
Abstract
Herbivory by barnacle geese (Branta leucopsis) alters the vegetation cover and reduces ecosystem productivity in high-Arctic peatlands, limiting the carbon sink strength of these ecosystems. Here we investigate how herbivory-induced vegetation changes affect the activities of peat soil microbiota using metagenomics, metatranscriptomics and targeted metabolomics in a comparison of fenced exclosures and nearby grazed sites. Our results show that a different vegetation with a high proportion of vascular plants developed due to reduced herbivory, resulting in a larger and more diverse input of polysaccharides to the soil at exclosed study sites. This coincided with higher sugar and amino acid concentrations in the soil at this site as well as the establishment of a more abundant and active microbiota, including saprotrophic fungi with broad substrate ranges, like Helotiales (Ascomycota) and Agaricales (Basidiomycota). A detailed description of fungal transcriptional profiles revealed higher gene expression for cellulose, hemicellulose, pectin, lignin and chitin degradation at herbivory-exclosed sites. Furthermore, we observed an increase in the number of genes and transcripts for predatory eukaryotes such as Entomobryomorpha (Arthropoda). We conclude that in the absence of herbivory, the development of a vascular vegetation alters the soil polysaccharide composition and supports larger and more active populations of fungi and predatory eukaryotes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Arctic terrestrial peatlands store 30–40% of the world`s soil organic carbon (SOC) (Tarnocai et al. 2009), the fate of which is determined by the balance between plant growth, herbivory and microbial decomposition (Ping et al. 2015). Herbivory has a large impact on the composition of plant communities (Zacheis et al. 2001; Maron and Crone 2006), which in turn affects the quality of litter input to the soil (Bardgett and Wardle 2003; Wardle et al. 2004; Van der Heijden et al. 2008; Fivez et al. 2014). Plant cell wall polymers, such as cellulose, hemicelluloses, pectins and lignin, are the major constituents of photosynthetically fixed organic carbon in peatlands (Chesworth et al. 2008; Gilbert 2010) and the cell walls of different plant lineages have characteristic macromolecular organization and polymer composition (Sarkar et al. 2009). Thus, the vegetation is assumed to determine the SOC composition and its decomposability (Davidson and Janssens 2006; Ping et al. 2015).
Our knowledge of soil ecosystem functioning is largely based on studies that focus on one or a few components of these complex ecosystems, but in order to thoroughly understand processes like decomposition it is important to study the microbial food web structures and linkages (Crotty et al. 2014). Fungi degrade detritus organic matter, consisting of a variety of plant polysaccharides and lignin (Thormann 2006). Bacterial communities in soils are known to be involved in numerous activities including degradation of detritus polymers (Tveit et al. 2013), microbial necromass (Müller et al. 2018) and active predation (Davidov et al. 2006; Morgan et al. 2010). Groups of non-fungal eukaryotes are involved in both predation and degradation of microbial and plant polymers (Crotty et al. 2011). The numbers of omnivorous eukaryotes such as Collembola (orders: Entomobryomorpha, Poduromorpha, Symphypleona) have been shown to positively and negatively correlate with microbial biomasses (Sabais et al. 2011; Thakur et al. 2015), making it difficult to reconstruct the linkages of microbial food webs based on abundances of taxa alone. Geisen et al. (2016) described different groups of protists and arthropods, so far believed to be bacteriovorous, as facultative mycophagous, refining our understanding of protists and arthropods as key players that control the biomass of both bacteria and fungi in soil food webs.
In a productive ecosystem, herbivory can lead to higher productivity by maintaining the dominant plant species while excluding less productive species (Bardgett et al. 1998; Bardgett and Wardle 2003). In less productive ecosystems, herbivory can lead to decreased plant and root biomass like shown for a high-Arctic peatland, Svalbard (Sjögersten et al. 2011) or in the coastal marshes of Hudson Bay, Canada (Jefferies et al. 2006). The effect of increased plant species richness and productivity resulted in larger microbial biomasses and rates of respiration (Zak et al. 2003; Zak and Kling 2006), and an overall increase in the density and diversity of microorganisms (Eisenhauer et al. 2013). In western Svalbard, increased peatland herbivory by barnacle geese (Branta leucopsis) has led to a suppression of vascular plant growth and dominance of mosses within the family Amblystegiaceae (brown mosses) (Kuijper et al. 2006), possibly changing these peatlands from carbon sources into carbon sinks (Sjögersten et al. 2011).
Here we compared peat protected from grazing for 18 years (exclosure; experimental condition) with grazed peat (grazed sites; natural condition), studying the differences in soil, soil polysaccharide composition, microbial community composition and microbial activities directed at polysaccharide decomposition, targeting all three domains of life. For this, we have sequenced eight metagenomes and eight metatranscriptomes, and performed targeted metabolomics, antibody staining of polysaccharides and extracellular enzyme assays in addition to describing the peat soil and the vegetation.
Materials and methods
Study site and sampling
The fieldwork was carried out in the high-Arctic peatland Solvatn, situated close to the research settlement Ny-Ålesund, Svalbard (78° 55′ N, 11° 56′ E), in August 2016. In the Solvatn peatland, experimental exclosures had been maintained since 1998 by 0.5-m tall fences prohibiting Barnacle geese grazing from areas of 0.7 × 0.7 m (Sjögersten et al. 2011) (Fig. 1). A total of four replicates from the grazed areas and four replicates for the exclosed areas were collected for DNA and RNA extractions, enzyme assays and plant polymer profiling while 12 replicates from each condition were collected for pore water amino acid and sugar measurements. Samples were collected at 1–2 cm depth and immediately frozen in liquid nitrogen or processed for pore water extraction and filtration; 400 µl of pore water was filtered with Whatman™ Mini-UniPrep™ G2 Syringeless Filters (GE Healthcare, Buckinghamshire, UK). The O2 [%] and temperature T [°C] of the sites were measured at four different depths using an optical O2 sensor and thermometer (Fibox 4, PreSens Precision Sensing GmbH, Regensburg, Germany) (Table S1—online resource 2). The water [%] and total organic matter (TOM) contents were estimated as described in Tveit et al. (2013) (Table S2—online resource 2). The pH was measured in pore water using a portable field pH meter (Multi 350i, WTW, Weilheim, Germany). Plant material was collected for plant species characterizations.

