
Abstract
This paper presents an analytical framework for analyzing polymorphisms created by two mutation events in samples of DNA sequences modeled in the general coalescent tree setting. I developed the framework by deriving analytical formulas for the numbers of the topologies of the genealogies with two mutation events. This approach gives an advantage to analyze polymorphisms in large samples of DNA sequences at a non-recombining locus under vicarious evolutionary scenarios. Particularly the framework allows to estimate the probability of polymorphism data created by two mutation events as well as the ages of the events. Based on these results I extended the definition of the site frequency spectrum by classifying pairs of polymorphic sites into groups and presented analytical expressions for computing the expected sizes of these groups. Within the framework I also designed a Bayesian approach for inferring the haplotype of the most recent common ancestor at two polymorphic sites. Lastly, the framework was applied to polymorphism data from human APOE gene region under various demographic scenarios for ancestral human population and explored the signature of linkage disequilibrium for inferring the ancestral haplotype at two polymorphic sites. Interestingly enough, the results show that the most frequent haplotype at two completely linked polymorphic sites is not always the most likely candidate for the haplotype of the most recent common ancestor.





Similar content being viewed by others


References
Cann R, Stoneking M, Wilson A (1987) Mitochondrial DNA and human evolution. Nature 325:31–6
Coop G, Griffiths RC (2004) Ancestral inference on gene trees under selection. Theor Popul Biol 66(3):219–232
Corbo RM, Scacchi R (1999) Apolipoprotein E (APOE) allele distribution in the world. Is APOE*4 a ’thrifty’ allele? Ann Hum Genet 63(Pt 4):301–310
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261(5123):921–923
Davignon J, Gregg RE, Sing CF (1988) Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis 8(1):1–21
de Knijff P, van den Maagdenberg AM, Frants RR, Havekes LM (1994) Genetic heterogeneity of apolipoprotein E and its influence on plasma lipid and lipoprotein levels. Hum Mutat 4(3):178–194
Evans SN, Shvets Y, Slatkin M (2007) Non-equilibrium theory of the allele frequency spectrum. Theor Popul Biol 71(1):109–119
Feller W (1970) An introduction to probability and its applications, 3rd edn. Wiley, New York
Felsenstein J, Kuhner MK, Yamato J, Beerli P (1999) Likelihoods on coalescents: a Monte Carlo sampling approach to inferring parameters from population samples of molecular data. In: Statistics in Molecular Biology and Genetics, IMS Lecture Notes Monogr. Ser., vol 33. Institute of Mathematical Statistics, Hayward, pp 163–185
Forster P (2004) Ice ages and the mitochondrial DNA chronology of human dispersals: a review. Philos Trans R Soc Lond B Biol Sci 359(1442):255–264 discussion 264
Forster P, Matsumura S (2005) Evolution. Did early humans go north or south? Science 308(5724):965–966
Fu YX (1995) Statistical properties of segregating sites. Theor Popul Biol 48(2):172–197
Fullerton SM, Clark AG, Weiss KM, Nickerson DA, Taylor SL, Stengrd JH, Salomaa V, Vartiainen E, Perola M, Boerwinkle E, Sing CF (2000) Apolipoprotein E variation at the sequence haplotype level: implications for the origin and maintenance of a major human polymorphism. Am J Hum Genet 67(4):881–900
Griffiths RC (2003) The frequency spectrum of a mutation, and its age, in a general defusion model. Theor Popul Biol 64:241–251
Griffiths RC, Tavaré S (1994) Sampling theory for neutral alleles in a varying environment. Philos Trans R Soc Lond B 344:403–410
Griffiths RC, Tavaré S (1995) Unrooted genealogical tree probabilities in the infinitely-many-sites model. Math Biosci 127:77–98
Griffiths RC, Tavaré S (1998) The age of a mutation in a general coalescent tree. Commun Stat Stoch Models 14:273–295
