
Abstract
Standing dead trees (snags) are recognized for their influence on methane (CH4) cycling in coastal wetlands, yet the biogeochemical processes that control the magnitude and direction of fluxes across the snag-atmosphere interface are not fully elucidated. Herein, we analyzed microbial communities and fluxes at one height from ten snags in a ghost forest wetland. Snag-atmosphere CH4 fluxes were highly variable (− 0.11–0.51 mg CH4 m−2 h−1). CH4 production was measured in three out of ten snags; whereas, CH4 consumption was measured in two out of ten snags. Potential CH4 production and oxidation in one core from each snag was assayed in vitro. A single core produced CH4 under anoxic and oxic conditions, at measured rates of 0.7 and 0.6 ng CH4 g−1 h−1, respectively. Four cores oxidized CH4 under oxic conditions, with an average rate of − 1.13 ± 0.31 ng CH4 g−1 h−1. Illumina sequencing of the V3/V4 region of the 16S rRNA gene sequence revealed diverse microbial communities and indicated oxidative decomposition of deadwood. Methanogens were present in 20% of the snags, with a mean relative abundance of < 0.0001%. Methanotrophs were identified in all snags, with a mean relative abundance of 2% and represented the sole CH4-cycling communities in 80% of the snags. These data indicate potential for microbial attenuation of CH4 emissions across the snag-atmosphere interface in ghost forests. A better understanding of the environmental drivers of snag-associated microbial communities is necessary to forecast the response of CH4 cycling in coastal ghost forest wetlands to a shifting coastal landscape.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The spatial footprint of ghost forests is expanding regionally and globally as a result of surface-(i.e., sea level rise) and subsurface (i.e., saltwater intrusion)-based salinization of coastal freshwater ecosystems [1,2,3]. As forest retreat rates are accelerating [1], Kirwan and Gedan predict that saltwater-driven land conversion will lead to broad-scale changes in coastal ecosystem structure and function in the coming years. Coastal forested wetlands provide feedback mechanisms to climate through the balance of carbon sequestration and emissions of greenhouse gases, particularly CH4 [4]. Therefore, a better understanding of how the conversion of coastal forested wetlands from live to ghost forests will be necessary to predict how climate-change associated stressors will influence carbon dynamics in these systems.
While the physical and biogeochemical processes that influence deadwood decomposition in upland systems has been well studied (e.g., [5,6,7,8]), much less is known about factors that influence deadwood decomposition in lowland systems, where sediment anoxia is more prevalent and deadwood is more prone to water saturation [9]. Differences in physical and biogeochemical parameters between upland and lowland systems, particularly as they relate to the presence and prevalence of oxic and anoxic microenvironments, may influence microbial community structure and, thus, the balance of aerobic vs. anaerobic decomposition pathways in deadwood. A product of the anerobic degradation of carbon compounds is CH4, a powerful greenhouse gas with a global warming potential ca. 28–36× that of CO2 over a 100-year time period [10]. As wetlands are the single largest source of global CH4 emissions [11], an improved understanding of factors that influence dominant decomposition pathways, and thus CH4 emissions, from forested wetland systems is important.
Methane dynamics in wetlands are controlled by microbial communities, more specifically the balance between methanogenesis and methanotrophy. Methanogenesis is the terminal step in the anaerobic degradation of carbon, which occurs in nutrient-depleted, anoxic microsites [12, 13]. The majority of CH4 production in wetlands is mediated by methanogenic archaea [14]. Recent evidence indicates that both fungi [15] and cyanobacteria [16] have the capacity to produce CH4 in aerobic conditions, though the quantitative importance of these taxa in methane production remains unresolved. Methane produced in wetlands has a variety of fates including: escape to the atmosphere via sediment-[17, 18], water-[19, 20], or plant-atmosphere interface [21,22,23] and consumption by methanotrophic bacteria and archaea (Table S1 and citations therein) that are widely distributed in the environment [24,25,26].
Approximately 15–30% of estimated total global wetlands are forested [27, 28], and woody vegetation has been shown to be a pathway for CH4 flux [23, 29]. Research showing the importance of living trees in CH4 emissions from Amazon floodplain forests [30] promoted interest on the role of plants in CH4 exchange. Covey and Megonigal [31] provide a comprehensive review of plant CH4 exchange in trees and forests, suggesting that CH4 fluxes through live trees might be an important and understudied pathway. The trunks of snags have also been recognized as both a source and sink of methane emissions in lowland coastal systems [21, 32, 33]. Much of the prior literature on deadwood carbon dynamics focused on coarse woody debris and felled trees in upland forests [34, 35], thus there is a need to more fully understand processes that affect deadwood decomposition in lowland systems, particularly in deadwood stocks that are standing or suspended [36, 37]. As a result of this recent expansion of research on live and dead trees, CH4 emissions from tree stems have been identified as a new frontier in the global carbon cycle [38], with calls for more research to better elucidate the biophysical mechanisms that modulate CH4 flux across this understudied atmospheric interface.
Both methanogens [39,40,41] and methanotrophs [42,43,44] have been identified in the phyllosphere (i.e., the above-ground parts of a plant [45]). The relative balance between these two functional guilds has the potential to drive plant CH4 exchange. Microbial communities in live trees have been shown to modulate plant CH4 exchange from lowland systems, reducing CH4 emissions across the plant-atmosphere interface by 30–40% [43, 46]. However, there is limited knowledge on the role of phyllosphere-associated microbial communities in modulating carbon fluxes from snags in lowland systems, despite isotopic evidence that microbial oxidation of CH4 occurs within the trunks of snags [47].
In this study, we used high-throughput Illumina sequencing of the V3/V4 region of the 16S rRNA gene to survey the microbial community in the trunks of ten snags in a ghost forest wetland at Gull Rock State Game Lands in Hyde County, North Carolina. Static flux chambers and snag core incubations were used to assess CH4 dynamics across the plant-atmosphere interface. A better understanding of the presence, prevalence, and activity of methanogens and methanotrophs in these phyllosphere-associated communities can support our ability to more accurately forecast the response of CH4 dynamics in forested wetland ecosystems to a shifting coastal landscape [34, 39, 48].
Materials and Methods
Site Description and Snag Selection
The Albemarle Pamlico Peninsula (Fig. 1a), located in eastern North Carolina, is surrounded by two brackish bodies of water: the Albemarle Sound to the north and the Pamlico Sound to the east and south [49]. Fifty percent of the peninsula is within 1.5 m of mean sea-level [50]. Due to the potential for high productivity, much of the peninsula was converted into croplands in the late 1970s [51]. In order to maintain aerated soils, an artificial drainage network was installed. The low-lying elevation of the peninsula [49] and the extensive drainage infrastructure makes the Albemarle Pamlico peninsula vulnerable to saltwater intrusion [50, 52, 53]. As a result, ghost forests have expanded in coastal North Carolina over the past few decades [2].

