
Abstract
Seabirds and their response to climate perturbations are important bioindicators of changes in Antarctic ecosystems. During 30 years of observations of two chinstrap penguin (Pygoscelis antarcticus) colonies, one on King George Island and the other on Penguin Island (South Shetland Islands, Antarctica), the size of the breeding populations decreased by 84 and 41 %, respectively. We applied analyses of amplified fragment length polymorphisms to study the genetic structure of the two populations and to evaluate the influence of the sudden population decrease. Our data indicate that there were only weak genetic differences between the populations, which were not strong enough to support the hypothesis of population differentiation. Weak genetic differences observed between the two populations seem not to be determined by selection processes. We hypothesize that the very low level of between-population genetic structure can be explained by some extent of genetic drift, which is largely compensated by gene flow. Moreover, the two populations seem to remain in a stationary state. Our results support the hypothesis of limited natal philopatry in chinstrap penguins. The observed decrease in population size is probably caused by emigration or a rise in juvenile mortality due to the increasing krill limitation of the marine food web. However, detailed research is required to address this issue.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In the nineteen and mid-twentieth centuries, hunters depleted many seal and whale populations in the Southern Ocean. It had been suggested that these populations had competed with penguins for the same food sources, mainly krill, and that the decreases in the populations of marine mammals allowed some species of seabirds, including penguins, to increase in abundance (Trivelpiece and Volkman 1979). This so-called krill surplus hypothesis has been challenged and does not appear to be valid for minke whales (Ruegg et al. 2010). However, it is still invoked to explain the population dynamics of other krill predators. For example, chinstrap penguin (Pygoscelis antarcticus) populations experienced significant changes from the 1930s to the 1970s (Conroy 1975). Many colonies increased 6–10 % per annum (Laws 1985), and at some localities fivefold increases occurred during the mid-twentieth century (Rootes 1988). However, during the last three decades of observations the number of chinstrap penguin nests on King George Island and Penguin Island (South Shetland Islands) showed strong declining tendencies (Ciaputa and Sierakowski 1999; Hinke et al. 2007; Sander et al. 2007a, b; Korczak-Abshire 2010; Trivelpiece et al. 2011). A similar trend has been observed at Livingston Island (South Shetland Islands) since the mid-1970s (Hinke et al. 2007), as well as in almost the entire western Antarctic Peninsula region (Trathan et al. 1996; Forcada et al. 2006). This might have resulted from a decrease in sea ice cover followed by reduced krill abundances (Fraser et al. 1992; Hinke et al. 2007; Trivelpiece et al. 2011) or due to the long-term protection (ATCM XXIX 2006) of whale and fur seal (Arctocephalus spp.) populations that started to rebound from past exploitation, increasing the competition for the same food as preferred by penguins (Wilkinson and Bester 1990; Bester et al. 2009). Furthermore, some penguin nesting areas in the vicinity of the Arctowski research station were lost due to human activity (Chwedorzewska and Korczak 2010; Korczak-Abshire 2010).
According to some authors, chinstrap penguins had an estimated global population of 6.5–7.5 million pairs (Croxall et al. 1984; Woehler 1993; Williams 1995). Even if those data are overestimated, the fact is that this species is the most numerous pygoscelid penguins, which also include Adélie (P. adeliae) and gentoo (P. papua) penguins. Chinstrap penguins breed in the northern regions of Antarctica, mainly on the Antarctic Peninsula and associated archipelagos. During winter, chinstrap penguins leave their breeding colonies and spend the winter in open water. All pygoscelid species exhibit natal philopatry, the likelihood that individuals breed at/or near their place of origin. However, chinstrap penguins are generally less philopatric than the other two pygoscelid species (Ainley et al. 1995; Macdonald et al. 2002). During the breeding season, chinstrap penguins typically forage 30–90 km from their colonies (Trivelpiece et al. 1987; Ainley et al. 1995). Their diet is dominated by Antarctic krill (Euphausia superba) (Volkman et al. 1980), but various fish species and amphipods were also occasionally recorded in their diet (Rombola et al. 2003).
Satellite telemetry studies conducted to determine the winter movements of chinstrap penguins from King George Island showed differences in winter migratory behaviour of this species (Wilson et al. 1998; Trivelpiece et al. 2007). These differences may reflect individual ties to two different ancestral epicentres of chinstrap penguin populations: a well-established site on the South Shetland Islands and relatively recent one that arose from emigration during the expansion of this species in the mid-1900s (Trivelpiece et al. 2007).