Griffiths RC, Tavaré S (1999) The ages of mutations in gene trees. Ann Appl Prob 9(3):567–590
Griffiths RC, Tavaré S (2003) The genealogy of a neutral mutation. In: Green PJ, Hjort NL, Richardson S (eds) Highly Structured Stochastic Systems, Oxford Statistical Science. Oxford University Press, Oxford, pp 393–413
Hammer MF (1995) A recent common ancestry for Human Y chromosomes. Nature 378:376–8
Hobolth A, Uyenoyama M, Wiuf C (2008) Importance sampling for the infinite sites model. Stat Appl Genet Mol Biol 7:32
Hobolth A, Wiuf C (2009) The genealogy, site frequency spectrum and ages of two nested mutant alleles. Theor Popul Biol 75:260–265
Hudson RR (1983) Testing the constant-rate neutral allele model with protein sequence data. Evolution 37:203–217
Hudson RR (1991) Gene genealogies and the coalescent process. In: Futuyma D, Antonovics J (eds) Oxford Surveys in Evolutionary Biology, vol 7. Oxford University Press, Oxford, pp 1–44
Ingman M, Kaessmann H, Pääbo S, Gyllensten U (2000) Mitochondrial genome variation and the origin of modern humans. Nature 408:708–13
Jenkins PA, Song Y (2011) The effect of recurrent mutation on the frequency spectrum of a segregating site and the age of an allele. Theor Popul Biol 80(2):158–173
Jobling M, Tyler-Smith C (2003) The human Y chromosome: an evolutionary marker comes of age. Nature Rev Genet 4:598–612
Kimmel M, Chakraborty R, King JP, Bamshad M, Watkins WS, Jorde LB (1998) Signatures of population expansion in microsatellite repeat data. Genetics 148:1921–30
Kimura M, Ohta T (1973) The age of a neutral mutant persisting in a finite population. Genetics 75:199–212
Kingman JFC (1982a) Exchangeability and the evolution of large populations. In: Koch G, Spizzichino F (eds) Exchangeability in Probability and Statistics. North Holland Publishing Company, Amsterdam, pp 97–112
Kingman JFC (1982b) On the genealogy of large populations. J Appl Prob 19A:27–43
Kingman JFC (1982c) The coalescent. Stoch Process Appl 13:235–248
Kuhner MK, Yamato J, Felsenstein J (1995) Estimating effective population size and mutation rate from sequence data using Metropolis-Hastings sampling. Genetics 140:1421–1430
Kuhner MK, Yamato J, Felsenstein J (1998) Maximum likelihood estimation of population growth rates based on the coalescent. Genetics 149:429–434
Maca-Meyer N, Gonzalez A, Larruga J, Flores C, Cabrera V (2001) Major genomic mitochondrial lineages delineate early human expansions. BMC Genet 2:13
Machado CA, Kliman RM, Markert JA, Hey J (2002) Inferring the history of speciation from multilocus DNA sequence data: the case of Drosophila pseudoobscura and close relatives. Mol Biol Evol 19(4):472–488
Mellars P (2004) Neanderthals and the modern human colonization of europe. Nature 432(7016):461–465
Mellars P (2006) A new radiocarbon revolution and the dispersal of modern humans in eurasia. Nature 439(7079):931–935
Merriwether DA, Clark AG, Ballinger SW, Schurr TG, Soodyall H, Jenkins T, Sherry ST, Wallace DC (1991) The structure of human mitochondrial DNA variation. J Mol Evol 33:543–555
Nee S, May RM, Harvey PH (1994) The reconstructed evolutionary process. Philos Trans R Soc B 344:305–311
Nielsen R (2000) Estimation of population parameters and recombination rates from single nucleotide polymorphisms. Genetics 154(2):931–942
Nordborg M (2001) Coalescent theory. In: Balding D, Bishop M, Cannings C (eds) Handbook of Statistical Genetics. Wiley, Chichester
Pakendorf B, Stoneking M (2005) Mitochondrial DNA and human evolution. Annu Rev Genomics Hum Genet 6:165–183
Pritchard JK, Seielstand MT, Perez-Lezaun A, Feldman MW (1999) Population growth of human Y chromosomes: a study of Y chromosome. Mol Biol Evol 16:1791–1798
Rannala B (1997) Gene genealogy in a population of variable size. J Hered. 78:417–423
Sargsyan O (2006) Analytical and simulation results for the general coalescent. PhD dissertation, University of Southern California