Sampling sites and sample collection. a In the picture one of the exclosures, EX1, and grazed sites, GR1, are shown, displaying the different vegetation. b The top peat layer at 1–2 cm below the living vegetation was sampled as indicated by the boxes across the peat profile figures. c Location of the Solvatn peatland, within the settlement Ny-Ålesund, Svalbard
Analysis of pore water amino acids and sugars
Free amino acids in the pore water samples were analyzed using a Thermo UPLC system consisting of an Accucore HILIC column (150 mm × 2.1 mm, 2.6 μm particle size) coupled to an Orbitrap Exactive mass spectrometer. The separation was carried out using a gradient from 95% eluent B (acetonitrile, 0.1% v/v formic acid) to 40% eluent A (water, 0.1% v/v formic acid) (Hu et al. 2017). The mass resolution was set to 50.000 and the injection volume was 25 μL. Sugar concentrations in pore water were measured with an HPLC (Dionex ICS-5000) using a Thermo CarboPac column (0.4 mm × 150 mm; pre-column 0.4 mm × 35 mm) and an electrochemical detector. The eluent was 3 mM KOH and samples were processed at 30 °C at a flow rate of 9 µl min−1, with an injection volume of 40 μL. All of the peaks were integrated by Xcalibur 2.2.
Comprehensive microarray polymer profiling (CoMPP)
The polysaccharide composition of Arctic peat soils and of three different plant types were measured using Comprehensive Microarray Polymer Profiling (CoMPP) at the University of Copenhagen as described in Moller et al. (2008). For further details, see supplementary materials and methods section 1 (online resource 1).
Enzyme assays
Polysaccharide degradation enzyme assays were carried out using the GlycoSpot™ technology (Copenhagen/Denmark). The substrates used were xylan (beechwood), arabinoxylan (wheat), 2HE-cellulose (synthetic), arabinan (sugar beet), pectic galactan (lupin), galactomannan (carob), xyloglucan (tamarind) and rhamnogalacturonan (soy bean). For further details, see supplementary materials and methods section 2 (online resource 1).
Nucleic acids extraction and rRNA depletion
Nucleic acids were extracted and quantified as described previously (Urich et al. 2008; Tveit et al. 2013). Total RNA samples were processed with the Ribo-Zero Magnetic Kit for Bacteria from Illumina (San Diego, CA/USA) to remove 16 and 23S rRNA molecules and enrich the mRNA fraction of the metatranscriptome. For further details, see supplementary materials and methods section 3 (online resource 1).
Sequencing and sequence preprocessing
Sequencing was performed at the “Norwegian High Throughput Sequencing Centre” NSC Oslo, Norway (http://www.sequencing.uio.no). DNA samples were prepared for sequencing with the TruSeq Nano DNA Library Prep Kit (Illumina, San Diego, CA/USA), with an input mass of 100 ng DNA. RNA samples were prepared with the TruSeq Stranded mRNA Library Prep Kit (Illumina, San Diego, CA/USA) with random primers and an input mass of 10 ng RNA. Single reads were sequenced using the HiSeq 4000 with a read length of 150 bp, resulting in approximately 38–55 Mio reads per library. Trimmomatic (Bolger et al. 2014) was used for an initial quality filtering of the sequences to remove low quality reads. Sequences were further processed with SortMeRNA v. 2.0 to separate reads into SSU rRNA, LSU rRNA and non-rRNA (Kopylova et al. 2012).
Analysis of SSU rRNA coding reads
Blastn searches against the SILVA SSU reference database (v. 128) were performed to taxonomically classify the SSU rRNA gene reads (-evalue 10–1—num_alignments 50—num_descriptions 50). The Blast outputs were analyzed using MEGAN (Huson et al. 2011) v. 6.13.1 (parameters: min bit score 100.0; top percent 2.0; min support percent 0.01, 25 best hits) as described previously (Söllinger et al. 2018).
Taxonomic and functional annotation of mRNA and mRNA coding genes
Randomly selected subsamples of 5 million nucleotide reads from the non-RNA datasets were taxonomically and functionally classified using NCBI nr (as of March 2017) and KEGG (v. 81.0) databases, respectively. NCBI nr was used for taxonomic annotation with DIAMOND v. 0.9.17 (Buchfink et al. 2015) applying an e-value threshold of 10–3. The output was uploaded in MEGAN v. 6.13.1 (parameters: min score 50.0; top percent 2.0; min support percent 0.01, 25 best hits), as described previously (Söllinger et al. 2018). Reads that had been taxonomically classified using the NCBI nr database were used as queries in a blastx search against the KEGG database with e-value threshold 10–10 as described previously (Tveit et al. 2015).
Functional and taxonomic annotation of CAZyme encoding genes and transcripts
Randomly selected subsamples of 5 million nucleotide reads from each of the DNA and non-rRNA datasets (for non-rRNA the same subsets were used as above) were translated into open reading frames (ORFs) of 30 amino acids or longer by the program Open Reading Frame (ORF) finder (Wheeler et al. 2003). The ORFs were screened for Protein families using the Pfam (protein family) database (v. 31) (Finn et al. 2014) and HMMsearch, a tool within the hidden markov models (HMMs) package (v 3.1b2) (Finn et al. 2015). All database hits with e-values below a threshold of 10−4 were counted. The resulting Pfam annotations were screened for CAZymes using Pfam models of previously identified CAZymes (Tveit et al. 2015; Söllinger et al. 2018) including starch, cellulose, hemicellulose, pectin and lignin degrading enzymes. Translated reads assigned to the selected CAZymes were extracted, followed by blastp against the NCBI nr database with an e-value threshold of 10–1 to obtain taxonomic information (as of March 2017) and analyzed using MEGAN (parameters: min score 50; top percent 2.0; min support percent 0.01; 25 best hits). In order to provide more depth in the analysis of the eukaryotic taxa, the full datasets were taxonomically annotated and reads assigned to Helotiales, Agaricales and Entomobryomorpha were extracted and functionally annotated as described above.
Statistical analyses and data visualization
Significance testing was performed with the Wilcoxon rank sum test using the R package (R Core Team 2014). The results from statistical tests are summarized in Table S3 (online resource 2). Correspondence analyses (Greenacre 2017) were performed as described previously (Tveit et al. 2015). The R packages ggplot2 and heatmap3 were used for plotting. Spearman correlation analysis was performed, using the function cor() in R. The geographical map of Svalbard was created using QGIS (v. 3.18.0), the base map was provided by the Norwegian Polar Institute (https://geodata.npolar.no).