Long-term observations of chinstrap penguin populations from the South Shetland Islands and their well-documented reproductive ecology and behaviour make these populations some of the best described colonies with currently strong decreasing tendencies (e.g. Volkman and Trivelpiece 1981, Jabłoński 1986; Trivelpiece et al. 1987, 2011; Sierakowski 1991; Hinke et al. 2007). Thus, biological responses of these populations may serve as bioindicators of Antarctic ecosystem changes (Croxall et al. 2002; Kato et al. 2004; Hinke et al. 2007; Sander et al. 2007a, b; Ballerini et al. 2009; Lynch et al. 2009).
The International Union for the Conservation of Nature and Bird Life International currently list the conservation status of chinstrap penguins as “least concern” because of their estimated large global population and large range (IUCN 2011). However, given that at least some populations of this species experienced a sudden decrease in size (Hinke et al. 2007; Sander et al. 2007a, b; Trivelpiece et al. 2011), investigation into the potential effects of such a sudden decreases on the genetic structure of chinstrap penguins should provide information on the condition of local populations.
The aim of this study was to explore the genetic diversity and population structure of two declining chinstrap penguin populations from the South Shetland Islands using amplified fragment length polymorphism (AFLP) analyses (Busch et al. 2000; Milot et al. 2008; Chwedorzewska et al. 2010; Hoffman et al. 2012). Falling population sizes can lead to loss of neutral genetic variation, fixation of mildly deleterious alleles, and thereby reduced population fitness (Kalinowski and Waples 2002; Baker 2006; Markert et al. 2010). The presence of genetic differences between the two populations, separated by only short distances, can be interpreted as evidence of a limited between-population gene flow and, hence, natal philopatry.
Materials and methods
Population census
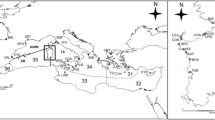
The investigated chinstrap penguin colonies are located on the western shore of Admiralty Bay, Antarctic Specially Protected Area (ASPA 128), King George Island (62°10′S, 58°30′W) (population A = 1,116 breeding pairs) and Penguin Island (62°06′S, 57°56′W) (population B = 4,161 breeding pairs). These colonies were investigated during the austral summer of 2006/2007. The distance between the two study colonies is approximately 32 km (Fig. 1).

King George Island and Penguin Island, South Shetland Islands, Antarctica. Places of penguin feather samples collection for DNA analysis marked in grey
Molecular analysis and scoring
Body feathers from 167 individuals (n = 90 for population A and n = 77 for population B) were collected. Three of the smallest tail feathers were taken from each individual with tweezers. Samples were immediately stored in 70 % ethanol. To avoid resampling the same penguin, feathers were collected during egg incubation when birds stayed on their nests. Individuals were chosen randomly from each breeding group from both colonies. DNA extraction and AFLP analysis were performed in laboratory of the Plant Breeding and Acclimatization Institute National Research Institute, Radzików, Poland.
DNA was extracted from fully developed body feathers not contaminated with penguin guano (n = 89 for population A and n = 75 for population B). From two or three individual feathers, a 1-cm segment was cut from the calamus end and fragmented using a scalpel. Penguin feather quills contain soft tissue and some blood sufficient for DNA extraction. Lysis was performed at 56 °C overnight with gentle shaking in a water bath. DNA was extracted by DNeasy Tissue Kit (Qiagen, Hilden, Germany). The purity and quantity of each sample were determined spectrophotometrically. DNA integrity and RNA impurities were tested by agarose gel electrophoresis. After quality and quantity tests, only 122 (n = 79 for population A and n = 43 for population B) DNA samples were sufficiently pure to be used for further analysis.
The amplified fragment length polymorphism (AFLP) technique was performed according to the procedure described by Vos et al. (1995) with minor modifications (Chwedorzewska et al. 2005, 2008, 2010). Briefly, 250 ng of genomic DNA was digested simultaneously with two restrictive enzymes EcoRI and MseI following ligation of the appropriate adaptors, preselective and finally, selective amplification steps. For the selective amplification, four primer pair combinations were used (E-AGT/M-CTT, E-ATG/M-CGA, E-AGG/M-CTT, E-AAA/M-CGT). The EcoRI compatible primers were labelled at their 5′-ends with γ-32P-ATP. PCR products were separated on 7 % PAGE, and X-ray films were exposed to the gels at −70 °C overnight. Reproducible (i.e. clearly distinguishable) AFLP fragments were scored twice by two independent persons and stored in the form of a binary matrix where 1 indicates the presence of a band and 0 its absence.