Sargsyan O (2010) Topologies of the conditional ancestral trees and full-likelihood-based inference in the general coalescent tree framework. Genetics 185:1355–68
Sargsyan O, Wakeley J (2008) A coalescent process with simultaneous multiple mergers for approximating the gene genealogies of many marine organisms. Theor Popul Biol 74:104–114
Sawyer SA, Hartl DL (1992) Population genetics of polymorphism and divergence. Genetics 132(4):1161–1176
Slatkin M, Hudson RR (1991) Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129(2):555–562
Slatkin M, Rannala B (1997) Estimating the age of alleles by use of interaallelic variability. Am J Hum Genet 60:447–458
Stengrd JH, Zerba KE, Pekkanen J, Ehnholm C, Nissinen A, Sing CF (1995) Apolipoprotein E polymorphism predicts death from coronary heart disease in a longitudinal study of elderly Finnish men. Circulation 91(2):265–269
Stephens M (2000) Times on trees, and the age of an allele. Theor Popul Biol 57:109–119
Stephens M, Donnelly P (2000) Inference in molecular population genetics. J R Stat Soc B 62:605–655
Stephens M, Donnelly P (2003) Ancestral inference in population genetics models with selection (with discussion). Aust N Z J Stat 45:395–430
Stringer C (2002) Modern human origins: progress and prospects. Philos Trans R Soc Lond B 357:563–579
Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD (1993) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 90(5):1977–1981
Tajima F (1983) Evolutionary relationship of DNA sequences in finite populations. Genetics 105:437–460
Takahata N (1993) Allelic genealogy and human evolution. Mol Biol Evol 10(1):2–22
Tavaré S, Zeitouni O (2004) Ancestral inference in population genetics. In: Picard J (ed) Lectures on Probability Theory and Statistics, Ecole d’Ets de Probabilit de Saint-Flour XXXI - 2001, Lecture Notes in Mathematics, vol 1837. Springer, New York, pp 1–188
Thompson EA (1975) Humman evolutionary trees. Cambridge University Press, Cambridge
Thomson R, Pritchard JK, Shen P, Oefner PJ, Feldman MW (2000) Recent common ancestry of human Y chromosomes Evidence from DNA sequence data. Proc Natl Acad Sci USA 97:7360–7365
Vigilant L, Stoneking M, Harpending H, Hawkes K, Wilson A (1991) African populations and the evolution of human mitochondrial DNA. Science 253:1503–7
Wakeley J (2008) An introduction to coalescent theory. Roberts & Co, Boulder
Watterson GA (1975) On the number of segregating sites in genetical models without recombination. Theor Popul Biol 7:256–276
Weiss G, von Haeseler A (1998) Inference of population history using a likelihood approach. Genetics 149:1539–1546
Wiuf C, Donnelly P (1999) Conditional genealogies and the age of a neutral mutant. Theor Popul Biol 56:183–201
Xie X (2011) The site-frequency spectrum of linked sites. Bull Math Biol 73(3):459–494
Zannis VI, Nicolosi RJ, Jensen E, Breslow JL, Hayes KC (1985) Plasma and hepatic apoE isoproteins of nonhuman primates. Differences in apoE among humans, apes, and New and Old World monkeys. J Lipid Res 26(12):1421–1430
Zivkovic D, Wiehe T (2008) Second-order moments of segregating sites under variable population size. Genetics 180(1):341–357
Acknowledgments
I would like to thank Professor Simon Tavaré for his advice on the early stage of this work and Professor John Wakeley for helpful discussion on the early version of this manuscript. I would like also to thank the two anonymous reviewers for their comments and suggestions for improving the presentation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
1.1 The proof of Lemma 1
I first derive expressions for the numbers \(N_{1}(k,m;b,c,a)\) and \(N_{2}(k,m;a,c,b)\) when \(a, b, c\) are positive. In the derivations I use an additional condition that the the sequences in the sample are labeled and in a particular order. To remove this condition for completing the proof, the derived numbers must be multiplied by \(\genfrac(){0.0pt}0{a+b+c}{a\ \ b\ \ c}.\)