Results
The effects of herbivore grazing on peat vegetation and soil O2 availability
The exclosures (EX) were established 18 years prior to sampling, preventing the access of herbivores to the vegetation (Fig. 1a). During this time, a vascular plant community dominated by Poa arctica (up to 90% coverage) and Cardamine pratensis (up to 20% coverage) had developed. The vascular plants Ranunculus hyperborus, Saxifraga cernua and Saxifraga cespitosa were also present. Eight moss species within the brown moss family Amblystegiaceae were identified, making up the entire moss community within the exclosures (Table 1). At the grazed sites (GR), a total of seven moss species dominated the vegetation (up to 100% coverage), while two vascular plant species were detected (P. arctica and R. hyperborus) (Table 1).
Due to the higher coverage of vascular plants in the exclosures, these sites contained much higher root densities, previously quantified by Sjögersten et al. (2011) to be 12 times higher. The O2 [%] within the first two centimeters of peat was ~ 20% in both the exclosures and grazed sites. The O2 concentration decreased with depth, being ~ 19–20% at 5 cm depth within the exclosures and 13–16% in the grazed sites (Table S1—online resource 2). We observed surface temperatures in the range of 10.4–10.8 °C (EX) and 8.3–14 °C (GR), while at 10 cm soil depth the temperature was approximately 5–6 °C in both sites (Table S1—online resource 2). The water content of the peat soils was lower in the exclosures (mean: 84.5%) compared to the grazed sites (mean: 92.4%) (Table S2—online resource 2). The total soil organic matter percentage of the fresh weight was higher in the exclosures (mean EX: 13.0%; mean GR: 6.5%) (Table S2—online resource 2). The pH of the upper soil layer in both the exclosures and grazed sites was between 7.0 and 7.1.
Soil polysaccharide, sugar and amino acid content vary between exclosures and grazed sites
To identify the relationship between vegetation and soil polysaccharide composition we mapped the polysaccharide content in three different plant types—P. arctica, S. cespitosa and a mixture of Amblystegiaceae mosses. The same polysaccharide identification was done for the peat soil from the exclosures and grazed sites. A correspondence analysis confirmed that all three plant polysaccharide profiles were distinctly different from one another (Figures S1a and S1b—online resource 3). Saxifraga cespitosa was richer in the pectins homogalacturonan, rhamnogalacturonan and arabinan as well as cellulose and the hemicelluloses xyloglucan, mixed linkage glucans (MLG) and xylan. Poa arctica was richer in glucan, arabinoxylan and the glycoprotein AGP (arabinogalactan protein), while the mosses were richer in mannan-containing hemicelluloses, homogalacturonan (HG) with an intermediate degree of methyl esterification (DE) and galactan (Table S4—online resource 2).
Correspondingly we found that cellulose, glucan, xyloglucan, arabinoxylan and rhamnogalacturonan were more abundant in the soils from the exclosures while homogalacturonan was equally abundant at both sites. For arabinan and mannan-containing hemicelluloses, only some tested antibodies had significantly higher abundances in the exclosures (Table 2 and Figure S1c and S1d—online resource 3).
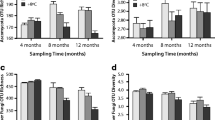
The pore water concentrations of sugars, including glucose, fructose, mannose, xylose and galactose, were significantly higher in the peat soils from the exclosures (Fig. 2a). Glucose was the most abundant sugar in the pore water of the exclosures (mean: 40.3 µM) and the grazed sites (mean: 9.9 µM), followed by fructose (EX mean: 18.3 µM and grazed site mean: 5.4 µM). There was less mannose, xylose and galactose in the peat, but the concentrations were always higher in the exclosures (mean EX: mannose 5.8 µM, xylose 2.2 µM, galactose 0.4 µM) than the grazed sites (mean GR: mannose 0.8 µM, xylose b.d., galactose 0.02 µM).

Sugar and amino acid concentrations, masses of nucleic acids and polysaccharide degrading enzyme rates in peat soil samples exclosures (red) and grazed sites (blue). a Pore water concentrations [µM] of the sugars glucose, fructose, mannose, xylose and galactose. b Pore water concentrations [µM] of amino acids. c Masses of DNA and RNA in micrograms per gram of dry peat soil [µg gDW−1 peat]. d Enzymatic potential for polysaccharide degradation on eight polysaccharide substrates. Individual dots show the signal strength relative to the sum of measured signal strength for each substrate. The values thus indicate the potential enzyme activity in the eight samples relative to each other. The rates were normalized by dry weights (DW). All compounds marked with an ‘*’ show a significant difference of the grazed and the exclosed. For statistical testing the Wilcoxon rank sum test was used with R (v3.6.1), values can be found in Table S3
We also observed significantly higher concentrations of a broad range of amino acids in the exclosures (Fig. 2b and Table S3—online resource 2). Aspartic acid and alanine had the highest concentrations with mean values of 10.5 and 13.2 µM, respectively, in the exclosures, while the concentrations in the grazed sites were much lower (0.9 and 2.3 µM). The amino acids glycine, leucine, proline, serine and threonine ranged from 4.0 to 6.8 µM in the exclosures and 0.6 to 1.3 µM in the grazed sites. Only glutamic acid was found at lower concentrations in the exclosures (mean EX: 0.9 µM) compared to the grazed sites (mean GR: 2.0 µM). The other amino acids were below 1.8 µM (mean values) but always at higher concentrations in the exclosures than the grazed sites.
Microbial activities in the peat soil
To study the link between the vegetation, soil chemistry and the composition and activity of the microbial community we extracted total nucleic acids for the analysis of DNA and RNA from four replicate samples of the upper 2 cm oxic layer of peat soil in the exclosures and four from the grazed sites. As depicted in Fig. 2c we observed higher DNA amounts in the exclosures than the grazed sites (mean: EX 92.5 µg DNA gDW peat−1, GR 58.3 µg DNA gDW peat−1), while the amount of RNA per gram dry weight was equally high at the two sites (mean: EX 71.2 µg RNA gDW peat−1, GR 69.6 µg RNA gDW peat−1) (Fig. 2c and Table S5—online resource 2). Next, we investigated the potential enzyme activities for decomposition of some of the most common plant polysaccharides—cellulose, mannan, xyloglucan, xylan, arabinoxylan, galactan, arabinan and rhamnogalacturonan (Sarkar et al. 2009). This confirmed that the potential for polysaccharide degradation was significantly higher in the exclosures than the grazed sites (Fig. 2d and Table S3—online resource 2).