Statistical analyses
Non-redundant markers were evaluated by means of AFLpop 1.1 (Duchesne and Bernatchez 2002). GenAlex 5.3 (Peakall and Smouse 2001) was used to evaluate allele frequencies, number of markers shared among individuals with a frequency ≥5 %, number of private markers, number of different alleles (Na), number of effective alleles (Ne), Shannon’s information index (I), expected heterozygosity (He), percentage of polymorphic alleles (P %), Nei’s genetic distance (Nei GD). Molecular variance (Φ PT ) values, computed with GenAlex (Halliburton 2004), were used to estimate gene flow rates using the following formula: Nm = (1–Φ PT )/4Φ PT (Wright 1969). The polymorphism information content (PIC) was calculated based on allele frequencies using the following formula: PIC = 2xf i x(1–f i ), where f i states for the i allele frequency (Roldán-Ruiz et al. 2000). The pop-assign excel add-in (Bernatchez and Duchesne 2000) allowed approximation of the minimum number of loci required for population differentiation success. The linkage disequilibrium (LD) between loci within samples (Excoffier and Lischer 2010) was tested using 10,000 permutations in Arlequin 3.5 (Excoffier and Lischer 2010). Tests of LD were performed for all pairs of loci. The percentage of pairs of loci in LD using polymorphic markers was estimated for each population. The difference in LD values between populations was tested using χ2 test. AFLP binary data sets of the two separate populations, as well as the entire selection of individuals, were analysed for LD using a modified index of association (I A ) equation in MultiLocus v.1.3b. Significance of I A was determined by randomization (1,000 times) procedures by comparing the observed value of I A with that expected under the null hypothesis of complete panmixia.
To visualize the genetic differences between the populations, a NeighborNet was constructed using SplitsTree 4.6 (Huson and Bryant 2006). In addition, analyses of the population structure were performed by Bayesian clustering of the AFLP data using structure 2.2.3 (Pritchard and Wen 2003) in two separate runs: (1) on the AFLP data matrix containing all the loci and (2) on the data matrix without loci showing significant LD. Results of both runs were analysed, and the ΔK(K) functions were calculated in order to estimate the modal values of ΔK(K). Each simulation was performed using the length of burn-in and MCMC (Markov chain Monte Carlo) (500,000 each) at Bioportal (Kumar et al. 2009) to quantify the amount of variation of the likelihood for each K. The range of possible Ks tested was 1–10. The maximum ΔK(K) values were used as estimates of Ks (Evanno et al. 2005).
Deviation from selection neutrality was tested using Tajima’s D and Fu’s F S tests using the Arlequin 3.5 software (Schneider et al. 2000).
Historical demographic expansions were tested by means of analysis of mismatch distribution (Excoffier and Laurent 2004). The parameters of the demographic expansion θ 0 and θ 1 were estimated by a generalized nonlinear least-square approach, and confidence intervals of the parameters were computed using a parametric bootstrap approach (Schneider and Excoffier 1999). The mismatch distribution was performed to distinguish between a smooth unimodal distribution and a multimodal or ragged distribution (Rogers 1995). The raggedness (r) statistic was calculated to quantify the smoothness of the mismatch distribution (Harpending et al. 1993). The significance of raggedness index was calculated to assess the goodness-of-fit of expansion model. All analyses were calculated in Arlequin 3.5.
The presence of putative loci under positive and balancing selection was evaluated with the Mcheza software (http://popgen.eu/soft/mcheza/user.html). “Neutral” mean F ST , force mean F ST options and infinite allele model were used for computations.
Results
Census data
The analysis of these short-term, from 1978 to 2009, changes show that the investigated penguin colonies at King George Island represent different dynamics compared with those on Penguin Island (Fig. 2). From 1978 to 1981, a rapid (27.3 %) decrease of chinstrap penguin populations was observed at King George Island, while the penguins at Penguin Island increased by 24.6 %. However, a comparison of the recent census data with those from 1978 showed that over the extended period both breeding populations decreased dramatically. A loss of 84 and 41 % were recorded at King George Island (population A) and Penguin Island (population B), respectively.