Since \(N_{1}(k,m;b,c,a)\) represents possible topologies of the genealogies with two mutations that are consistent with the data \((a,b,c)\) when the ancestral haplotype at the two sites is \(TT,\) transition history at early and late mutation events is \(H_1=(TT\rightarrow TC\rightarrow CC)\), and the number of ancestral lineages at the early and late mutation events are \(k\) and \(m\), respectively; from the definition of \((k,m)\) follows that \(3 \le m \le b+a+1\), \(k\) is less than or equal to \(m-1\), and \(2\le k \le a+1\), consequently \(2 \le k \le \min (a+1,m-1).\)
The genealogies satisfying the above conditions can be represented as a combination of three parts determined by the early and late mutation events. The first part of the genealogy is between sampling time point and the late mutation event; the second part is the part of the genealogy between the late and early mutation events; the third part is between the early mutation event and the time of the most recent common ancestor of the sample. The numbers of possible topologies in the three parts of the genealogy determine \(N_{1}(k,m;b,c,a)\).
For this purpose I consider the sample as combination of three groups of sequences determined by the haplotypes \(TT, TC, CC.\) In the first part of the genealogy the lineages coalesce only with the lineages of their groups, in other words the groups do not share an ancestor up to the time of the late mutation event. At this time point the number of the lineages associated with the group \(CC\) is 1. Let the numbers of the lineages for the groups of \(TC\) and \(TT\) label by \(m_1\) and \(m_2,\) respectively. The number of topologies of the genealogies conditioned on \(\{m_1, m_2\},\) is a product of the numbers of possible topologies in the three parts of the genealogy. I provide in details the derivation of the number of possible topologies in the first part of the genealogy.
The coalescent events occur one at a time in consecutive order back in time, and the total number of such events on this part of the genealogy is \(a+b+c-m.\) These coalescent events are distributed among the genealogies of the groups \(CC, TC\), and \(TT\) as \(c-1\), \(b-m_1\), and \(a-m_2,\) respectively. Consequently, the number of ways of distributing the coalescent events for the groups is
For a given sequence of the coalescent events among the groups, the topologies of the genealogies of the groups have random joining structure, therefore the numbers of the possible topologies of the genealogies of the groups \(TT, TC, CC\) are computed based on the following formula: If \(i\) lineages are traced back in time and stopped when the number of lineages is \(j\) by randomly joining two lineages at each coalescent event, the number of all possible generated topologies is equal to
From this observation follows that the number of all possible topologies for the first part of the genealogy conditional on \(\{m_1, m_2\},\) is equal to
The same type of approach one can use to compute the numbers of the topologies for the second and third parts of the genealogy (details not shown). These numbers are equal to
and
respectively. Thus, after multiplying the above three expressions and simplifying the product, the following formula holds for the number of the topologies of the genealogies conditional on \(m_1, m_2\):
where
From the definition of \(m_1\) and \(m_2,\) the following inequalities hold
and from these conditions follows that \(m_2\) satisfies
and
consequently
From the above derivations and the boundary conditions in (33) follows that the labeled and particularly ordered version of \(N_{1}(k,m;b,c,a)\) can be represented as the sum
Note that changing the range for \(m_2\) in the above sum to \(\max (k-1,m-b-1) \le m_2 \le \min (a,m-1)\) does not change the result of the sum because the term corresponding to \(m_2=m-1\) is zero.