Location of study site in Hyde County, North Carolina, USA: (a) Location of Gull Rock State Game Lands in relation to the state of North Carolina and the Albemarle-Pamlico Peninsula, (b) ghost forest landscape where standing dead trees were sampled, (c) representative snag with static chamber and Gasmet DX4040 tubing attached. Panel (a) was created using Google Earth; image is copyrighted by DigitalGlobe (2016)
Our study took place in the growing season during June 2019 at Gull Rock State Game Lands (Fig. 1a), a 28,400 acre preserve in Hyde County, North Carolina that is more extensively described in Martinez and Ardón and Martinez et al. [33, 47]. The ghost forest where samples were taken (Fig. 1b) is located on the access road to the E. Merle Edwards/Loop Road Impoundment. The snags at the site were all pines, most likely Pinus serotina Michx., in various stages of decay. Snags were systematically selected (n = 10) to ensure that they were structurally sound enough to support static flux chambers and withstand drilling [21]. The mean height and diameter at breast height (DBH) of the snags were 8.3 ± 4.5 m (range 2.5–15.1 m) and 0.26 ± 0.09 m (range 0.12–0.38 m), respectively. A detailed description of the vegetation at the site and how it has changed over recent decades in response to saltwater intrusion can be found in Taillie et al. [54].
Snag-Atmosphere Methane Fluxes
We used static chambers [55] to measure snag-atmosphere CH4 fluxes on all ten snags at heights ranging from 60–120 cm. Semi-rigid, vented chambers were designed after Siegenthaler et al. [56] and temporarily installed on snags in a location on each trunk that allowed for optimal sealing of the chamber [33]. Snag-atmosphere CH4 fluxes were measured using two methods, as described previously [33, 47]. A Gasmet DX4040 portable gas analyzer (Gasmet Technologies, Finland) was used to quantify CH4 flux over a 15 min interval, with measurements taken continuously every 20 s over the duration of the incubation and validated using an Agilent 7890A gas chromatograph (Agilent Systems, Santa Clara, CA) as described previously [33]. Linear regression models were used to calculate CH4 fluxes. An incubation met the assumption of linearity when r2 > 0.85 [21] and was considered significantly different from 0 when P < 0.05.
Methane Sampling from Trunk Airspace
To confirm the presence of CH4 in trunk airspace, a protocol inspired by Covey et al. [57] and described in detail by Carmichael et al. [21] and Martinez et al. [47] was utilized on the ten snags selected for sampling. Sampling occurred immediately after the static flux chambers were removed from the snags. Three holes (30 cm, 60 cm, and 120 cm above ground level) were drilled horizontally from the bark to the center of the trunk using a 1.27 cm drill bit. Immediately after drilling, each hole was plugged with a 15 mm SubaSeal stopper (Sigma-Aldrich, St. Louis, MO), which was then pierced with a sampling wand connected to the Gasmet for 5 min. Air temperature, wind speed, and barometric pressure were noted at 0 and 5 min to ensure there were no major fluctuations over the duration of the sampling period. In addition, ambient air immediately outside of each drilled hole was sampled for 2 min after each trunk sample for comparison and to flush the trunk gases out of the Gasmet system before each subsequent sample.
For gas samples obtained from trunk airspace, t-tests were used to evaluate measured differences in CH4 concentrations between trunk airspace and ambient air next to the trunk at each measured height. A one-way analysis of variance was used to evaluate the comparison between CH4 concentrations in trunk airspace and ambient air next to the trunk at all heights above water level. A Shapiro–Wilk test for normality was run on each statistical analysis. If normality assumptions were violated, a non-parametric equivalent of the test (i.e., a Mann–Whitney Rank Sum test or Kruskal–Wallis one-way ANOVA on ranks with a Tukey test for multiple comparisons) was used to test for significance (P < 0.05). All statistical analyses were conducted using Sigma Plot v.14.5 (Systat Software, Palo Alto, CA).
Potential CH4 Production and Oxidation Incubations
After CH4 sampling from trunk airspace was completed, duplicate cores were taken from each snag at 60 cm height to measure potential methane production (PMP) and potential methane oxidation (PMO) following Chan and Parkin [58]. Cores were extruded and sealed in a plastic straw that was duct taped on both ends, then stored on ice until receipt in the lab. One core was used for PMP assays, and the second was used to measure PMO as described in detail in Martinez et al. [47]. PMP was measured under anaerobic conditions, using ultra high purity (UHP) N2 as a headspace gas. PMO was measured under aerobic conditions, with the headspace of vials spiked above ambient CH4 to ca. 45 ppm using UHP CH4. Snag core CH4 fluxes were calculated as described in Martinez et al. [47]. The sample size was too small to run statistical analyses on the rates of CH4 production and oxidation in snag cores.
Microbial Community Sampling and DNA Extraction
Deadwood was sampled using a protocol similar to Tláskal et al. [59]. A 1.27 cm drill bit was sterilized using ethanol and then used to drill a hole horizontally to the center of each snag at the midpoint of the location where the static flux chamber was installed. Three replicates were taken at each location. Shavings were collected aseptically using an ethanol-sterilized scoopula and deposited into a sterile 50 mL Falcon tube (Thermo Fisher Scientific, Waltham, MA). Samples were immediately placed on ice in the field and stored in a freezer until they were transported back to the lab. Upon receipt in the lab, samples were held at − 80 °C [60] before processing for DNA extraction.
Prior to DNA extractions, samples were ground independently using liquid nitrogen and a sterile mortar and pestle; this process was repeated three times per sample [40] to obtain a finely ground product. Genomic DNA was extracted from each sample following the protocol in the MP Biomedicals FastDNA Spin Kit for Soil (Santa Ana, CA). A NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA) was used to quantify DNA in each extraction. Samples were subsequently stored at − 15 °C before shipment to North Carolina State University’s Genomic Sciences Laboratory in Raleigh, NC, for Illumina sequencing.
Microbial Sequence Analysis and Bioinformatics
Library construction and DNA sequencing were performed at North Carolina State University’s Genomic Sciences Laboratory. DNA sequencing was performed on an Illumina MiSeq platform (Illumina, San Diego, CA) using the V3/V4 region of the 16S rRNA gene. The 341F/805R primer pair, which targets bacteria and archaea [61], was selected from Klindworth et al. [62]. The sequence data from this study are archived in the NCBI Sequence Read Archive database under the BioProject number PRJNA994044.
Forward and reverse sequences were 300 bp long. The DADA (version 1.21.1) pipeline was used to generate an ASV table for the samples [63]. Initial steps in the pipeline included removing forward and reverse primers, filtering and trimming the reads, where the forward reads were trimmed to 275 bases and the reverse reads to 250 bases. The DADA2 algorithm inferred the sequence variants in each sample after learning the base pair transition error rates. The forward and reverse reads were merged to generate the denoised sequences, and merged sequences that were outside of the range of 383–433 bp were removed as potential non-target hits. Chimeric sequences were removed and taxa were assigned to the sequence variants using the Silva reference database (v. 132) [64, 65]. A phylogenetic tree was constructed using the R package phangorn [66], where the multiple sequence alignment was performed using the R package DECIPHER [67]. The tree construction was based on the steps in Callahan et al. [68].