Numbers of breeding pairs of Pygoscelis antarcticus on investigated King George Island colony (population A) and on Penguin Island colony (population B) from 1978 to 2007. Census on King George Island (A) colony on 1978/1979 taken from Jabłoński (1986), 1979/1980–Jabłoński (1984), 1980/1981–Trivelpiece et al. (1987), 1988/1989—Sierakowski (1991), 1989/1990–Rakusa-Suszczewski and Sierakowski (1993), 1990/1991, 1992/1993, 1994/1995, 1995/1996, 1996/1997–Ciaputa and Sierakowski (1999). Census on Penguin Island (B) colony on1979/1980 taken from Jabłoński (1980), 1980/1981–Trivelpiece et al. (1987), 1999/2000, 2000/2001–Pfeiffer and Peter (2004), 2003/2004–Sander et al. (2007a)
Polymorphism
At total of 228 markers were amplified by 4 selective primer pairs; only 82 markers identified for both populations were non-redundant. Only a few private bands for each population were identified, three for population A and one for population B. It was estimated that 27 markers should be sufficient to differentiate populations with 0.99 probability of success. The majority of markers shared between populations appeared with frequencies exceeding 5 %. More than 97.6 % of the non-redundant markers identified among samples from population A and 86.6 % from population B were polymorphic, in total 92.1 % (Table 1).
The linkage disequilibrium (LD) was examined for 5,792 pairs of loci of which 528 were significant, at least at the 5 % level in the case of the population A and 4,480 combinations for the population B with 490 significant LDs. Percentage of pairs of loci in LD for each population was 9.12 and 10.94 %, respectively, and was higher than expected simply by chance. No significant difference was detected in the proportion of significant LD values between the two populations (χ2 = 0.165, df = 1, p > 0.68).
When populations were considered separately, the observed index of association (I A ) across loci in population A (I A = 0.03) indicated no significant correlation (p = 0.4) of alleles across loci. The same was true for population B (I A = 0.26, p = 0.1). Thus, the null hypothesis of complete panmixia within populations could not be rejected. The observed I A value (I A = 14.96) for all analysed accessions from both populations was not significantly higher (p = 0.2) than the I A calculated from 1000 artificially recombined data sets, which suggests that complete panmixia for all analysed accessions, between both populations can also not be rejected.
Genetic structure and gene flow
The NeighborNet analysis indicated only weak genetic differences between the two populations (Fig. 3).

NeighborNet derived from non-redundant AFLP markers and based on the matrix of p distances
The Bayesian clustering of the AFLP data yielded K = 1 for both runs (encompassing either all loci or without loci showing significant LD) and did thus also suggest no significant between-population differences.
The other employed methods also indicated only small genetic distances between both populations, Nei’s genetic distance = 0.025, Φ PT = 0.077.
The Nm (gene flow) value was estimated to be 2.997.
Neutrality tests and demography
Thirty-five of 82 non-redundant markers appeared to be under selection pressure: three loci under positive selection and 32 loci under balancing selection.
Tajima’s D neutrality tests for populations A and B were insignificant but slightly positive (not shown) while Fu’s F S statistics were significantly negative in both populations (Table 2).
Harpending’s raggedness indexes were lower than 0.05 for both populations (A = 0.0039, p = 0.07 and B = 0.0046, p = 0.43). The tests of the goodness-of-fit of the observed mismatch distribution to the expected under demographic expansion using the sum of squared deviations (SSD) statistics were insignificant for both populations (A = 0.0021, p = 0.06 and B = 0.0014, p = 0.497). Spatial expansion was tested using mismatch distribution. Raggedness indexes were r = 0.0039 (p = 0.11) for population A and r = 0.0046 (p = 0.44) for population B. The SSD statistics were 0.0021 (p = 0.07) for population A and 0.0014 (p = 0.51) for population B (Fig. 4).

Mismatch distribution, demographic and spatial expansion of King George Island (A) and Penguin Island (B) populations
Discussion
The analysis of changes over the past 30 years show that the penguin colonies at King George Island represent different dynamics compared with those on Penguin Island. However, comparison of the recent census data with those from 1978 shows that over the extended period these two chinstrap penguin breeding populations are dramatically decreasing. A loss of 84 and 41 % were recorded at King George Island and Penguin Island, respectively (Fig. 2).
One of the most obvious effects of climate change in the Antarctic Peninsula region is the fluctuation of sea ice coverage (Curran et al. 2003). This strongly affects the abundance of food for penguin and other sea predator populations (Loeb et al. 1997; Hinke et al. 2007; Ballerini et al. 2009). Also, human activity influences changes in Antarctic seabird and marine mammal populations (Chwedorzewska and Korczak 2010). Reported decreases of chinstrap penguin populations from the South Shetland Islands indicate necessity of providing information about the genetic diversity of local population. Two declining chinstrap penguin populations from the South Shetland Islands were investigated to answer the question how a sudden decrease in population size affect genetic structure of local penguin populations.