To compute this sum the following identity is used:
After applying this to the above sum, it can be expressed as
For computing \(J_i,\ i=1,2,3,\) the following formula [see Feller (1970)] is used:
After applying this formula to \(J_1\) the following formula holds
Applying the same formula to \(J_2\) and \(J_3\) the following equations hold:
and
Combining \(J_i,\ i=1,2,3,\) together completes the proof of the formula for \(N_{1}(k,m;b,c,a).\)
Derivation of the formula for \(N_{2}(k,m;a,c,b)\) is very similar to the proof of the previous case. I again consider the genealogy as a combination of three parts determined by the early and late mutation events. The first part of the genealogy is between the sampling time point and the late mutation event, and it is a combination of the genealogies for the groups \(TT, TC, CC\) that do not share ancestor between the groups. The numbers of the ancestral lineages of these groups at the late mutation event are equal to \(m_2, m_1, 1,\) respectively. Note that \(m_2\) and \(m_1\) satisfy \(m=1+m_1+m_2\) and
As a result of these conditions, the following boundary conditions hold for \(m_2\!:\)
\(m\) and \(k\) satisfy conditions \(3 \le m \le a+b+1\) and \(2 \le k \le m.\)
Based on the similar arguments as in the previous case, the number of the topologies of the genealogies with two mutations conditional on \((m_1, m_2)\) is
The sum of these numbers is equal to \(N_{2}(k,m;a,c,b)\!:\)
The range for \(m_2\) in the above sum can be changed to \(\max (1,m-1-b)\le m_2 \le \min (m-k+1,a)\) because \(2 \le k\) and the term corresponding to \(m_2=m-1\) is zero. Using the following identity
the sum in the expression (36) can be represented as
To compute \(I_i,\ i=1,2,3,\) I use the same approach as for \(J_i,\ i=1,2,3\). Thus, the following formulas hold
and
For \(I_2\) and \(I_3\) the following formulas hold
and
Combining the formulas for \(I_i\), \(i=1,2,3\), completes the proof of the formula for \(N_{2}(k,m;a,c,b).\) The arguments for deriving the formulas when \(b=0\) are very similar. The details are not provided.
1.2 The proof of Theorem 1
The probabilities of \(r_n\) and \(h_n\): To show that \(h_n\) and \(r_n\) are independent of the topology of the genealogy, it is enough to show that the statement holds for \(r_n.\) I use an induction with respect to \(n\). Obviously, the statement holds for \(n=2\) and \(n=3\). Assuming that the statement holds for any \(k\) less than \(n, n> 3\), it needs to be shown that the statement also holds for \(n\).
For \(n\) sequences, let \(E_{2r}(G_n)\) be the set of genealogies \(G_n\) with two mutation events occurred on same lineage. This set \(E_{2r}(G_n)\) can be partitioned into disjoint sets \(E_{2r}(G_n, \ell )\) determined by the condition that the late mutation event occurred on branch \(\ell .\) Each of these genealogies can be considered as a result of a branching event at time \(T_n\) from a lineage of a genealogy \(G_{n-1}\) that has \(n-1\) leaves and coalescence waiting times equal to \((T_{n-1}+T_n, T_{n-2},\ldots , T_2).\) Let \(\ell _0\) be the branch of \(G_{n-1}\) on which the branching event occurred. I label the three branches of \(G_n\) created by the branching event as \(\ell _1, \ell _2, \ell _3\) and their lengths (for which I use the same notation as for the branches) satisfy the equations \(\ell _1=\ell _2=T_n\) and \(\ell _3=\ell _0-T_n\). Since mutation events occur independently on different branches of the genealogy according to a Poisson process with rate \(\theta /2,\) it is easy to see that \(\mathrm{I}\!\mathrm{P}(E_{2r}(G_n, \ell )) = \mathrm{I}\!\mathrm{P}(E_{2r}(G_{n-1}, \ell ))\) if \(\ell \ne \ell _i, i=1,2,3,\) and
and
where, \(Z_i\) are defined as \(\sum _{j=2}^i T_j.\)
After combining all the equations above the following equation holds
Since the first term in the above sum is independent from the topology of the genealogy based on the inductive assumption and the second term is also independent from the topology, it follows that the probability of \(E_{2,r}(G_n)\) is independent from the topology of \(G_n\).