Analyses were performed in R [69] with extensive use of the R package Phyloseq (version 1.30.0) [70]. The average sample abundance was 293,061. One sample replicate for T-4 had a low count (21,374) as well as a lower sequencing quality compared to the other samples and was thus removed. There were 12,021 taxa for the 29 remaining samples. The taxa were filtered such that the taxa Kingdom classification assignment was to either ‘Archaea’ or ‘Bacteria’. Taxa with a Family classification to ‘Mitochondria’ or an Order classification to ‘Chloroplast’ were also removed. Any ASVs that had fewer than 10 reads in total were removed. After the taxa filtering steps, 9,139 ASVs remained.
The relative abundance (RA) of the taxa was examined for the individual snag replicates on the Kingdom, Phylum, Class, Order, and Family levels. RA is a metric that represents the proportion of total reads and is measured on a scale of 0.0–1.0. The average RA was also calculated for the snag replicates where the RA values were averaged over the snag replicates for each taxonomical assignment. The taxonomical assignments that are ‘NA’ were excluded. The relative abundances were also examined for ASVs that were assigned to cultivated taxa with confirmed methanogenic and methanotrophic capacities (Table S1). The average RA for each ASV was calculated for the snag replicates. The ASVs were then assigned to the categories of Methanogens, Methanotrophs, or Other. The Methanotrophs category was replaced by three subcategories based on their abundance, where methanotroph ASVs with a RA ≤ 0.01 were assigned to category Methanotroph_L1, 0.01 < RA ≤ 0.02 were assigned to category Methanotroph_L2, and RA > 0.02 were assigned to category Methanotroph_L3. Methanogenic ASVs in the data included Methanobacteriales and Methanomassiliicoccales; whereas methanotrophic ASVs included Methylocapsa, Methylocella, and Methylovirgula. The relative abundance of the 56 ASVs that were assigned to taxonomic ranks known to contain methanotrophs (i.e., Methylocapsa, Methylocella, and Methylovirgula) were then summed for each snag, averaged over the snag replicates, and scaled to represent relative abundance within the methanotrophic community of each snag. A limitation of 16S rRNA data is that functions cannot be reliably known, especially for methane oxidation because the trait is polyphyletic. Therefore, all lineages identified herein as methanotrophs or methanogens are conservatively considered putative methane-oxidizers or -producers for the purpose of analysis.
Alpha diversity measures of the Shannon Index and the complement of Simpson’s Diversity (1-D) were calculated after the dataset was rarefied to 30,000 reads. There were 8,730 ASVs in the rarefied set. All additional analyses used the non-rarefied dataset. The beta diversity of the snag replicates was examined with principal component analysis (PCA) [71]. Initially, the ASV count data were transformed using the centered log-ratio (clr) transformation using the CoDaSeq R package (version 0.99.4) [72]. Before applying the transformation, any existing zero counts were replaced with non-zero values and this calculation was performed with the zCompositions R package (version 1.3.3.1) using the count zero multiplicative method [71]. The Euclidean distance matrix was calculated for the clr transformed ASV counts using the distance function in phyloseq. The ordinate function (method = “RDA”) in phyloseq was used to generate the PCA plot.
Hierarchical clustering was performed to visualize sample clustering in a method similar to that described by Gloor and Reid [73]. Initially, the ASV read counts were agglomerated on the class level. The zero value replacement and clr transformation steps were applied to the class level count table as described above. The distance matrix was calculated in R using the dist function (method = “euclidean”) and hierarchical clustering was performed with hclust (method = ”ward.D2″) to generate a dendrogram of the 29 snag samples.
Results
Snag-Atmosphere Methane Fluxes
Of the ten static flux chambers that were used to measure CH4 fluxes across the snag-atmosphere interface, five passed quality control standards (Table 1). Methane production was measured in three out of ten chambers, with an average flux of 0.21 ± 0.15 mg CH4 m−2 h−1. Snags 5, 8, and 9 had measured fluxes of 0.08, 0.51, and 0.03 mg CH4 m−2 h−1, respectively. Methane consumption was measured in two out of ten chambers, with an average flux of − 0.07 mg CH4 m−2 h−1. Snags 4 and 10 had measured fluxes of − 0.11 and − 0.03 mg CH4 m−2 h−1, respectively.
Methane Sampling from Trunk Airspace
There were no significant differences in CH4 concentrations in ambient air by height among the 30, 60, and 120 cm measurements, which is indicative of atmospheric mixing. The mean concentration of CH4 in trunk airspace at 30, 60, and 120 cm decreased by height (Table 1): 23.7 ± 7.5, 16.7 ± 5.3, and 6.3 ± 1.9 µL L−1, respectively. However, there were no significant differences among heights, possibly due to high variability in the dataset. Snag 7 was unique in that the trunk CH4 concentration at 60 cm was 1.5 × higher than at 30 cm, and the trunk CH4 concentration dropped 15% between 60 and 120 cm. This indicates a hot spot of CH4 in this snag at the 60 cm sample height, the location where the sample for trunk microbial community analysis was obtained. Trunk airspace CH4 concentrations were significantly elevated (P = 0.01) compared to the air immediately outside of the trunk (6.3 ± 2.0 µL L−1) at 30 cm (23.7 ± 7.5 µL L−1), but not at 60 or 120 cm (Table 1).
Potential CH4 Production and Oxidation Incubations
One core from each snag was incubated under anoxic conditions to measure potential CH4 production, and one core from each snag was incubated under oxic conditions to measure potential CH4 oxidation. Of the 20 cores, 6 passed quality control standards (Table 1). A single core from snag 2 produced CH4 under anoxic conditions at a measured rate of 0.7 ng CH4 g−1 h−1. A single core from snag 6 produced CH4 under oxic conditions at a measured rate of 0.6 ng CH4 g−1 h−1. Four cores, taken from snags 4, 5, 9, and 10, showed evidence of CH4 oxidation under oxic conditions, with an average rate of − 1.13 ± 0.31 ng CH4 g−1 h−1 (range − 0.5 to − 2.0 ng CH4 g−1 h−1). Snag 9 had the strongest rate of CH4 oxidation at − 2.0 ng CH4 g−1 h−1.
Trunk-Associated Microbial Communities in Snags
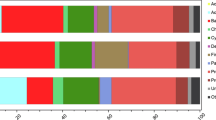
In nine of the ten snags (Fig. 2a), Proteobacteria, Acidobacteria, and Actinobacteria were the top three most abundant phyla, with mean relative abundances of 28%, 28%, and 26%, respectively. Verrucomicrobia and Planctomycetes were the fourth and fifth most abundant phyla, with mean relative abundances of 6% and 4%, respectively. Individual snags had absolute abundances of phyla that varied from the mean, but the pattern in mean relative abundance of phyla was consistent among 9 out of 10 snags. Snag 7 differed from this pattern in that the top five most abundant phyla were Proteobacteria (39%), Acidobacteria (18%), Bacteroidetes (11%), Actinobacteria (11%), and Verrucomicrobia (7%). Across all ten snags, the distribution of the relative abundance of all phyla was consistent among all snag replicates (Fig. S1).