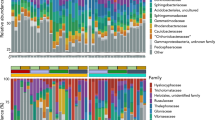
In order to study the microbial communities and their patterns of gene transcription, we removed ribosomes from the total RNA by ribodepletion and sequenced the remaining RNA as well as the total DNA from four replicates collected in the exclosures, and four in the grazed sites, giving eight metatranscriptomic and eight metagenomic libraries in total. Each of the 16 libraries contained ~ 38–55 million sequence reads with a length of ~ 150 bp. The ribodepleted RNA libraries contained 48–83% non-rRNA sequences, ~ 30% of which could be taxonomically classified and 10–15% of which could be functionally classified (Table S6—online resource 2). The microbial community composition based on taxonomic annotation of mRNA was overall similar in the exclosures and grazed sites, but notable differences were also observed (Figs. 3 and S2—online resource 3). Bacterial genes and transcripts comprised approximately 90% of total mRNA and 92% of total rRNA genes at exclosed sites, compared to approximately 96% of total mRNA and 95% of total rRNA genes at grazed sites. Correspondingly, the abundances of reads assigned to eukaryotic kingdoms (fungi, Protista and Metazoa) were particularly high in the exclosures relative to the grazed sites (Figs. 3 and S3—online resource 3). However, the 16S rRNA gene abundance and transcriptional activity were dominated by Actinobacteria, followed by Alpha-, Beta-, Delta- and Gamma-proteobacteria, Chloroflexi, Acidobacteria, Verrucomicrobia and Bacteriodetes in both grazed sites and exclosures. There were also considerable numbers of reads assigned to Ciliophora, fungi and Nematoda (Figs. 3 and S2—online resource 3). Overall, the most abundant taxa (SSU rRNA gene abundance) were the transcriptionally (mRNA abundance) most active (r = 0.86–0.94; only the taxa displayed in the boxes considered: Fig. 3a and b).

Microbial community composition a based on mRNA sequences and therefore representing the transcriptionally active microbial community. b Based on SSU rRNA gene sequences reflecting the potential microbial community in peat soil from exclosures and grazed sites. The size of the boxes reflect the relative abundances of taxa. Taxonomy profiles are displayed at phylum-level (class-level for Proteobacteria) and are generated by averaging data sets from four replicates
To identify which microbial taxa were responsible for polymer degradation we extracted from the metagenomes and metatranscriptomes genes and transcripts encoding enzymes for polysaccharide and lignin degradation (Table S7—online resource 2). In the exclosures, the microbial transcription for cellulose degradation was highest (0.40%), followed by hemicellulose (0.36%), pectin (0.24%) and lignin (0.16%) (Fig. 4a). In the grazed sites, the transcription for hemicellulose degradation was highest (0.31%), followed by cellulose (0.21%), pectin (0.07%) and lignin (0.04%). Corresponding to the higher abundance of fungi (Figs. 3 and S3—online resource 3), a larger fraction of the genes and transcripts for polymer decomposition were assigned to fungi in the exclosures (18.95%) than the grazed sites (1.38%) (Fig. 4a). The majority of these transcripts were assigned to the fungal phyla Basidiomycota (6.32% EX & 0.60% GR) and Ascomycota (10.56% EX & 0.48% GR). Actinobacteria (30.92% EX & 20.14% GR), Proteobacteria (11.28% EX & 14.51% GR) and Bacteriodetes (8.46% EX & 13.82% GR) were the most transcriptionally active bacterial polysaccharide degraders, while the majority of transcripts for lignin degradation were assigned to Proteobacteria (28.15% EX & 33.03% GR) (Fig. 4b). The taxonomic distribution of genes for polymer degradation was similar at exclosed and grazed sites, with the exception that the transcript to gene ratio was much higher for fungal than bacterial taxa (Fig. 4b).

Genes and transcripts for polysaccharide and lignin degradation in prokaryotes and eukaryotes. a Number of transcripts for cellulose, hemicellulose, pectins and lignin degradation relative to the total number of reads assigned to a function for all microorganisms (left) and fungi (right). b Taxonomic assignment of metatranscriptomic (mRNA) and metagenomic (gDNA) sequences encoding enzymes involved in polysaccharide and lignin degradation. The functional annotation is at phylum-level, comparing exclosures (EX) and grazed (GR) sites
Taxa that had different transcriptional activities in the exclosures and grazed sites were identified by correspondence analysis (Figure S4a—online resource 3). The transcriptional profiles from the exclosures were separated from the grazed sites along the first axis, explaining 42.7% of the inertia. By their contribution to the first axis inertia we identified the major eukaryotic and prokaryotic orders with different transcriptional activities in the exclosures and grazed sites. Out of the 20 taxa contributing most to inertia in each direction (Figure S4b—online resource 3; and Table S8—online resource 2), 15 taxa had higher numbers of transcripts in either the exclosures or the grazed sites, and an average relative abundance of mRNA transcripts above 0.5%. Among these, three eukaryotic (Agaricales, Helotiales and Entomobryomorpha) and seven bacterial orders (Lactobacillales, Chitinophagales, Burkholderiales, Sphingobacteriales, Nakamurellales, Corynebacteriales, Micrococcales) were more active in the exclosures, whereas five bacterial orders (Methylococcales, Anaerolineales, Solibacterales, Thiotrichales, Desulfobacterales) were more active in the grazed sites. These 15 orders accounted for 7 to 18% of the total number of microbial mRNA transcripts. We then compared the transcriptional activity and relative abundance of SSU rRNA genes for these 15 taxa (Figure S4b—online resource 3), finding similar patterns in the SSU rRNA genes and the mRNA for some of the taxa. Particularly interesting were the much higher relative abundances of the three eukaryotic orders in the exclosures: Agaricales was on average 7.7-fold (SSU rRNA genes) and 71.4-fold (mRNA) higher, Helotiales was 29.8-fold (SSU rRNA genes) and 46.9-fold (mRNA) higher, while Entomobryomorpha was 20.4-fold (SSU rRNA genes) and 11.8-fold (mRNA) more abundant and transcriptionally active (Fig. 5).

