be better defined; human validation of the utility of hypoxia imaging is sparse at best. Anti-hypoxia therapies exist in the clinic and more are on their way. Either they don’t work very well or we don’t know how to use them optimally. Hypoxia imaging may allow better definition of a sub-population of cancer patients that would benefit for novel anti-hypoxia directed therapies.
References
Gray LH, Conger AD, Ebert M (1953) The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br J Radiol 26:638–648
Vaupel P (2004) The role of hypoxia-induced factors in tumor progression. Oncologist 9:10–17
Brown JM (1999) The hypoxic cell: A target for selective cancer therapy-Eighteenth Bruce F. Cain Memorial Award Lecture. Cancer Res 59:5863–5870
Vaupel P, Harrison L (2004) Tumor hypoxia: Causative factors, compensatory mechanisms, and cellular response. Oncologist 9:4–9
Vaupel P, Kelleher DK, Hockel M (2001) Oxygen status of malignant tumors: pathogenesis of hypoxia and significance for tumor therapy. Semin Oncol 28:29–35
Kallinowski F, Zander R, Hoeckel M, Vaupel P (1990) Tumor tissue oxygenation as evaluated by computerized-pO2-histography. Int J Radiat Oncol Biol Phys 19:953–961
Vaupel P, Schlenger K, Knoop C, Hockel M (1991) Oxygenation of human tumors: evaluation of tissue oxygen distribution in breast cancers by computerized O2 tension measurements. Cancer Res 51:3316–3322
Brizel DM, Rosner GL, Harrelson J, Prosnitz LR, Dewhirst MW (1994) Pretreatment oxygenation profiles of human soft tissue sarcomas. Int J Radiat Oncol Biol Phys 30:635–642
Hockel M, Schlenger K, Aral B, Mitze M, Schaffer U, Vaupel P (1996) Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res 56:4509–4515
Nordsmark M, Bentzen SM, Rudat V, Brizel D, Lartigau E, Stadler P, Becker A, Adam M, Molls M, Dunst J, Terris DJ, Overgaard J (2005) Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother Oncol 77:18–24
Powis G, Kirkpatrick L (2004) Hypoxia inducible factor-1{alpha} as a cancer drug target. Mol Cancer Ther 3:647–654
Belozerov VE, Van Meir EG (2005) Hypoxia inducible factor-1: a novel target for cancer therapy. Anticancer Drugs 16:901–909
Hockel M, Vaupel P (2001) Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst 93:266–276
Caldwell JH, Revenaugh JR, Martin GV, Johnson PM, Rasey JS, Krohn KA (1995) Comparison of fluorine-18-fluorodeoxyglucose and tritiated fluoromisonidazole uptake during low-flow ischemia. J Nucl Med 36:1633–1638
Hall EJ, Giaccia AJ (2006) Oxygen effect and reoxygenation. In: Hall EJ, Giaccia AJ (eds) Radiobiology for the Radiologist. Lippincott Williams & Wilkins, Philadelphia, pp 85–105
Ciocca DR, Calderwood SK (2005) Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 10:86–103
Bussink J, Kaanders JH, van der Kogel AJ (2003) Tumor hypoxia at the micro-regional level: clinical relevance and predictive value of exogenous and endogenous hypoxic cell markers. Radiother Oncol 67:3–15
Williams KJ, Parker CA, Stratford IJ (2005) Exogenous and endogenous markers of tumour oxygenation status: definitive markers of tumour hypoxia? Adv Exp Med Biol 566:285–294
Rajendran JG, Krohn KA (2005) Imaging hypoxia and angiogenesis in tumors. Radiol Clin North Am 43:169–187
Koh WJ, Bergman KS, Rasey JS, Peterson LM, Evans ML, Graham MM, Grierson JR, Lindsley KL, Lewellen TK, Krohn KA et al (1995) Evaluation of oxygenation status during fractionated radiotherapy in human nonsmall cell lung cancers using [F-18]fluoromisonidazole positron emission tomography.Int J Radiat Oncol Biol Phys 33:391-398
Rajendran JG, Wilson DC, Conrad EU, Peterson LM, Bruckner JD, Rasey JS, Chin LK, Hofstrand PD, Grierson JR, Eary JF, Krohn KA (2003) [(18)F]FMISO and [(18)F]FDG PET imaging in soft tissue sarcomas: correlation of hypoxia, metabolism and VEGF expression. Eur J Nucl Med Mol Imaging 30:695–704
Ng P, Rajendran JG, Schwartz DL, Patterson LM, Scharnhorst J, Krohn KA (2003) Can [F-18] fluoromisonidazole PET imaging predict treatment response in head and neck cancer? J Nucl Med 44:128P