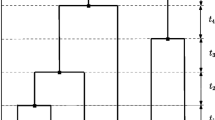
Derivation of the expression for \(r_n\): Since the above results show that the value of \(r_n\) is the same for any topology of the genealogy, I derive the expression for \(r_n\) by considering a genealogy in which the all \(n-1\) coalescent events occur along a single lineage. The branch lengths of this genealogy are equal to \(T_n, T_{n-1}\ldots , T_2, S_{n},\ldots , S_2,\) where \(S_i=\sum _{j=i}^n T_j\). Figure 6 shows an example of a such genealogy for the case of \(n=5.\)

The genealogy of five sequences with a specific topology in which the 4 coalescent events occur on the same lineage. The coalescence times and waiting times between consecutive coalescent events \((S_i\) and \(T_i, i=2,\ldots , 5)\) represent the branch lengths of the genealogy
Using the same partitioning for \(E_{2r}(G_n)\) as described in the above proof, all possibilities for \(E_{2r}(G_n, \ell )\) can be easily explored for the special type of genealogy: If the late mutation event is on the branch \(\ell =T_i\), the early mutation event can be on that branch or on the part of the lineage connecting the beginning of that branch to the root of the genealogy. The probability of \(E_{2r}(G_n, \ell )\) in this case is
Similarly, if the late mutation event is on the branch \(\ell = S_i,\) the probability of \(E_{2r}(G_n, \ell )\) is
Adding the above expressions for all the cases, the probability of two mutation events occurring on same lineage is equal to
After simplifying the above expression, the following equation holds
Combining the above expression with the representation
the following formula holds
The proof of the inequality \(h_n > r_n, n>2\): I use a mathematical induction to prove that \(h_n-r_n >0\) if \(n>2\). From the previous results follows that the difference \(h_n - r_n\) can be represented as
The following notations are needed \(Z_{i}=\sum _{j=2}^{i} T_j\) and \(\phi _n=L_n^2-2nZ_n^2+2\sum _{i=2}^{n-1}Z_{i}^2,\) which is equal to \(L_n^2-2nS_2^2+2\sum _{i=2}^{n}(S_2-S_{i})^2.\) For \(n=3\), it is easy to check that
Assume that \(\phi _k>0\) if \(k=n-1,\) I prove that this inequality also holds if \(k=n.\) Since \(L_n=\sum _{j=2}^n jT_j\), it can be represented as \(nT_n+L_{n-1}\) as well as \(nZ_n-\sum _{i=2}^{n-1}Z_i.\) Thus, the following formula holds
As a result, the inequality
holds since \(\phi _{n-1}\) is greater than 0 and
Derivations of the formulas for \(\mathrm{I}\!\mathrm{P}(E_{2r,s}\mid E_2)\) and \(\mathrm{I}\!\mathrm{P}(E_{2h,c}\mid E_2)\): I derive the formulas using the following representations for the probabilities \(\mathrm{I}\!\mathrm{P}(E_{2r,s}\mid E_2)\) and \(\mathrm{I}\!\mathrm{P}(E_{2h,c}\mid E_2)\!:\)
and
which I modify based on the Eq. (9) and changing the orders of the terms in sums. Thus, the following equations hold
and
The above sums I simplify further using the formulas for \(N_1()\) and \(N_2()\) and the identity
Hence, the following formulas hold
and
The proof of Theorem 1 is complete.