Average relative abundance of taxa in each snag at the level of (a) phylum and (b) class. Relative abundances were averaged over the snag replicates. Taxonomic assignments for ASVs that were not resolved (i.e., coded as N/A) were not included in the bar plots, which can result in total abundance values being less than 1
In seven of the ten snags (Fig. 2b), Acidobacteria, Alphaproteobacteria, and Actinobacteria were the top three most abundant classes, with mean relative abundances of 28%, 23%, and 12%, respectively. Individual snags had absolute abundances of bacterial classes that varied from the mean, but the pattern in mean relative abundance of classes was consistent among 7 out of 10 snags. In snag 6, the top three most abundant classes were Alphaproteobacteria (37%), Acidobacteria (28%), and Thermolephilia (11%). In snag 7, the top three most abundant classes were Alphaproteobacteria (26%), Acidobacteria (17%), and Bacteroidia (11%). In snag 10, the top three most abundant classes were Acidobacteria (31%), Alphaproteobacteria (22%), and Thermolephilia (11%). Across all ten snags, the distribution of the relative abundance of classes of bacteria was consistent among all snag replicates (Fig. S2).
Snags contained diverse microbial communities (Fig. 3a), with no distinct similarities in community structure that were evident following PCA (Fig. 3b). Hierarchical clustering (Fig. 3c) revealed three distinct clusters of similar communities: a cluster solely represented by all three replicates from snag 7, a second cluster including the snag replicates from snags 8 and 9, and a third cluster including all remaining snags.

Alpha (a) and beta (b) diversity of microbial communities in standing dead trees in Gull Rock State Game Lands. Each snag replicate is indicated by an individual point in panels (a) and (b). Alpha diversity measures (a) were calculated after the dataset was rarefied to 30,000 reads; there were 8,730 ASVs in the rarefied set. Principal component analysis (b) was used to examine beta diversity. Hierarchical clustering was performed to generate a dendrogram of the 29 snag samples (c), identifying three clusters: a cluster solely represented by snag 7, a second cluster including snags 8 and 9, and a third cluster including all remaining snags
An analysis of the top 20 ASVs contained in each sample (Table 2, Supplementary File Traits Associated with Top 20 ASVs) revealed that nine of the ten snags had microbial communities that can be broadly characterized as acid-tolerant aerobes, with a number of species that have been previously documented in association with plants and/or have the capacity to degrade plant polymers (Table 3). Snag 7 was an outlier (Fig. 3) and had the most diverse microbial community of all snags, and it was the only snag with a ca. 1:1 ratio of aerobes:facultative aerobes/anaerobes among the top 20 ASVs. Eight of the top 20 ASVs in snag 7 had cultured representatives that are facultative anaerobes, microaerophiles, or facultative aerobes (Supplementary File Traits Associated with Top 20 ASVs). One of the top 20 ASVs in snag 7, Telmatospirillum, is a genus that has been isolated from a methanogenic enrichment culture (Table 3). In addition, several of the top 20 ASVs, including Acidothermus, Granulicella, and Terracidiphilus, are species that are capable of degrading plant polymers; Endobacter and Myoavidus have been isolated from plants (Table 3).
CH4-Cycling Communities in Snags
An analysis of the most abundant phyla in each snag revealed that the relative abundance of taxa that contain known methanogens was extremely low. If present, potential methanogens were represented by the archaeal orders Methanobacteriales (snags 8 and 9) and Methanomassiliicoccales (snags 8 and 9); there were no cyanobacterial lineages in the sequencing data that indicated an expanded capacity for methanogenesis beyond the Archaea. Eighty percent of the snags (snags 1–7 and 10) did not have any detectable putative methanogenic Archaea. In snags 8 and 9 putative methanogenic Archaea were not in the top 20 most abundant phyla for each snag, with relative abundances of 4.6 × 10–5 and 1.7 × 10–5, respectively. Overall, if methanogens were present, they represented a mean relative abundance of 3.2 × 10–5 within each snag.
Members of the genera Methylocapsa, Methylocella, and Methylovirgula were identified in snags (Table S1). Each of these taxa contain cultured representatives that demonstrate methanotrophic activity, though not all members from these lineages are obligate methanotrophs and/or capable of methanotrophy (e.g., [44, 74, 75]). Methylocapsa aurea is a facultative methanotroph that is capable of using other C1 compounds, such as methanol and formate, as well as the multicarbon compound acetate (C2) [75]. Members of the genus Methylocella are capable of using methanol (C1), as well as other multicarbon compounds such as acetate (C2), ethanol (C2), pyruvate (C3), malate (C4), and succinate (C4) [74]. Methylovirgula ligni are facultative methylotrophs, capable of utilizing other C1 compounds such as methanol, as well as the multicarbon compounds ethanol (C2), pyruvate (C3), and malate (C4) [44]. An analysis of the top 20 ASVs found in each snag (Supplementary File Traits Associated with Top 20 ASVs) revealed that snags 2, 3, 4, 6, and 10 contained taxa with cultivated representatives that are capable of methanotrophy (Table 2, Table 3) within the top 20 ASVs, all with relative abundances of 1–2%. Methylovirgula was found in snags 2, 3, 4, and 10, and was the second most abundant ASV in snag 6. Brycoella and Edaphobacter, genera that have been co-cultured with methanotrophs (Table 3 and citations therein), were identified among the top 20 ASVs in 5 out of 10 snags (Table 2). The methylotrophic genera Hyphomicrobium and Methylovirgula (Table 3 and citations therein) were found within the top 20 ASVs in 6 out of 10 snags. There were no putative methanogens identified in the top 20 ASVs in any of the snags (Supplementary File Traits Associated with Top 20 ASVs); however, the 16th most abundant ASV in snag 7, Telmatospirillum (Table 3) has been co-cultured with methanogens [76].
Taxa associated with methanotrophy (Fig. 4) were identified in all of the snags, representing a range of 1–4% of all of the ASVs identified in each snag, with a mean relative abundance of 2% (Fig. 4a). In contrast, taxa associated with methanogenesis (Fig. 4a and Fig. 5) were represented by 2 ASVs that were identified in 2 of the 10 snags (snags 8 and 9), with a mean relative abundance in each snag of < 0.0001%. In snags where taxa associated with methanotrophy and methanogenesis were both present (i.e., snags 8 and 9), methanotrophs were, on average, two orders of magnitude more abundant than methanogens. A neighbor-joining tree inferring the phylogenetic relationship between representative SSU rRNA sequences retrieved from snag cores that were also assigned to groups that have cultivated representatives known to oxidize or produce methane can be seen in Fig. 5.