Rajendran JG, Mankoff DA, O’Sullivan F, Peterson LM, Schwartz DL, Conrad EU, Spence AM, Muzi M, Farwell DG, Krohn KA (2004) Hypoxia and glucose metabolism in malignant tumors: evaluation by [18F]fluoromisonidazole and [18F]fluorodeoxyglucose positron emission tomography imaging. Clin Cancer Res 10:2245–2252
Hicks RJ, Rischin D, Fisher R, Binns D, Scott AM, Peters LJ (2005) Utility of FMISO PET in advanced head and neck cancer treated with chemoradiation incorporating a hypoxia-targeting chemotherapy agent. Eur J Nucl Med Mol Imaging 32:1384–1391
Fujibayashi Y, Taniuchi H, Yonekura Y, Ohtani H, Konishi J, Yokoyama A (1997) Copper-62-ATSM: A new hypoxia imaging agent with high membrane permeability and low redox potential. J Nucl Med 38:1155–1160
Dearling JLD, Lewis JS, Mullen GED, Rae MT, Zweit J, Blower PJ (1998) Design of hypoxia-targeting radiopharmaceuticals: Selective uptake of copper-64 complexes in hypoxic cells in vitro. Eur J Nucl Med 25:788–792
Dearling JLJ, Lewis JS, Welch MJ, McCarthy DW, Blower PJ (1998) Redox-active complexes for imaging hypoxic tissues: Structure-activity relationships in copper(II)bis(thiosemicarbazone) complexes. Chem Commun 22:2531–2533
Lewis JS, McCarthy DW, McCarthy TJ, Fujibayashi Y, Welch MJ (1999) The evaluation of 64Cu-diacetyl-bis(N 4-methylthiosemicarbazone)(64Cu-ATSM) in vivo and in vitro in a hypoxic tumor model. J Nucl Med 40:177–183
Lewis JS, Sharp TL, Laforest R, Fujibayashi Y, Welch MJ (2001) Tumor uptake of copper-diacetyl-bis(N4-methylthiosemicarbazone): Effect of changes in tissue oxygenation. J Nucl Med 42:655–661
Dearling JL, Lewis JS, Mullen GE, Welch MJ, Blower PJ (2002) Copper bis(thiosemicarbazone) complexes as hypoxia imaging agents: structure-activity relationships. J Biol Inorg Chem 7:249–259
Lewis JS, Herrero P, Sharp T, Engelbach JA, Fujibayashi Y, Laforest R, Kovacs A, Gropler RJ, Welch MJ (2002) Delineation of hypoxia in canine myocardium using PET and copper(II)-diacetyl-bis(N4-methylthiosemicarbazone). J Nucl Med 43:1557–1569
Maurer RI, Blower PJ, Dilworth JR, Reynolds CA, Zheng Y, Mullen GED (2002) Studies on the mechanism of hypoxic selectivity in copper bis(thiosemicarbazone) radiopharmaceuticals. J Med Chem 45:1420–1431
Blower PJ, Lewis JS, Zweit J (1996) Copper radionuclides and radiopharmaceuticals in nuclear medicine. Nucl Med Biol 23:957–980
Dehdashti F, Mintun MA, Lewis JS, Bradley J, Govindan R, Laforest R, Welch MJ, A SB (2003) In vivo assesment of tumor hypoxia in lung cancer with 60Cu-ATSM. Eur J Nucl Med Mol Imaging 30:844–850
Dehdashti F, Grigsby PW, Mintun MA, Lewis JS, Siegel BA, Welch MJ (2003) Assessing tumor hypoxia in cervical cancer by positron emission tomography with 60Cu-ATSM: relationship to therapeutic response-a preliminary report. Int J Radiat Oncol Biol Phys 55:1233–1238
Howe FA, Robinson SP, McIntyre DJ, Stubbs M, Griffiths JR (2001) Issues in flow and oxygenation dependent contrast (FLOOD) imaging of tumours. NMR Biomed 14:497–506
Robinson SP, Rijken PF, Howe FA, McSheehy PM, Van Der Sanden BP, Heerschap A, Stubbs M, Van Der Kogel AJ, Griffiths JR (2003) Tumor vascular architecture and function evaluated by non-invasive susceptibility MRI methods and immunohistochemistry. J Magn Reson Imaging 17:445–454
Kostourou V, Robinson SP, Whitley GS, Griffiths JR (2003) Effects of overexpression of dimethylarginine dimethylaminohydrolase on tumor angiogenesis assessed by susceptibility magnetic resonance imaging. Cancer Res 63:4960–4966
Carnell DM, Taylor NJ, Smith RE, Hoskin PJ, Stirling JJ, d’Arcy J, Walker-Samuel S, Collins DJ, Leach MO, Padhani AR (2005) Evaluation of prostate gland hypoxia with quantified BOLD MRI: updated results from a correlated histological study. Miami, pp 270
Taylor NJ, Baddeley H, Goodchild KA, Powell ME, Thoumine M, Culver LA, Stirling JJ, Saunders MI, Hoskin PJ, Phillips H, Padhani AR, Griffiths JR (2001) BOLD MRI of human tumor oxygenation during carbogen breathing. J Magn Reson Imaging 14:156–163
Rijpkema M, Kaanders JH, Joosten FB, van der Kogel AJ, Heerschap A (2002) Effects of breathing a hyperoxic hypercapnic gas mixture on blood oxygenation and vascularity of head-and-neck tumors as measured by magnetic resonance imaging. Int J Radiat Oncol Biol Phys 53:1185–1191