All measures of genetic variation (He, N, Ne, I, PIC) (Table 1) showed that the versatile method AFLP was sufficient for genetic diversity studies. Although analyses of LD revealed numerous significant linkages between pairs of loci of individuals representing each population (ca 10 %), they appeared to be sporadic. There was no specific locus pair, which would have been in disequilibrium in each population, and the level of LD for the two populations was comparable. Analyses of associations demonstrated that there are numerous associated loci between both populations (I A = 14.96), but they were not significant. Thus, the two investigated populations may exchange, at least sporadically, genetic information, which is in agreement with the high level of gene flow identified between them. Analyses of LD demonstrated that individuals within each population (A and B) and also among populations mate randomly and panmixia cannot be rejected.
Analysis of outliers also show evidence of positive and balancing selection (35 polymorphic loci were candidate for selection). It is assumed that positive selection is responsible for adaptive traits. There are only 3 putative loci under positive selection. Thus, the presence of only three loci under positive selection of total 35 may indicate that adaptive selection is probably not the driving force that is responsible for population differentiation. The presence of numerous loci under balancing selection (n = 32) is in agreement with random distribution of pairs of loci in LD.
Both NeighborNet analysis and Bayesian clustering of our AFLP data suggested no significant population differentiation. A low level of genetic population structure was further supported by low values of Nei’s genetic distance and Φ PT . The lack of or the presence of only a very weak population genetic structure between the two investigated chinstrap penguin populations could be explained by gene flow between the colonies. This finding supports the hypothesis that the natal philopatry is less in chinstrap penguins than in the other two species of pygoscelids (Ainley et al. 1995; Macdonald et al. 2002). An abatement in the philopatric behaviour could also be an adaptive response to the significant population decrease to minimize adverse effects on genetic diversity. However, this hypothesis cannot be tested on the basis of our data. The hypothesis that there is some gene exchange between populations A and B is in accordance with the findings of Hinke et al. (2007) who recorded ca 1 % emigrants between two adjacent colonies in Admiralty Bay, King George Island, during 30 years of continuous monitoring.
One of the most intriguing questions concerning the analysed penguin populations is their diminishing population size in the monitored region. There might be several reasons for this. First of all, a lack of sufficient food may result in increased mortality of juveniles; unsuccessful attempts to hunt krill in the vicinity of their habitats may force them to forage elsewhere where they may fail and die (Hinke et al. 2007; Trivelpiece et al. 2011), thereby reducing the population. Alternatively, adults may move to new locations while seeking food. Additionally, the fact that chinstrap penguins are less philopatric than the other two pygoscelids suggests some population “dispersion”.
Selection neutrality tests revealed significant negative values of Fu’s F S for both populations, indicating the presence of demographic processes, positive selection or genetic hitchhiking (i.e. changes in an allele’s frequency due to any form of selection operating upon linked genes). Demographic processes were excluded based on the analysis of the mismatch distribution, which demonstrated that the mutation-drift equilibrium hypothesis was close to rejection only for population A. When spatial expansion was tested, neither population seemed to fit to the model indicating a stationary population state. Analysis of outliers demonstrated that positive selection was also not evident. However, the presence of numerous loci under LD within each population may be partly explained by genetic hitchhiking. Some demographic changes and subsequent effects on the genetic structure can also be expected, especially because the investigated populations, albeit declining (population A more than population B), are still part of a very large global chinstrap penguin population (IUCN 2011). To observed population declines could be a beginning of such demographic changes.
In summary, our data revealed only weak genetic differences between the chinstrap penguin populations on King George Island and Penguin Island. The differences were not strong enough to support the hypothesis of population differentiation. We hypothesize that the very low level of population genetic structure can be explained by some extent of genetic drift that is largely compensated by considerable gene flow between populations. Moreover, some extent of genetic hitchhiking seems to be present. Our results support the hypothesis of rather limited natal philopatry in chinstrap penguins. The question is whether the exemptions from a strong natal philopatry of penguin populations already existed before the population declines or whether it is an adaptive response to the significant population decreases to minimize adverse effects on genetic diversity? We hypothesize that the observed decrease in population size is probably caused by emigration and/or rising juvenile mortality due to increasing krill limitation in the marine food web. However, further research is required to address this issue. It seems that the use of codominant genetic markers (such as microsatellites) would be desirable in future research on chinstrap penguins. This methodological approach would be probably more useful in separating the effects of selection and demographic process on population differentiation. Our results, obtained employing AFLP methodology, seem to be a good base for further genetic studies of penguin populations from different localities supported by the data from long-term monitoring.