Diversity of taxa known to contain CH4-cycling microorganisms in each snag. (a) ASVs were assigned to the categories of Methanogens, Methanotrophs, and Other. Methanogens include Methanobacteriales and Methanomassiliicoccales. Methanotrophs include Methylocapsa, Methylocella, and Methylovirgula. Methanotrophs were divided into three subcategories based on their abundance, where methanotroph ASVs with a relative abundance ≤ 0.01 were assigned to category Methanotroph_L1, methanotrophs with a relative abundance 0.01 < x ≤ 0.02 were assigned to category Methanotroph_L2, and methanotrophs with a relative abundance > 0.02 were assigned to category Methanotroph_L3. All remaining ASVs, including putative uncultivated methanogenic and methanotrophic taxa, are categorized as Other. (b) Relative abundances of the 56 ASVs that were assigned to taxonomic ranks known to contain methanotrophs were summed for each snag, averaged over the snag replicates, and then scaled to represent a proportion of the methanotrophic community in each snag

Neighbor-joining tree inferring the phylogenetic relationship between representative SSU rRNA sequences retrieved from snag cores that were also assigned to groups known to oxidize or produce methane (shown by ASV number and classification) and related cultured and uncultured sequences from GenBank. Order names are abbreviated as follows: Methanobacteriales (MB), Methanomassiliicoccales (MM), Hyphomicrobiales (HM), and Methylacidiphilales (MA). Nodes with bootstrap values between 80–100 that were also supported by maximum likelihood analysis are marked with filled circles and those with bootstrap values between 50–75 that were also supported by maximum likelihood analysis are marked with unfilled circles. There were no nodes with bootstrap values between 75–80 that were also supported by maximum likelihood. The scale bar indicates fractional differences in nucleotide sequences
The genus Methylovirgula dominated the potential CH4-consuming community in each snag (Fig. 4b), representing 62% of the methanotrophic ASVs in each snag (range 24–88%). Methylocella were also identified in each snag, representing 37% of the putative methanotrophic ASVs (range 11–70%). ASVs assigned to Methylocapsa were identified 80% of the snags (snags 1–8), but with a much lower mean relative abundance (1%, range 0–7%).
Discussion
Microbial communities have been shown to modulate CH4 fluxes from live trees in lowland forests [43, 46], yet very little is known about the role of these communities in modulating CH4 fluxes from snags in lowland forests. To our knowledge, this study reports the first high-throughput characterization of the trunk-associated microbial community in snags in a ghost forest. Our results indicate that the snag-associated community is diverse and indicative of oxidative decomposition of deadwood. When present, taxa associated with methanogens were found in very low relative abundance in snags. In contrast, taxa associated with methanotrophs were identified in all snags and represented the sole CH4-cycling community in a majority of the snags. These data show that methanotrophs dominate the putative CH4-cycling community inside snags. As such, the phyllosphere-associated microbial community in snags has the potential to attenuate CH4 emissions across the plant-atmosphere interface in ghost forests, as previously suggested using isotopic and concentration information [33, 47].
Phyllosphere-Associated Microbial Communities and Microbial Decomposition of Deadwood
In this study, the top three most abundant phyla and the most abundant classes were similar in most snags, corroborating data from Moll et al. [6] who measured deadwood logs in an upland forest in the Nationalpark Hainich in Central Germany. Our results did reveal some differences in individual snags, which could be a result of differences in the stage of decay and/or successional state of the community [5, 7, 59]. An analysis of the top 20 ASVs in each snag revealed that snags contain diverse microbial communities whose structure was indicative of oxidative decomposition of deadwood. The single exception to this pattern was snag 7, which contained a unique, highly diverse community that was indicative of a community shift towards adaptation to lower O2 concentrations. This assertion is corroborated by data from trunk airspace CH4 sampling that provided evidence of a more reducing microenvironment within the snag that would be more conducive to anaerobic decay; although differences in the microbial community have also been shown to vary due to differences in in C:N ratio [77, 78], pH and water content [6, 59, 79], wood density [80], stage of decay or tree age [7, 59, 81], and/or the phyllosphere microbial community composition [8, 35, 80]. Hierarchical clustering analysis grouped all three replicates of snag 7 together as a single cluster. Replicates from snags 8 and 9, which were the only two snags with standing water at their base, formed a second, distinct cluster and may serve as a larger conduit for methane emissions (Table 1, [82]). The third cluster contained replicates from all of the remaining snags, which were located on raised hummocks. Taken together, these data support research from upland systems [37, 83] indicating that microenvironmental conditions, either within decaying wood or the immediate surrounding, influence microbial community structure in deadwood stocks.
Snags as an Interface for Methane Emissions from Ghost Forests
Previous research has demonstrated that, although the spatial footprint of snags over the soil/sediment is small, greenhouse gas emissions from snags can substantially increase the total ecosystem CO2equivalent [33]. Both methanogens and methanotrophs have been isolated from [41, 42, 44] and identified in metagenomic analyses [39, 40, 43] of the phyllosphere communities of live trees. Yet, much less is known about the presence and prevalence of CH4-cycling microorganisms in standing deadwood stocks such as snags [37].
In order for snags to act as either a source or sink for the atmospheric flux of CH4, two conditions must be met: (1) gas accumulation within the trunk airspace and (2) flux of the gas across the snag-atmosphere interface. The data provided evidence that CH4 accumulated within the trunk airspace of snags. With the exception of snag 7, mean CH4 concentrations within the trunk followed an expected pattern [21, 47, 84], decreasing by height, an observation that supports a soil-based origin of trunk CH4. After the CH4 is produced by methanogens in wetland soils/sediments, it diffuses into the trunk of a snag, where it has the potential to be oxidized by trunk-associated microbial communities or escape to the atmosphere across the snag-atmosphere interface. Interestingly, snag 7 was the only snag with bark intact up the trunk, which likely helped to hold moisture in and decreased gas exchange with the atmosphere, both conditions that would increase anoxic microsites within the trunk and influence microbial community structure.
The results of the core incubations and static flux measurements indicate that snags can be a source and sink of atmospheric CH4 fluxes and support recent findings in snags in similar systems [21, 33, 47]. Snag CH4 emissions in this study were well within the range recorded in static flux measurements across 83 snags in a similar study across five different ghost forests in the Albemarle Pamlico Peninsula [33]. The quantitative difference in emissions between this study and Martinez and Ardón’s 2021 study are likely a result of the high degree of spatiotemporal variability that has been noted in several studies of plant-atmosphere CH4 fluxes [21, 48, 85,86,87]. In contrast, a study of live tree stems in seasonally flooded Amazonian forests measured much higher values ranging from 10 to 270 mg CH4 m−2 h−1 [30], indicating that edaphic factors that influence the sediment redox environment may impact the magnitude and direction of plant-based CH4 emissions. Potential methane production incubations of snag cores in this study indicated that small amounts of CH4 may be produced under either oxic or anoxic conditions for a low proportion of snags. These values were lower than those in a similar study that demonstrated CH4 production in snags across ghost forests in the Albemarle Pamlico Peninsula [47]. Finally, methane consumption incubations in this study demonstrated methane consumption in a much greater proportion of snags, compared to those measured by Martinez et al. [47], although maximum rates measured were approximately equal.