Transcriptional profiles for polymer degradation by Helotiales, Agaricales and Entomobryomorpha. a Functional profile of the two fungal orders Helotiales and Agaricales displaying relative abundances of transcripts in exclosures (red) and grazed sites (blue) for enzymes involved in polymer degradation as described in Table S9. The fraction of total transcripts (mRNA) and rRNA genes assigned to these two fungal orders are shown in the lower box. b Functional profile of the order Entomobryomorpha showing its transcription for polymer degradation in the replicates of peat soils from the exclosures (red) and grazed sites (blue). In the lower square, the fraction of total transcripts (mRNA) and rRNA genes assigned to Entomobryomorpha is displayed. Oligo.Hydro—oligosaccharide hydrolysis; MLGs—mixed linkage glycans; Galactoman.—galactomannan; Arabinoxyl.—arabinoxylan
The number of polymer degradation transcripts assigned to Helotiales (of fungal transcripts: 21.5% EX, 4.0% GR) and Agaricales (of fungal transcripts: 37.6% EX, 4.3% GR) show that these are the major fungal polysaccharide degraders in the exclosures. Their profiles included transcripts for cellulose, xylans, glucans, mannans, pectins, lignin, chitin and bacterial cell walls (Fig. 5a and Table S9—online resource 2). Helotiales expressed genes for a broader range of substrates than Agaricales (Table S9—online resource 2), but for both fungal orders the relative abundance of transcripts for most polymers were higher in the exclosures (Fig. 5a). Similarly, the transcriptional activity of the arthropod order Entomobryomorpha was much higher in the exclosures (Fig. 5b). It had a narrower substrate range than the fungi, lacking transcripts for xylan, pectin and lignin decomposition (Table S9—online resource 2). However, Entomobryomorpha transcripts for bacterial cell wall, chitin, cellulose and oligosaccharide degradation were present, and consistently more abundant in the exclosures than the grazed sites (Fig. 5b). Finally we assessed whether the above patterns of transcription for polymer degradation were reflected in the transcription for central metabolisms in these three orders. We functionally annotated the Agaricales, Helotiales and Entomobryomorpha transcripts using KEGG (Kyoto encyclopedia of genes and genomes). This revealed that the transcript abundances for central metabolisms were highest in the exclosures for all three taxa (Figure S5—online resource 3), but with considerable variation between samples, especially for the fungi.
Discussion
In this study we have compared the microbial communities in soils below high-Arctic peat vegetation exposed to grazing and soils below peat vegetation protected from grazing for 18 years. The investigation of both the above- and belowground biology has allowed us to study the relationship between the vegetation and the soil microorganisms. Our comparison of the two soil treatments demonstrated that the removal of herbivores and altered vegetation leads to a different soil polysaccharide composition, lower potential extracellular enzymatic activities and a different microbial community with lower abundances and activities of fungi and putative predatory eukaryotes.
Reduced herbivore grazing alters the vegetation, soil structure and soil composition
We observed that a different vegetation with an 8 times larger coverage of vascular plants had established after 18 years of protection inside the exclosures. This shift, primarily caused by increased abundance of P. arctica, was already observed some years earlier as evidenced by a 12 times larger root biomass and 28 times larger vascular plant biomass in the exclosures than the grazed sites (Sjögersten et al. 2011). The vascular vegetation was associated with reduced soil water contents and higher oxygen levels in deeper layers, presumably due to the increased root formation. Similar observations were made in a study of a temperate peatland, where lower water table heights were observed in areas with greater vascular plant biomass (Murphy et al. 2009). The lowering of the water table and the diffusion of gases through the roots of vascular plants might be the reasons for the higher oxygen availabilities (Colmer 2003). However, at the surface (top ~ 0–2 cm) sampled for our molecular and chemical analyses, the oxygen levels were the same in exclosures and grazed sites, suggesting that oxygen limitation is mostly relevant in deeper layers.
The increased input of organic matter from P. arctica and other vascular plants to the soil inside the exclosures can explain the higher organic matter content and abundance of polysaccharides compared to the grazed sites. Previously, geese grazing and grubbing were found to reduce carbon stocks in Arctic wetlands from Svalbard and Canada (Van der Wal 2006; Speed et al. 2010; Sjögersten et al. 2011). In particular, we identified higher soil content of xyloglucan, arabinoxylan and pectins in the exclosures, matching the higher concentrations of monosaccharides. However, determining the exact origin of the monosaccharides in soils is challenging, as they might be root exudates (Bertin et al. 2003; Bais et al. 2006), polysaccharide hydrolysis products (Tveit et al. 2013) or both. The release of low-molecular weight compounds like sugars through root exudation varies e.g. with the plant type, its age and environment (Uren 2000; Bertin et al. 2003; Bais et al. 2006; McNear 2013). Monosaccharide stocks also depend on the kinetics of their usage, for which we do not have estimates. Nevertheless, the occurrence of more roots, more polysaccharides and higher potential rates of extracellular polysaccharide degrading enzymes suggests a combination of root exudation and polysaccharide hydrolysis as sources for the monosaccharides.
Increased input from vascular plants supports a more abundant and active microbiota
The higher availability of carbohydrates fueled a more abundant and active microbiota in the exclosures, judging by the amount of DNA per gram of dry soil. Although not a direct measure of total microbial biomass or the number of cells, nucleic acids provide good estimates for relative differences in the size of the microbiota between samples with similar properties for nucleic acids extraction (Tveit et al. 2015; Söllinger et al. 2018). Considering the higher organic matter content in the exclosures, the more abundant microbiota might be the result of higher substrate availability. This could explain the higher rates for extracellular polysaccharide degradation in the exclosures as more organisms generally would produce more enzymes. We also observed a considerably higher relative abundance of transcripts for extracellular polymer degrading enzymes in the exclosures, supporting the view that increased substrate availabilities leads not only to increased number of microorganisms but also to larger investments into extracellular polymer decomposing enzymes by microorganisms (Zak and Kling 2006; Wallenstein and Weintraub 2008). The altered organic matter quality may also be the reason for the increased relative abundance of fungi as they are believed to be specialists for the decomposition of, e.g., lignocellulose (Baldrian 2008). The effect of substrate quality on microbial activities was also seen in the higher potential enzyme rates for degradation of multiple different polysaccharides, in the exclosures, matching the higher abundances of these polysaccharides in the exclosures.
Altered structure and activity of the microbial communities
In line with the considerable changes in vegetation and soil chemistry we observed two-fold higher eukaryote to prokaryote gene ratios and five-fold higher transcript ratios in the exclosures than the grazed sites. A handful of taxa were instrumental to this shift; saprotrophic fungi of the orders Helotiales (Ascomycota) and Agaricales (Basidiomycota), and microbial predators and plant litter consumers of the order Entomobryomorpha (Arthropoda). In previous studies, fungal abundances were shown to be positively correlating with root exudation (Broeckling et al. 2008) and more generally the availabilities of easily degradable compounds like sugars (Edgecombe 1938; Thormann 2006) or increased nitrogen availability (Koranda et al. 2014). Increased monosaccharide concentrations could also be products of fungal degradation of polysaccharides. Thus, in our case, it is not possible to conclude on the causality between monosaccharide concentrations and fungal abundances. Nevertheless, Helotiales and Agaricales have very broad substrate ranges that includes cellulose and all major hemicelluloses and might thus have benefitted from the altered substrate quality and contributed to a variety of carbon decomposition activities once established in the exclosures.