References
Ainley DG, Nur N, Woehler J (1995) Factors affecting the distribution and size of pygoscelid penguin colonies in Antarctica. Auk 112:171–182
ATCM XXIX (2006) Proposal to de-list Antarctic fur seals as specially protected species. www.scar.org/treaty/atcmxxix/atcm29_wp039.pdf
Baker AJ (2006) Population declines and the risk of extinction in waders: genetic and ecological consequences of small population size. In: Boere GC, Galbraith CA, Stroud DA (eds) Waterbirds around the world. Edinburgh, UK, pp 668–671
Ballerini T, Tavecchia G, Olmastroni S, Pezzo F, Focardi S (2009) Nonlinear effects of winter sea ice on the survival probabilities of Adélie penguins. Oecologia 161:253–265
Bernatchez L, Duchesne P (2000) Individual-based genotype analysis in studies of parentage and population assignment: how many loci, how many alleles ? Can J Fish Aquat Sci 57:1–12
Bester MN, Ryan PG, Visagie J (2009) Summer survey of fur seals at Prince Edward Island, southern Indian Ocean. Afr J Mar Sci 31:1–5
Busch JD, Miller MP, Paxton EH, Sogge MK, Keim P (2000) Genetic variation in the endangered southwestern willow flycatcher. Auk 117:586–595
Chwedorzewska KJ, Korczak M (2010) Human impact upon the environment in the vicinity of Arctowski Station, King George Island, Antarctica. Pol Polar Res 31:45–60
Chwedorzewska KJ, Wojtuń B, Bednarek PT (2005) Genetic and morphological variation of Saxifraga cespitosa L. from Spitsbergen (Svalbard)—a preliminary study. Pol Biol 28:802–804
Chwedorzewska KJ, Kośiński I, Galera H (2008) Plantations of Convallaria majalis L. as a threat to the natural stands of the species: the genetic variability of the cultivated plants and the natural populations. Biol Conserv 141:2619–2624
Chwedorzewska KJ, Korczak M, Bednarek PT, Markowska-Potocka M (2010) Low genetic differentiation between two morphotypes of the gastropod Nacella concinna from Admiralty Bay, Antarctica. Pol Polar Res 31:195–200
Ciaputa P, Sierakowski K (1999) Long-term population changes of Adélie, chinstrap, and gentoo penguins in the regions of SSSI No. 8 and SSSI No. 34, King George Island, Antarctica. Pol Polar Res 20:55–365
Conroy JWH (1975) Recent increases in penguin populations in Antarctica and the Sub Antarctic. In: Stonehouse B (ed) The biology of penguins. Macmillan, London, pp 321–336
Croxall JP, Prince PA, Hunter I, McInnes SJ, Copestake PG (1984) The seabirds of the Antarctic Peninsula, islands of the Scotia Sea, and Antarctic continent between 80°W and 20°W: their status and conservation. In: Croxall JP, Evans PGH, Schreiber RW (eds) Status and conservation of the world’s seabirds, ICBP technical publication no. 2. ICBP, Cambridge, pp 637–666
Croxall JP, Trathan PN, Murphy EJ (2002) Environmental change and Antarctic seabird populations. Science 297:1510–1514
Curran MAJ, Van Ommen TD, Morgan VI, Phillips KL, Palmer AS (2003) Ice core evidence for Antarctic sea ice decline since the 1950s. Science 302:1203–1206
Duchesne P, Bernatchez L (2002) AFLPOP: a computer program for simulated and real population allocation based on AFLP data. Mol Ecol Notes 3:380–383
Evanno G, Regnaut VS, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Laurent HL (2004) Patterns of DNA sequence diversity and genetic structure after a range expansion: lessons from the infinite-island model. Mol Ecol 13:853–864
Excoffier L, Lischer HL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 3:564–567
Forcada J, Trathan PN, Reid K, Murphy EJ, Croxall JP (2006) Contrasting population changes in sympatric penguin species in association with climate warming. Glob Change Biol 12:411–423
Fraser W, Trivelpiece WZ, Ainley DG, Trivelpiece SG (1992) Increases in Antarctic penguin populations: reduced competition with whales or a loss of sea ice due to environmental warming? Polar Biol 11:525–531
Halliburton R (2004) Introduction to population genetics. Pearson Education Int, Upper Saddle River
Harpending HC, Sherry ST, Rogers AR, Stoneking M (1993) The genetic structure of ancient human populations. Curr Anthropol 34:483–496