Microbial Communities in Snags May Modulate Methane Emissions from Ghost Forests
Our sequencing data show that taxa that contain known methanogens are a very small proportion of the total community. Though methanogenesis has historically been associated with members of the Archaea, the capacity for methane production has recently been identified in other domains [15, 16]. Our sequencing primers did not target fungi, so we cannot comment on the capacity for fungal methane production in our sequencing data. However, a single core from snag 6 showed detectable methane production under oxic conditions, indicating that fungal CH4-producers may be present in snag phyllosphere communities. None of the methane-producing cyanobacterial lineages identified by Bižić et al. [16] were identified in the dataset. Instead, the methanogens in the dataset were associated with the archaeal orders Methanobacteriales and Methanomassiliicoccales. The low methanogen abundance across all snags does not appear to be an artifact of sequencing and/or primer section, as the primers used in this study have been shown to provide moderate coverage for Archaea [61] and used to amplify Archaea in other studies [88,89,90].
In contrast, taxa that contain known methanotrophs were identified in every snag. The presence of methanotrophic populations in the trunks of snags has been suspected for years [46, 47], but was confirmed for the first time in this study. Methylovirgula [91] were detected in the top 20 ASVs in 50% of the snags. The methanotrophic genera Methylocella [92,93,94] and Methylocapsa [95, 96] were found in lower relative abundance in our dataset. Methylocapsa are known to contain high-affinity enzymes that are capable of CH4 oxidation at atmospheric concentrations [95]; therefore, despite their low relative abundance, they may have a substantial impact on CH4 consumption within snags. In this study, mean trunk airspace CH4 concentrations were ca. 3–4× elevated above background atmospheric CH4, indicating an abundant CH4 supply for high-affinity oxidation.
Metagenomic surveys reveal that the uncultivated diversity in methanotrophic populations is likely extensive [25, 26, 39, 97, 98] and that many of these uncultivated lineages fall within taxa of bacteria (e.g., Alphaproteobacteria, Gammaproteobacteria, and Verrucomicrobia) that are known to contain methanotrophs and were identified in a high relative abundance in snags in this study. Genes associated with methanotrophy can move via horizontal gene transfer [99,100,101]. Most recently, Al-Shayeb et al. [102] identified a novel mobile extrachromosomal genetic element in Methanoperedens that supercharges methane oxidation capacity. The process of horizontal gene transfer allows for expanded functional capacities beyond what is transmitted vertically to an organism and our sequencing approach would not capture this level of functional diversity. Therefore, it is likely that the relative abundance of methanotrophic populations in our study represents a conservative estimate and that our study has not revealed the full extent of methanotrophic capacity in the microbial communities associated with snags.
Our data indicate that the microenvironment at the sample height was likely oxic and well-mixed with the atmosphere, conditions that would favor oxidative decomposition of deadwood and the proliferation of methane-consuming populations of microorganisms. However, if our stand was located in standing water, where waterlogging of woody tissues could lead to more extensive anoxic microsites within snags [82], microbial community structure could shift to favor methanogenesis over methanotrophy. Therefore, more research is needed to more fully resolve the abiotic factors that influence microbial community structure in standing deadwood stocks like snags [37, 83], and thus control carbon dynamics [38] in ghost forest systems.
Conclusions
Methanogens and methanotrophs have previously been identified as members of phyllosphere-associated microbial communities, and prior studies demonstrate that the relative abundance of the two functional guilds within individual trees has the capacity to influence ecosystem-level CH4 dynamics [39, 43]. In this study, we show that the balance of these guilds in snags is tipped in the favor of oxidative decomposition and CH4 consumption. These results support multiple lines of evidence from prior work at the site [33, 47] and suggest that trunk-associated microbial communities in snags may attenuate CH4 emissions across the plant-atmosphere interface.
Although characterization of community structure is a necessary first-step, broadening 16S rRNA-based surveys to include metagenomic surveys that target the pmoA and amoA functional genes could expand our knowledge of the presence and prevalence of uncultivated lineages of methanotrophs in these environments [25, 48] and provide insight into metabolically active members of the community [39]. In addition, employing a multi-tiered biogeochemical approach (e.g., Jeffrey et al. [43]) could better resolve the quantitative importance of snag-associated methanotrophic communities in attenuating methane emissions from ghost forest wetlands.
Data Availability
The Illumina sequence data underlying this article are available in GenBank under BioProject accession number PRJNA994044. Summarized data underlying this article are available in the article and in its online supplementary material. At the time of publication, the raw data the support this article are not publicly available from the corresponding author, but the data are available upon request.