1.3 The proof of Theorem 2
I represent the probability \(\mathrm{I}\!\mathrm{P}( D_{n}\mid \alpha , H_{\alpha }, E_2)\) as a ratio of probabilities:
To derive expressions for the probabilities in the numerator and denominator, I use the framework for generating polymorphisms in samples of DNA sequences in the general coalescent tree setting. The coalescence waiting times are determined by the random variables \(T_n,\ldots , T_2,\) and mutation events are added along the lineages of the genealogy according to a Poisson process with rate \(\theta _0/2.\) Because of these properties, the probability in the denominator can be represented as
where \(L_n\) is the total branch length of the genealogy. Using a similar approach described by Sargsyan (2010), the probability in the numerator can be represented as the sum
where \({\varGamma }(k,m)\) represent genealogies with two mutation events satisfying the following conditions: (1) The number of ancestral lineages at the early and late mutation events are \(k\) and \(m\), respectively. (2) The \(\alpha \) is the haplotype of the most recent common ancestor, and \(H_{\alpha }\) is the transition history at early and late mutation events. The symbol “\(\cong \)” means that the genealogy with two mutations is consistent with \(D_n.\) Each term of this sum can be represented as a sum based on the definition of the conditional expectation. Thus,
where the probability of \({\varGamma }(k,m)\) is equal to
and
the values of \(j\) and \(e\) are determined by \(\alpha , H_{\alpha }\) and the frequencies \((a,b,c)\) of the haplotypes \(TT, TC, CC.\) The quantity N(n) represents the number of all possible topological trees with \(n\) leaves and it is equal to \(=n!(n-1!)/2^{n-1},\) which can be easily derived based on tracing and joining two lineages at each coalescent event. Combining the above equations completes the proof.
1.4 The proof of Lemma 2
First I derive the asymptotic formula when \(TT\) is the ancestral haplotype. Factoring out some of the terms in Eq. (7), the following holds
and
To approximate this ratio under the limits \(a/n\rightarrow x,\) \(b/n\rightarrow y,\) \(c/n\rightarrow z\) as \(a,b,c\) tend to \(\infty ,\) first the following limit is derived
which follows from the representation
Using similar approach the following limits hold:
and
After expanding the expression
to
it is asymptotically equal to \((a+b+c)^2\frac{y}{2}(m-k)(2x+y(m-k+1)))\). After combining these results the following limit holds
Note that the above approach can be applied similarly to derive the asymptotic formula for \(N_{2}(k,m;a,c,b)\). The proof of the lemma is complete.
1.5 The proof of Theorem 3
In the standard coalescent setting \(T_i\), \(i=2,\ldots ,n\), are pairwise independent exponential random variables with rates \(2/(i(i-1))\), therefore the following equations hold:
and
Using these formulas and the approximations in Lemma 2, I compute the limit of the expression
under the conditions \(a/n\rightarrow x,\) \(b/n\rightarrow y,\) \(c/n\rightarrow z\) as \(a,b,c\) tend to \(\infty ,\) by substituting the ratio
with
and showing that the following identity holds
Similar to this approach the following equations hold for the other cases
Note that if \(m=k\) then
For the derivations of these formulas I used Wolfram Mathematica Software and the formulas
and
Under the limits \(\frac{a}{n}\rightarrow x, \frac{b}{n}\rightarrow y, c/n\rightarrow z\), as \(a,b,c\) tend to \(\infty \) the following limits hold in the standard coalescent setting:
where
and
Based on Eq. (6) and (17) the following holds
Combining these equations with the above limits the proof of the theorem is complete.