Hinke JT, Salwicka K, Trivelpiece SG, Watters GM, Trivelpiece WZ (2007) Divergent responses of Pygoscelis penguins reveal a common environmental driver. Oecologia 153:845–855
Hoffman JI, Clark MS, Amos W, Peck LS (2012) Widespread ampliffation of ampliffed fragment length polymorphisms (AFLPs) in marine Antarctic animals. Polar Biol 35:919–929. doi:10.1007/s00300-011-1139-2
Huson DH, Bryant D (2006) Application of phylogenetic networks in Evolutionary studies. Mol Biol Evol 23:254–267
IUCN 2011. IUCN red list of threatened species. version 2011.2. www.iucnredlist.org downloaded on 24 Nov 2011
Jabłoński B (1980) Distribution and numbers of birds and pinnipeds on Penguin Island (South Shetland Islands) in January 1979. Pol Polar Res 1:109–116
Jabłoński B (1984) Distribution, number and breeding preferences of penguins in the region of the Admiralty Bay (King George Islands, South Shetland Islands) in the season 1979/80. Pol Polar Res 5:5–16
Jabłoński B (1986) Distribution, abundance and biomass of a summer community of birds in the region of the Admiralty Bay (King George Islands, South Shetland Islands, Antarctica) in 1978/79. Pol Polar Res 7:217–260
Kalinowski ST, Waples RS (2002) The ratio of effective to census size in fluctuating populations. Conserv Biol 16:129–136
Kato A, Watanabe K, Naito Y (2004) Population changes of Adélie and emperor penguins along the Prince Olav Coast and on the Riiser-Larsen Peninsula. Polar Biosci 17:117–122
Korczak-Abshire M (2010) Climate change influences on Antarctic bird populations. Pap Glob Change 17:43–53
Kumar S, Skjaeveland A, Orr RJ, Enger P, Ruden T, Mevik BH, Burki F, Botnen A, Shalchian-Tabrizi K (2009) AIR: a batch-oriented web program package for construction of supermatrices ready for phylogenomic analyses. BMC Bioinfo 10:357. doi:10.1186/1471-2105-10-357
Laws RM (1985) The ecology of the Southern Ocean. Am Sci 73:26–40
Loeb V, Siegel V, Holm-Hansen O, Hewitt R, Fraser W, Trivelpiece W, Trivelpiece S (1997) Effects of sea-ice extent and krill or salp dominance on the Antarctic food web. Nature 387:897–900
Lynch HJ, Fagan WF, Naveen R (2009) Population trends and reproductive success at a frequently visited penguin colony on the western Antarctic Peninsula. Polar Biol 33:493–503
Macdonald JA, Barton KJ, Metcalf P (2002) Chinstrap penguins (Pygoscelis antarctica) nesting on Sabrina Islet, Balleny Islands, Antarctica. Polar Biol 25:442–447
Markert JA, Champlin DM, Gutjahr-Gobell R, Grear JS, Kuhn A, McGreevy TJ, Roth A, Bagley MJ, Nacci DE (2010) Population genetic diversity and fitness in multiple environments. BMC Evol Biol 10:205. doi:10.1186/1471-2148-10-205
Milot E, Weimerskirch H, Bernatchez L (2008) The seabird paradox: dispersal, genetic structure and population dynamics in a highly mobile, but philopatric albatross species. Mol Ecol 17:1658–1673
Peakall R, Smouse PE (2001) GenALEx V5 Genetic analysis in excel. Population genetic software for teaching and research. Canberra, Australian. www.anu.edu.au/BOZo/GenALEx/
Pfeiffer S, Peter H-U (2004) Ecological studies toward the management of an Antarctic tourist landing site (Penguin Island South Shetland Islands). Polar Rec 40:1–9
Pritchard JK, Wen W (2003) Documentation for structure software: version 2. University of Chicago, Chicago. http://pritch.bsd.uchicago.edu/software/readme_2_1/readme.html
Rakusa-Suszczewski S, Sierakowski K (1993) Pinnipeds in Admiralty Bay, King George Island, South Shetlands (1988–1992). Pol Polar Res 14:441–456
Rogers AR (1995) Genetic evidence for a pleistocene population explosion. Evolution 49:608–615
Roldán-Ruiz I, Dendauw J, van Bockstaele E, Depicker A, De Loose M (2000) AFLP markers reveal high polymorphic rates in ryegrasses (Lolium spp.). Mol Breed 6:125–134
Rombola E, Marschoff E, Coria N (2003) Comparative study of the effects of the late pack-ice break-off on chinstrap and Adelie penguins’ diet and reproductive success at Laurie Island, South Orkney Islands, Antarctica. Polar Biol 26:41–48
Rootes DM (1988) The status of birds at Signy Island, South Orkney Islands. Brit Antarct Surv B 80:87–119