References
Kirwan ML, Gedan KB (2019) Sea-level driven land conversion and the formation of ghost forests. Nat Clim Chang 9:450–457
Smart LS, Taillie PJ, Poulter B, Vukomanovic J, Singh KK, Swenson JJ, Mitasova H, Smith JW, Meentemeyer RK (2020) Aboveground carbon loss associated with the spread of ghost forests as sea levels rise. Environ Res Lett 15:104028
White EE Jr, Ury EA, Bernhardt ES, Yang X (2021) Climate change driving widespread loss of coastal forested wetlands throughout the North American Coastal Plain. Ecosystems 25:812–827
McLeod E, Chmura GL, Bouillon S, Salm R, Björk M, Duarte CM, Lovelock CE, Schlesinger WH, Silliman BR (2011) A blueprint for blue carbon: toward an improved understanding of the role of vegetated coastal habitats in sequestering CO2. Front Ecol Environ 9:552–560
Hoppe B, Purahong W, Wubet T, Kahl T, Bauhus J, Arnstadt T, Hofrichter M, Buscot F, Krüger D (2016) Linking molecular deadwood-inhabiting fungal diversity and community dynamics to ecosystem functions and processes in Central European forests. Fungal Diversity 77:367–379
Moll J, Kellner H, Leonhardt S, Stengel E, Dahl A, Bässler C, Buscot F, Hofrichter M, Hoppe B (2018) Bacteria inhabiting deadwood of 13 tree species are heterogeneously distributed between sapwood and heartwood. Environ Microbiol 20:3744–3756
Kielak AM, Scheublin TR, Mendes LW, van Veen JA, Kuramae EE (2016) Bacterial community succession in pine-wood decomposition. Front Microbiol 7:231
Purahong W, Tanunchai B, Muszynski S, Maurer F, Fareed Mohamed Wahdan S, Malter J, Buscot F, Noll M (2022) Cross-kingdom interactions and functional patterns of active microbiota matter in governing deadwood decay. Proc R Soc B 289:20220130
Stagg CL, Baustian MM, Perry CL, Carruthers TJB, Hall CT (2018) Direct and indirect controls on organic matter decomposition in four coastal wetland communities along a landscape salinity gradient. J Ecol 106:655–670
Masson-Delmotte V, Zhai P, Pirani A, Connors SL, Péan C, Berger S, Caud N, Chen Y, Goldfarb L, Gomis MI, Huang M, Leitzell K, Lonnoy E, Matthews JBR, Maycock TK, Waterfield T, Yelekçi O, Yu R, Zhou B (eds), Intergovernmental Panel on Climate Change (2021) Climate Change 2021: The Physical Science Basis Contribution of Working Group to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change
Saunois M, Stavert AR, Poulter B, Bousquet P, Canadell JG, Jackson RB, Raymond PA, Dlugokencky EJ, Houweling S, Patra PK, Ciais P, Arora VK, Bastviken D, Bergamaschi P, Blake DR, Brailsford G, Bruhwiler L, Carlson KM, Carrol M, Castaldi S, Chandra N, Crevoisier C, Crill PM, Covey KR, Curry CL, Etiope G, Frankenberg C, Gedney N, Hegglin MI, Höglund-Isaksson L, Gugelius G, Ishizawa M, Ito A, Janssens-Maenhout G, Hensen KM, Joos F, Kleinen T, Krummel PB, Langenfelds RL, Lauruelle GG, Liu L, Machida T, Maksyutov S, McDonald KC, McNorton J, Miller PA, Melton JR, Morino I, Müller J, Murguia-Flores F, Naik V, Niwa Y, Noce S, O’Doherty S, Parker RJ, Peng C, Peters GP, Prigent C, Prinn R, Ramonet M, Reginer P, Riley WJ, Rosentreter JA, Segers A, Simpson IJ, Shi H, Smith SJ, Steele LP, Thornton B, Tian H, Tohjima Y, Tubiello FN, Tsuruta A, Viovy N, Voulgarakis A, Weber TS, van Weele M, van der Werf GR, Weiss RF, Worthy D, Wunch D, Yin Y, Zhang W, Zhang Z, Zhao Y, Zheng B, Zhu Q, Zhu Q, Zhuang Q (2020) The Global Methane Budget 2000–2017. Earth Syst Sci Data 12:1561–1623. https://doi.org/10.5194/essd-12-1561-2020
Angle JC, Morin TH, Solden LM, Narrowe AB, Smith GJ, Borton MA, Rey-Sanchez C, Daly RA, Mirfenderesgi G, Hoyt DW, Riley WJ, Miller CS, Bohrer G, Wrighton KC (2017) Methanogenesis in oxygenated soils is a substantial fraction of wetland methane emissions. Nat Commun 8:1567
Conrad R (2007) Microbial ecology of methanogens and methanotrophs. Adv Agron 96:1–63
Conrad R (2009) The global methane cycle: recent advances in understanding the microbial processes involved. Environ Microbiol Rep 1:285–292
Lenhart K, Bunge M, Ratering S, Neu TR, Schüttmann I, Greule M, Kammann C, Schnell S, Müller C, Zorn H, Keppler F (2012) Evidence for methane production by saprotrophic fungi. Nat Commun 3:1046
Bižić M, Klintzsch T, Ionescu D, Hindiyeh MY, Günthel M, Muro-Pastor AM, Eckert W, Urich T, Keppler F, Grossart H-P (2020) Aquatic and terrestrial cyanobacteria produce methane. Sci Adv. https://doi.org/10.1126/sciadv.aax5343
Chanton JP, Martens CS, Kelley CA (1989) Gas transport from methane-staturated, tidal freshwater and wetland sediments. Limnol Oceanogr 34:807–819
Morse JL, Ardón M, Bernhardt ES (2012) Greenhouse gas fluxes in southeastern U.S. coastal plain wetlands under contrasting land uses. Ecol Appl 22:264–280
Bastviken D, Tranvik LJ, Downing JA, Crill PM, Enrich-Prast A (2011) Freshwater methane emissions offset the continental carbon sink. Science 331:50
Helton AM, Bernhardt ES, Fedders A (2014) Biogeochemical regime shifts in coastal landscapes: the contrasting effects of saltwater incursion and agricultural pollution on greenhouse gas emissions from a freshwater wetland. Biogeochemistry 120:133–147. https://doi.org/10.1007/s10533-014-9986-x
Carmichael MJ, Helton AM, White JC, Smith WK (2018) Standing dead trees are a conduit for the atmospheric flux of CH4 and CO2 from wetlands. Wetlands 38:133–143
Dacey JWH, Klug MJ (1979) Methane efflux from lake sediments through water lilies. Science 203:1253–1255
Rusch H, Rennenberg H (1998) Black alder (Alnus glutinosa (L.) Gaertn.) trees mediate methane and nitrous oxide emission from the soil to the atmosphere. Plant Soil 201:1–7
Guerrero-Cruz S, Vaksmaa A, Horn MA, Niemann H, Pijuan M, Ho A (2021) Methanotrophs: discoveries, environmental relevance, and a perspective on current and future applications. Front Microbiol 12:678057
Knief C (2015) Diversity and habitat preferences of cultivated and uncultivated aerobic methanotrophic bacteria evaluated based on pmoA as a molecular marker. Front Microbiol 6:1346
Smith GJ, Wrighton KC (2019) Metagenomic approaches unearth methanotrophy phylogenetic and metabolic diversity. Curr Issues Mol Biol 33:57–84
Lehner B, Döll P (2004) Development and validation of a global database of lakes, reservoirs and wetlands. J Hydrol 296:1–22
Matthews E, Fung I (1987) Methane emission from natural wetlands: global distribution, area, and environmental characteristics of sources. Global Biogeochem Cycles 1:61–86
Schütz H, Schröder P, Rennenberg H (1991) Role of plants in regulating the methane flux to the atmosphere. In: Sharkey TD, Holland EA, Mooney HA (eds) Trace Gas Emissions by plants. Academic Press Inc, New York, pp 29–63