Geisen et al. (2016) found that many protists and arthropods are facultative mycophagous, making them key players in soil microbial food webs. Hence, the higher fungal abundances in the exclosures might have supported the increase in protists and metazoan taxa abundances.
Overall, the vascular vegetation seems to sustain a more complex food web of prokaryotic and fungal detritivores that are food sources for predatory eukaryotes. Whether this is what sustains the larger amino acid pools observed in the exclosures is unclear, but if the transcriptional activity and relative abundance of putative predators reflects predation rates, it may, as bacterial and fungal cells consist of 10–70% protein (Ritala et al. 2017). Our results suggest that Entomobryomorpha are able to degrade bacterial cell walls but also plant and fungal polymers. By regulating microbial turnover, Entomobryomorpha have previously been shown to enhance nutrient mineralization rates and increase nitrogen availability in soils (Cragg and Bardgett 2001; Thakur et al. 2015). Additionally, amino acids may be released through root exudations (Canarini et al. 2019), offering an alternative explanation for the amino acid pools. The origin of the nitrogen needed to support a larger microbial biomass and amino acid pool cannot be directly identified with our data. However, Solheim et al. (1996) showed that nitrogen fixation in Svalbard soils protected from grazing is limited compared to grazed soils where it was supported by epiphytic cyanobacteria. This indicates that nitrogen is lost from grazed systems and regained through fixation, while in the absence of herbivores the microbiota are able to recycle the nitrogen in decaying plants, supporting larger plant and microbial biomasses, and pools of free amino acids.
Low molecular weight nitrogen sources, like amino acids, are important for several microbial processes, hence fueling the microbial community in soils (Schimel and Weintraub 2003; Jones et al. 2004), but also the vegetation (Jones et al. 2004, 2005; Sauheitl et al. 2009). Thus, in the absence of herbivory, feedback effects between an increasingly active microbial community and vegetation might have accelerated the establishment of a new ecosystem state. An important future task will be to identify the fluxes of amino acids, sugars and other metabolites between plants and microorganisms during this ecosystem transition from a grazed to non-grazed state.
Conclusion
The establishment of a larger vascular plant biomass in the absence of herbivores led to higher soil concentrations of polysaccharides, monosaccharides and amino acids. We found that this corresponded with larger and more active populations of saprotrophic fungi and putative predatory eukaryotes. This study establishes a fundament for targeting molecular and microbial mechanisms that control the interactions between above and below ground biology in high-Arctic peatlands.
Data availability
Metagenomes and metatranscriptomes have been deposited in the Sequence Read Archive (SRA) database (BioProject Accession: PRJNA170725, https://www.ncbi.nlm.nih.gov/sra/PRJNA170725). For details about run accession numbers, see supplementary data availability (online resource 1).
References
Bais HP, Weir TL, Perry LG, Gilroy S, Vivanco JM (2006) The role of root exudates in rhizosphere interactions with plants and other organisms. Annu Rev Plant Biol 57:233–266. https://doi.org/10.1146/annurev.arplant.57.032905.105159
Baldrian P (2008) Wood-inhabiting ligninolytic basidiomycetes in soils: ecology and constraints for applicability in bioremediation. Fungal Ecol 1:4–12. https://doi.org/10.1016/j.funeco.2008.02.001
Bardgett RD, Wardle DA (2003) Herbivore-mediated linkages between aboveground and belowground communities. Ecology 84:2258–2268. https://doi.org/10.1890/02-0274
Bardgett RD, Wardle DA, Yeates GW (1998) Linking above-ground and below-ground interactions: how plant responses to foliar herbivory influence soil organisms. Soil Biol Biochem 30:1867–1878. https://doi.org/10.1016/S0038-0717(98)00069-8
Bertin C, Yang X, Weston LA (2003) The role of root exudates and allelochemicals in the rhizosphere. Plant Soil 256:67–83. https://doi.org/10.1023/A:1026290508166
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Broeckling CD, Broz AK, Bergelson J, Manter DK, Vivanco JM (2008) Root exudates regulate soil fungal community composition and diversity. Appl Environ Microbiol 74:738–744. https://doi.org/10.1128/AEM.02188-07
Buchfink B, Xie C, Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60. https://doi.org/10.1038/nmeth.3176
Canarini A, Kaiser C, Merchant A, Richter A, Wanek W (2019) Root exudation of primary metabolites: mechanisms and their roles in plant responses to environmental stimuli. Front Plant Sci 10:157. https://doi.org/10.3389/fpls.2019.00157
Chesworth W, Camps Arbestain M, Macías F, Spaargaren O, Mualem Y, Morel-Seytoux HJ, Horwath WR (2008) Carbon cycling and formation of soil organic matter. In: Chesworth W (ed) Encyclopedia of soil science. Encyclopedia of earth sciences series. Springer, Dordrecht. https://doi.org/10.1007/978-1-4020-3995-9_88
Colmer TD (2003) Long-distance transport of gases in plants: a perspective on internal aeration and radial oxygen loss from roots. Plant Cell Environ 26:17–36. https://doi.org/10.1046/j.1365-3040.2003.00846.x
Cragg RG, Bardgett RD (2001) How changes in soil faunal diversity and composition within a trophic group influence decomposition processes. Soil Biol Biochem 33:2073–2081. https://doi.org/10.1016/S0038-0717(01)00138-9
Crotty FV, Blackshaw RP, Murray PJ (2011) Tracking the flow of bacterially derived 13C and 15N through soil faunal feeding channels. Rapid Commun Mass Spectrom 25:1503–1513. https://doi.org/10.1002/rcm.4945
Crotty FV, Blackshaw RP, Adl SM, Inger R, Murray PJ (2014) Divergence of feeding channels within the soil food web determined by ecosystem type. Ecol Evol 4:1–13. https://doi.org/10.1002/ece3.905