1.6 The proof of Theorem 4
Derivations here are very similar to the approach used by Sargsyan (2010), for more details see that paper. First I represent the joint distribution of the ages of \(\xi _1, \xi _2,\) and \(\xi \) as the ratio of two probabilities:
The denominator in the above equation can be substituted by the expression in Theorem 2. To derive an expression for the probability in the numerator one can use very similar approach as in Theorem 2. After determining \(j\) and \(e\) based on \(\alpha , H_{\alpha }\) and \((a,b,c)\) (the frequencies of the haplotypes \(TT, TC, CC)\) the following equation holds:
where \({\varGamma }(k,m)\) has distribution
The probabilities in the above sum can be written as
Each of the factors in the above product are evaluated separately:
and conditional on \({\varGamma }(k,m)\), \(\xi _1\) and \(\xi _2\) and \(\xi \) are respectively equal to \(S_{k+1}+U_0 T_{k}\), \(S_{m+1}+U_1 T_{m}\), and \(S_2,\) in distribution, where \(S_i\) is \(\sum _{j=i}^n T_j\), and if \(k\ne m\) then \(U_1=U_{00}\) and \(U_2=U_{01}\) where \(U_{00}, U_{01}\) are independent uniform random variables on \((0,1)\), if \(k=m\) then \(U_2=\min (U_{00}, U_{01})\) and \(U_1=\max (U_{00}, U_{01})\). Thus, if \(k\ne m\) then the following equation holds
recall that \(1\wedge r = \min (1,r)\) and \((a)_{+}\) is equal to \(a\) if \(a>0\), otherwise it is 0. If \(k=m\) and \(t_1<t_2\)
if \(t_2<t_1\) then the following holds
From the steps in the proof of Theorem 2 the following equation holds
After combining the above results the proof of the theorem is complete.
1.7 The proof of Theorem 5
To derive expressions in the theorem, I use Eq. (26) under the limits \(\theta _0\rightarrow 0,\) \(a/n\rightarrow x, b/n\rightarrow y,\) \(c/n\rightarrow z\) as \(a,b,c\) tend to \(\infty .\) Using the properties of the coalescence waiting times in the standard coalescent setting, the following formulas hold
I use these formulas in Eq. (26) combined with the limits in Lemma 2 to derive analytical expressions for the expected ages of \(\xi _1\) and \(\xi _2,\) conditional on \(\alpha =TT,\) \(H_1,\) and \((x,y,z).\) In the case of \(\alpha =TC\) and transition histories \(H_2\) and \(H_3,\) two type of substations occur \(TC\rightarrow TT\) and \(TC\rightarrow CC.\) I infer the expected ages of these two substation events instead of the ages of the early and late mutation events. Before using Eq. (26) in this case, I first derive an expression for computing the expected age of \(\xi _{TC, TT}.\) Similar steps can be done for the expected age of \(\xi _{TC, CC}.\) Hence, the following holds
Note that
and
Now under the limits \(a/n\rightarrow x, b/n\rightarrow y,\) \(c/n\rightarrow z\) as \(a, b, c\) tend to \(\infty \) I derive the formulas for the experted conditional ages of \(\xi _1\) and \(\xi _2\) by using the limits in Lemma 2 for
and compute the following sums
where
If \(m=k\), the following formula holds
The derivations of the above formulas are tedious and are not shown in details. For this purpose I used Wolfram Mathematica 3.0 as helping tool. The derivations are based on the computation of the power series by transforming them into a geometric series. For example, to derive a formula for the series \(\sum \frac{t^i}{i^2}\) the coefficients \(1/i^2\) in the terms can be replaced by the integral \(\int _0^1\int _0^1 w^{i-1} v^{i-1}dwdv\) and the summation and integration order can be changed because the terms in the series are non-negative, that is
Thus, the series inside of the integral is a geometric series.
Rights and permissions
About this article

Cite this article
Sargsyan, O. An analytical framework in the general coalescent tree setting for analyzing polymorphisms created by two mutations. J. Math. Biol. 70, 913–956 (2015). https://doi.org/10.1007/s00285-014-0785-8
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00285-014-0785-8
Keywords
- Two mutations
- Haplotype frequency spectrum
- General coalescent trees
- Ages of two mutations
- Inferring the ancestral haplotype
- Bayesian inference
- Large sample size
- APOE polymorphisms