Ruegg KC, Anderson EC, Baker CS, Vant M, Jackson J, Palumbi SR (2010) Are Antarctic minke whales unusually abundant because of 20th century whaling? Mol Ecol 19:281–291
Sander M, Coelho Balbăo T, Schneider Costa E, Rodrigo dos Santos C, Petry MV (2007a) Decline of the breeding population of Pygoscelis antarctica and Pygoscelis Adeliae on Penguin Island, South Shetland Antarctica. Polar Biol 30:651–654
Sander M, Coelho Balbăo T, Polito MJ, Schneider Costa E, Bertoldi Carneiro AP (2007b) Recent decrease in chinstrap penguin (Pygoscelis antarctica) populations at two of Admiralty Bay’s islets on King George Island, South Shetland Islands, Antarctica. Polar Biol 30:659–661
Schneider S, Excoffier L (1999) Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics 152:1079–1089
Schneider MP, Erdmann J, Delles C, Fleck E, Regitz-Zagrosek V, Schmieder RE (2000) Functional gene testing of the Glu298Asp polymorphism of the endothelial NO synthase. J Hypertens 18:1767–1773
Sierakowski K (1991) Birds and mammals in the region of SSSI No. 8 in the season 1988/89 (South Shetlands, King George Island, Admiralty Bay). Pol Polar Res 12:25–54
Trathan PN, Croxall JP, Murphy EJ (1996) Dynamics of Antarctic penguin populations in relation to inter-annual variability in sea ice distribution. Polar Biol 16:321–330
Trivelpiece W, Volkman N (1979) Nest-site competition between adelie and chinstrap penguins: an ecological interpretation. Auk 96:675–681
Trivelpiece WZ, Trivelpiece SG, Volkman N (1987) Ecological segregation of Adelie, gentoo, and chinstrap penguins at King George Island, Antarctica. Ecology 68:351–361
Trivelpiece WZ, Buckelew S, Reiss C, Trivelpiece SG (2007) The winter distribution of chinstrap penguins from two breeding sites in the South Shetland Islands of Antarctica. Polar Biol 30:1231–1237
Trivelpiece WZ, Hinke JT, Miller AK, Reiss CS, Trivelpiece SG, Watters GM (2011) Variability in krill biomass links harvesting and climate warming to penguin population changes in Antarctica. PNAS 108:7625–7628
Volkman NJ, Trivelpiece W (1981) Nest-site selection among Adelie, chinstrap and gentoo penguins in mixed species rookeries. Wilson Bull 93:243–248
Volkman NJ, Presler P, Trivelpiece W (1980) Diets of Pygoscelid penguins at King George Island, Antarctica. Condor 82:373–378
Vos P, Hogers R, Bleeke M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zauber M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
Wilkinson IS, Bester MN (1990) Continued population increase in fur seals, Arctocephalus tropicalis and A. gazella, at the prince Edward Islands. S Afr T Nav Antarkt 20:58–63
Williams TD (1995) The penguins: Spheniscidae. Oxford University Press, Oxford
Wilson RP, Culik BM, Kosiorek P, Adelung D (1998) The over-winter movements of a chinstrap penguin (Pygoscelis antarctica). Polar Rec 34:107–112
Woehler EJ (1993) The distribution and abundance of Antarctic and Sub-Antarctic penguins. Scientific Committee on Antarctic Research, Cambridge
Wright S (1969) Evolution and the genetics of populations. The theory of gene frequencies, vol 2. University of Chicago Press, Chicago
Acknowledgments
The authors wish to thank the members of the 31st Polish Antarctic Expedition on Arctowski Station for logistical support during field season, especially Piotr Angiel M.Sc. who also helped in collecting feather samples. The authors would also like to thank Dr Anna Kidawa for her professional opinion. The authors would also like to thank the anonymous reviewers for their critical comments and valuable suggestions, which helped to improve this manuscript. This research was supported by the Ministry of Scientific Research and Higher Education grant IPY/268/2006.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Korczak-Abshire, M., Chwedorzewska, K.J., Wąsowicz, P. et al. Genetic structure of declining chinstrap penguin (Pygoscelis antarcticus) populations from South Shetland Islands (Antarctica). Polar Biol 35, 1681–1689 (2012). https://doi.org/10.1007/s00300-012-1210-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00300-012-1210-7