Davidov Y, Friedjung A, Jurkevitch E (2006) Structure analysis of a soil community of predatory bacteria using culture-dependent and culture-independent methods reveals a hitherto undetected diversity of Bdellovibrio-and-like organisms. Environ Microbiol 8:1667–1673. https://doi.org/10.1111/j.1462-2920.2006.01052.x
Davidson EA, Janssens IA (2006) Temperature sensitivity of soil carbon decomposition and feedbacks to climate change. Nature 440:165–173. https://doi.org/10.1038/nature04514
Edgecombe AE (1938) The effect of galactose on the growth of certain fungi. Mycologia 30:601–624. https://doi.org/10.2307/3754358
Eisenhauer N, Dobies T, Cesarz S, Hobbie SE, Meyer RJ, Worm K, Reich PB (2013) Plant diversity effects on soil food webs are stronger than those of elevated CO2 and N deposition in a long-term grassland experiment. Proc Natl Acad Sci USA 110:6889–6894. https://doi.org/10.1073/pnas.1217382110
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR et al (2014) Pfam: the protein families database. Nucleic Acids Res 42:D222–D230. https://doi.org/10.1093/nar/gkt1223
Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F et al (2015) HMMER web server: 2015 update. Nucleic Acids Res 43:W30–W38. https://doi.org/10.1093/nar/gkv397
Fivez L, Vicca S, Janssens IA, Meire P (2014) Western Palaearctic breeding geese can alter carbon cycling in their winter habitat. Ecosphere 5:1–20. https://doi.org/10.1890/ES14-00012.1
Geisen S, Koller R, Hünninghaus M, Dumack K, Urich T, Bonkowski M (2016) The soil food web revisited: Diverse and widespread mycophagous soil protists. Soil Biol Biochem 94:10–18. https://doi.org/10.1016/j.soilbio.2015.11.010
Gilbert HJ (2010) The biochemistry and structural biology of plant cell wall deconstruction. Plant Physiol 153:444–455. https://doi.org/10.1104/pp.110.156646
Greenacre M (2017) Correspondence analysis in practice. Interdisciplinary statistics series, 3rd edn. Chapman and Hall/CRC, Boca Raton
Hu Y, Zheng Q, Wanek W (2017) Flux analysis of free amino sugars and amino acids in soils by isotope tracing with a novel liquid chromatography/high resolution mass spectrometry platform. Anal Chem 89:9192–9200. https://doi.org/10.1021/acs.analchem.7b01938
Huson DH, Mitra S, Ruscheweyh H-J, Weber N, Schuster SC (2011) Integrative analysis of environmental sequences using MEGAN4. Genome Res 21:1552–1560. https://doi.org/10.1101/gr.120618.111
Jefferies RL, Jano AP, Abraham KF (2006) A biotic agent promotes large-scale catastrophic change in the coastal marshes of Hudson Bay. J Ecol 94:234–242. https://doi.org/10.1111/j.1365-2745.2005.01086.x
Jones DL, Shannon D, Murphy DV, Farrar J (2004) Role of dissolved organic nitrogen (DON) in soil N cycling in grassland soils. Soil Biol Biochem 36:749–756. https://doi.org/10.1016/j.soilbio.2004.01.003
Jones DL, Healey JR, Willett VB, Farrar JF, Hodge A (2005) Dissolved organic nitrogen uptake by plants - an important N uptake pathway? Soil Biol Biochem 37:413–423. https://doi.org/10.1016/j.soilbio.2004.08.008
Kopylova E, Noé L, Touzet H (2012) SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28:3211–3217. https://doi.org/10.1093/bioinformatics/bts611
Koranda M, Kaiser C, Fuchslueger L, Kitzler B, Sessitsch A, Zechmeister-Boltenstern S, Richter A (2014) Fungal and bacterial utilization of organic substrates depends on substrate complexity and N availability. FEMS Microbiol Ecol 87:142–152. https://doi.org/10.1111/1574-6941.12214
Kuijper DPJ, Bakker JP, Cooper EJ, Ubels R, Jónsdóttir IS, Loonen MJJE (2006) Intensive grazing by Barnacle geese depletes High Arctic seed bank. Can J Bot 84:995–1004. https://doi.org/10.1139/b06-052
Maron JL, Crone E (2006) Herbivory: effects on plant abundance, distribution and population growth. Proc R Soc B 273:2575–2584. https://doi.org/10.1098/rspb.2006.3587
McNear DH Jr (2013) The rhizosphere—roots, soil and everything in between. Nat Educ Knowl 4:1
Moller I, Marcus SE, Haeger A, Verhertbruggen Y, Verhoef R, Schols H et al (2008) High-throughput screening of monoclonal antibodies against plant cell wall glycans by hierarchical clustering of their carbohydrate microarray binding profiles. Glycoconj J 25:37–48. https://doi.org/10.1007/s10719-007-9059-7
Morgan AD, Maclean RC, Hillesland KL, Velicer GJ (2010) Comparative analysis of Myxococcus predation on soil bacteria. Appl Environ Microbiol 76:6920–6927. https://doi.org/10.1128/AEM.00414-10
Murphy MT, McKinley A, Moore TR (2009) Variations in above- and below-ground vascular plant biomass and water table on a temperate ombrotrophic peatland. Botany 87:845–853. https://doi.org/10.1139/B09-052
Müller AL, Pelikan C, de Rezende JR, Wasmund K, Putz M, Glombitza C et al (2018) Bacterial interactions during sequential degradation of cyanobacterial necromass in a sulfidic arctic marine sediment. Environ Microbiol 20:2927–2940. https://doi.org/10.1111/1462-2920.14297
Ping CL, Jastrow JD, Jorgenson MT, Michaelson GJ, Shur YL (2015) Permafrost soils and carbon cycling. Soil 1:147–171. https://doi.org/10.5194/soil-1-147-2015
R Core Team (2014) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/
Ritala A, Häkkinen ST, Toivari M, Wiebe MG (2017) Single cell protein—state-of-the-art, industrial landscape and patents 2001–2016. Front Microbiol 8:2009. https://doi.org/10.3389/fmicb.2017.02009
Sabais ACW, Scheu S, Eisenhauer N (2011) Plant species richness drives the density and diversity of Collembola in temperate grassland. Acta Oecol (Montrouge) 37:195–202. https://doi.org/10.1016/j.actao.2011.02.002
Sarkar P, Bosneaga E, Auer M (2009) Plant cell walls throughout evolution: towards a molecular understanding of their design principles. J Exp Bot 60:3615–3635. https://doi.org/10.1093/jxb/erp245
Sauheitl L, Glaser B, Weigelt A (2009) Uptake of intact amino acids by plants depends on soil amino acid concentrations. Environ Exp Bot 66:145–152. https://doi.org/10.1016/j.envexpbot.2009.03.009