Rodrigues LM, Howe FA, Griffiths JR, Robinson SP (2004) Tumor R2* is a prognostic indicator of acute radiotherapeutic response in rodent tumors. J Magn Reson Imaging 19:482–488
Neeman M (2002) Functional and molecular MR imaging of angiogenesis: Seeing the target, seeing it work. J Cell Biochem Suppl 39:11–17
Ling CC, Humm J, Larson S, Amols H, Fuks Z, Leibel S, Koutcher JA (2000) Towards multidimensional radiotherapy (MD-CRT): biological imaging and biological conformality. Int J Radiat Oncol Biol Phys 47:551–560
Brahme A (2003) Biologically optimized 3-dimensional in vivo predictive assay-based radiation therapy using positron emission tomography-computerized tomography imaging. Acta Oncol 42:123–136
Bentzen SM (2005) Theragnostic imaging for radiation oncology: dose-painting by numbers. Lancet Oncol 6:112–117
Chao C, Bosch WR, Mutic S, Lewis JS, Dehdashti FD, Mintun MA, Demsey JF, Perez CA, Purdy JA, Welch MJ (2001) A novel approach to overcome hypoxic tumor resistance: Cu-ATSM-guided intensity-modulated radiation therapy. Int J Radiat Biol Phys 49:1171–1182
Thorwarth D, Eschmann SM, Scheiderbauer J, Paulsen F, Alber M (2005) Kinetic analysis of dynamic 18F-fluoromisonidazole PET correlates with radiation treatment outcome in head-and-neck cancer. BMC Cancer 5:152
Ljungkvist AS, Bussink J, Rijken PF, Kaanders JH, van der Kogel AJ, Denekamp J (2002) Vascular architecture, hypoxia, and proliferation in first-generation xenografts of human head-and-neck squamous cell carcinomas. Int J Radiat Oncol Biol Phys 54:215–228
Collins DJ, Padhani AR (2004) Dynamic magnetic resonance imaging of tumor perfusion. Approaches and biomedical challenges. IEEE Eng Med Biol Mag 23:65–83
Koh WJ, Rasey JS, Evans ML, Grierson JR, Lewellen TK, Graham MM, Krohn KA, Griffin TW (1992) Imaging of hypoxia in human tumors with [F-18]fluoromisonidazole. Int J Radiat Oncol Biol Phys 22:199–212
Rasey JS, Koh WJ, Evans ML, Peterson LM, Lewellen TK, Graham MM, Krohn KA (1996) Quantifying regional hypoxia in human tumors with positron emission tomography of [18F]fluoromisonidazole: a pretherapy study of 37 patients. Int J Radiat Oncol Biol Phys 36:417–428
Eschmann SM, Paulsen F, Reimold M, Dittmann H, Welz S, Reischl G, Machulla HJ, Bares R (2005) Prognostic impact of hypoxia imaging with 18F-misonidazole PET in non-small cell lung cancer and head and neck cancer before radiotherapy. J Nucl Med 46:253–260
Piert M, Machulla HJ, Picchio M, Reischl G, Ziegler S, Kumar P, Wester HJ, Beck R, McEwan AJ, Wiebe LI, Schwaiger M (2005) Hypoxia-specific tumor imaging with 18F-fluoroazomycin arabinoside. J Nucl Med 46:106–113
Evans SM, Kachur AV, Shiue CY, Hustinx R, Jenkins WT, Shive GG, Karp JS, Alavi A, Lord EM, Dolbier WR Jr, Koch CJ (2000) Noninvasive detection of tumor hypoxia using the 2-nitroimidazole [18F]EF1. J Nucl Med 41:327–336
Acknowledgements
The hypoxia imaging studies in Seattle were initiated in collaboration with Janet Rasey. The work described in this review was by Joseph Rajendran and Janet Eary, whom the authors gratefully acknowledge, along with funding from NIH, CA-42045. The BOLD-MRI work was done in collaboration with Jane Taylor & Peter Hoskin (Mount Vernon Hospital, London) and Simon Robinson (Institute of Cancer Research, London). The Cu-ATSM studies described in this review was undertaken with Farrokh Dehdashti and her collaborators, Perry W. Grigsby, Jeffrey Bradley, Ramaswamy Govindan, Michael J. Welch. Mark Mintun and Barry A. Siegel. The development of Cu-ATSM as a hypoxia imaging agent was supported by the US Department of Energy (DE-FG02-87ER60512). The Image guided radiotherapy work in Tuebingen is done in collaboration with Daniella Thorwarth and SM Eschmann.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 2.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by-nc/2.0/.
About this article
Cite this article
Padhani, A.R., Krohn, K.A., Lewis, J.S. et al. Imaging oxygenation of human tumours. Eur Radiol 17, 861–872 (2007). https://doi.org/10.1007/s00330-006-0431-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00330-006-0431-y