Schimel JP, Weintraub MN (2003) The implications of exoenzyme activity on microbial carbon and nitrogen limitation in soil: a theoretical model. Soil Biol Biochem 35:549–563. https://doi.org/10.1016/S0038-0717(03)00015-4
Sjögersten S, van der Wal R, Loonen MJJE, Woodin SJ (2011) Recovery of ecosystem carbon fluxes and storage from herbivory. Biogeochemistry 106:357–370. https://doi.org/10.1007/s10533-010-9516-4
Solheim B, Endal A, Vigstad H (1996) Nitrogen fixation in Arctic vegetation and soils from Svalbard, Norway. Polar Biol 16:35–40. https://doi.org/10.1007/BF01876827
Speed JDM, Woodin SJ, Tømmervik H, van der Wal R (2010) Extrapolating herbivore-induced carbon loss across an arctic landscape. Polar Biol 33:789–797. https://doi.org/10.1007/s00300-009-0756-5
Söllinger A, Tveit AT, Poulsen M, Noel SJ, Bengtsson M, Bernhardt J et al (2018) Holistic assessment of rumen microbiome dynamics through quantitative metatranscriptomics reveals multifunctional redundancy during key steps of anaerobic feed degradation. mSystems 3:e00038-e118. https://doi.org/10.1128/mSystems.00038-18
Tarnocai C, Canadell JG, Schuur EAG, Kuhry P, Mazhitova G, Zimov S (2009) Soil organic carbon pools in the northern circumpolar permafrost region. Glob Biogeochem Cycles. https://doi.org/10.1029/2008GB003327
Thakur MP, Herrmann M, Steinauer K, Rennoch S, Cesarz S, Eisenhauer N (2015) Cascading effects of belowground predators on plant communities are density-dependent. Ecol Evol 5:4300–4314. https://doi.org/10.1002/ece3.1597
Thormann MN (2006) Diversity and function of fungi in peatlands: a carbon cycling perspective. Can J Soil Sci 86:281–293
Tveit AT, Schwacke R, Svenning MM, Urich T (2013) Organic carbon transformations in high-Arctic peat soils: key functions and microorganisms. ISME J 7:299–311. https://doi.org/10.1038/ismej.2012.99
Tveit AT, Urich T, Frenzel P, Svenning MM (2015) Metabolic and trophic interactions modulate methane production by Arctic peat microbiota in response to warming. Proc Natl Acad Sci USA 112:E2507-2516. https://doi.org/10.1073/pnas.1420797112
Uren NC (2000) Types, amounts, and possible functions of compounds released into the rhizosphere by soil-grown plants. In: Willig S, Varanini Z, Nannipieri P (eds) The rhizosphere: biochemistry and organic substances at the soil-plant interface, 1st edn. CRC Press, Boca Raton, pp 19–40
Urich T, Lanzén A, Qi J, Huson DH, Schleper C, Schuster SC (2008) Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS ONE 3:e2527. https://doi.org/10.1371/journal.pone.0002527
Van der Heijden MGA, Bardgett RD, van Straalen NM (2008) The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol Lett 11:296–310. https://doi.org/10.1111/j.1461-0248.2007.01139.x
Van der Wal R (2006) Do herbivores cause habitat degradation or vegetation state transition? Evidence from the tundra. Oikos 114:177–186. https://doi.org/10.1111/j.2006.0030-1299.14264.x
Wallenstein MD, Weintraub MN (2008) Emerging tools for measuring and modeling the in situ activity of soil extracellular enzymes. Soil Biol Biochem 40:2098–2106. https://doi.org/10.1016/j.soilbio.2008.01.024
Wardle DA, Bardgett RD, Klironomos JN, Setälä H, van der Putten WH, Wall DH (2004) Ecological linkages between aboveground and belowground biota. Science 304:1629–1633. https://doi.org/10.1126/science.1094875
Wheeler DL, Church DM, Federhen S, Lash AE, Madden TL, Pontius JU et al (2003) Database resources of the National Center for Biotechnology. Nucleic Acids Res 31:28–33. https://doi.org/10.1093/nar/gkg033
Zacheis A, Hupp JW, Ruess RW (2001) Effects of migratory geese on plant communities of an Alaskan salt marsh. J Ecol 89:57–71. https://doi.org/10.1046/j.1365-2745.2001.00515.x
Zak DR, Kling GW (2006) Microbial community composition and function across an arctic tundra landscape. Ecology 87:1659–1670. https://doi.org/10.1890/0012-9658(2006)87[1659:MCCAFA]2.0.CO;2
Zak DR, Holmes WE, White DC, Peacock AD, Tilman D (2003) Plant diversity, soil microbial communities, and ecosystem function: are there any links? Ecology 84:2042–2050. https://doi.org/10.1890/02-0433
Acknowledgements
We thank Maarten Loonen for access to the field sites. ArcBiont project Helmholtz Foundation-UiT for financial support and Edda Marie Rainer for help with the fieldwork. Sequencing was performed at The Norwegian Sequencing Centre. We thank Thomas Rattei for bioinformatics support. The computations were performed on resources provided by UNINETT Sigma2—the National Infrastructure for High Performance Computing and Data Storage in Norway, account nos. NN9639K and NS9593K. We would like to thank the two reviewers Josef Elster and Elie Verleyen for helpful comments.
Funding
Open access funding provided by UiT The Arctic University of Norway (incl University Hospital of North Norway).. A.T.T. was supported by the Research Council of Norway FRIPRO Mobility Grant Project Time and Energy 251027/RU, co-funded by ERC under Marie Curie Grant 608695, and Tromsø Research Foundation starting grant project Cells in the Cold 17_SG_ATT. K.M.B was supported by Tromsø Research Foundation starting grant project Cells in the Cold 17_SG_ATT.
Author information
Authors and Affiliations
Contributions
ATT, KMB and MMS conceived the study. KMB and ATT performed fieldwork. KMB, YH, JS and BJ performed experiments. AR contributed pore water analyses. KMB and ATT analyzed the data. KMB and ATT wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Research involving human and animal participants
The research was performed without involving human participants or animals subjects. We declare informed consent.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bender, K.M., Svenning, M.M., Hu, Y. et al. Microbial responses to herbivory-induced vegetation changes in a high-Arctic peatland. Polar Biol 44, 899–911 (2021). https://doi.org/10.1007/s00300-021-02846-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00300-021-02846-z