Pangala SR, Enrich-Prast A, Basso LS, Peixoto RB, Bastviken D, Hornibrook ERC, Gatti LV, Ribeiro H, Calzans LSB, Sakuragui CM, Bastos WR, Malm O, Gloor E, Miller JB, Gauci V (2017) Large emissions from flooplain trees close the Amazon methane budget. Nature 552:230–234
Covey KR, Megonigal JP (2019) Methane production and emissions in trees and forests. New Phytolo 222:35–51. https://doi.org/10.1111/nph.15624
Jeffrey LC, Reithmaier G, Sippo JZ, Johnston SG, Tait DR, Harada Y, Maher DT (2019) Are methane emissions from mangrove stems a cryptic carbon loss pathway? insights from a catastrophic forest mortality. New Phytol 224:146–154
Martinez M, Ardón M (2021) Drivers of greenhouse gas emissions from standing dead trees in ghost forests. Biogeochemistry 154:471–488
Baldrian P (2017) Forest microbiome: diversity, complexity and dynamics. FEMS Microbiol Rev 41:109–130
Johnston SR, Boddy L, Weightman AJ (2016) Bacteria in decomposing wood and their interactions with wood-decay fungi. FEMS Microbiol Ecol. https://doi.org/10.1093/femsec/fiw179
Gora EM, Kneale RC, Larjavaara M, Muller-Landau HC (2019) Dead wood necromass in a moist tropical forest: stocks, fluxes, and spatiotemporal variability. Ecosystems 22:1189–1205
Gora EM, Lucas JM, Yanoviak SP (2019) Microbial composition and wood decomposition rates vary with microclimate from the ground to the canopy in a tropical forest. Ecosystems 22:1206–1219
Barba J, Bradford MA, Brewer PE, Bruhn D, Covey KR, van Haren J, Megonigal JP, Mikkelsen TN, Pangala SR, Pihlatie M, Poulter B, Rivas-Ubach A, Schadt CW, Terazawa K, Warner DL, Zhang Z, Vargas R (2019) Methane emissions from tree stems: a new frontier in the global carbon cycle. New Phytol 222:18–28
Putkinen A, Silijanen HMP, Laihonen A, Paasisalo I, Porkka K, Tiirola M, Haikarainen I, Tenhovirta S, Pihlatie M (2021) New insight to the role of microbes in the methane exchange in trees: evidence from metagenomic sequencing. New Phytol 231:524–536
Yip DZ, Veach AM, Yang ZK, Cregger MA, Schadt CW (2018) Methanogenic Archaea dominate mature heartwood habitats of Eastern Cotton wood (Populus deltoides). New Phytol 222:115–121. https://doi.org/10.1111/nph.15346
Zeikus JG, Ward JC (1974) Methane formation in living trees: a microbial origin. Science 184:1181–1183
Iguchi H, Sato I, Sakakibara M, Yurimoto H, Sakai Y (2012) Distribution of methanotrophs in the phyllosphere. Biosci Biotechnol Biochem 76:1580–1583
Jeffrey LC, Maher DT, Chiri E, Leung PM, Nauer PA, Arndt SK, Tait DR, Greening C, Johnston SG (2021) Bark-dwelling methanotrophic bacteria decrease methane emissions from trees. Nat Commun. https://doi.org/10.1038/s41467-021-22333-7
Vorob’ev AV, de Boer W, Folman LB, Bodelier PLE, Doronina NV, Suzina NE, Trotsenko YA, Dedysh SN (2009) Methylovirgula ligni gen. nov., sp. nov., an obligately acidophilic, facultatively methylotrophic bacterium with a highly divergent mxaF gene. Int J Syst Evol Microbiol 59:2538–2545
Vorholt JA (2012) Microbial life in the phyllosphere. Nat Rev Microbiol 10:828–840
Jeffrey LC, Maher DT, Tait DR, Reading MJ, Chiri E, Greening C, Johnston SG (2021) Isotopic evidence for axial tree stem methane oxidation within subtropical lowland forests. New Phytol 230:2200–2212
Martinez M, Ardón M, Carmichael MJ (2022) Identifying sources and oxidation of methane in standing dead trees in freshwater forested wetlands. Front Environ Sci 9:737379
Ge M, Korrensalo A, Laiho R, Kohl L, Lohila A, Pihlatie M, Li X, Laine AM, Anttila J, Putkinen A, Wang W, Koskinen M (2024) Plant-mediated CH4 exchange in wetlands: a review of mechanisms and measurement methods with implications for modelling. Sci Total Environ 914:169662
Moorhead KK, Brinson MM (1995) Response of wetlands to rising sea level in the lower coastal plain of North Carolina. Ecol Appl 5:261–271
Titus JG, Richman C (2001) Maps of lands vulnerable to sea level rise: modeled elevations along the US Atlantic and Gulf coasts. Climate Res 18:205–228
Carter LJ (1975) Agriculture: a new frontier in coastal North Carolina. Science 189:271–275
Bhattachan A, Emanuel RE, Ardón M, Bernhardt ES, Anderson SM, Stillwagon MG, Ury EA, BenDor TK, Wright JP (2018) Evaluating the effects of land-use change and future climate change on vulnerability of coastal landscapes to salwater intrusion. Elementa Sci Anthr. https://doi.org/10.1525/elementa.316
Hauer ME, Evans JM, Mishra DR (2016) Millions projected to be at risk from sea-level rise in the continental United States. Nat Clim Change 6:691–695. https://doi.org/10.1038/nclimate2961
Taillie PJ, Moorman CE, Poulter B (2019) Decadal-scale vegetation change driven by salinity at leading edge of rising sea level. Ecosystems 22:1918–1930
Livingston GP, Hutchinson GL (2009) Enclosure-based measurement of trace gas exchange: applications and sources of error. In: Matson PA, Harriss RC (eds) Biogenic trace gases: measuring emissions from soil and water. Wiley-Blackwell, Cambridge, Massachusetts, pp 14–51
Siegenthaler A, Welch B, Pangala SR, Peacock M, Gauci V (2016) Technical note: semi-rigid chambers for methane gas flux measurements on tree stems. Biogeosciences 13:1197–1207
Covey KR, Wood SA, Warren RJ II, Lee X, Bradford MA (2012) Elevated methane concentrations in trees of an upland forest. Geophys Res Lett 39:L15705. https://doi.org/10.1029/2012GL052361
Chan ASK, Parkin TB (2001) Methane oxidation and production activity in soils from natural and agricultural ecosystems. J Environ Qual 30:1896–1903
Tláskal V, Zrůstová P, Vrška T, Baldrian P (2017) Bacteria associated with decomposing dead wood in a natural temperate forest. FEMS Microbiol Ecol. https://doi.org/10.1093/femsec/fix157
Lauber C, Zhou N, Gordon JI, Knight R, Fierer N (2010) Effect of storage conditions on the assessment of bacterial community structure in soil and human-associated samples. FEMS Microbiol Lett 307:80–86
Langille MGI, Comeau A, Gohil N, Quinn D, Kwawakume A, Desai D (2014) Integrated Microbiome Resource (IMR): Protocols. https://imr.bio/protocols.html. 2022
Klindworth A, Pruesse E, Schweeer T, Peplies J, Quast C, Horn M, Glöckner FO (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acid Res 41:1. https://doi.org/10.1093/nar/gks808
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583
Callahan B (2018) Silva taxonomic training data formatted for DADA2 (Silva version 132) [Data set]. Zenodo. https://doi.org/10.5281/zenodo.1172783
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web based tools. Nucleic Acid Res 41:D590–D596
Schliep KP (2011) phangorn: phylogenetic analysis in R. Bioinformatics 27:592–593
Wright ES (2015) DECIPHER: harnessing local sequence context to improve protein multiple sequence alignment. BMC Biofinform 16:322
Callahan BJ, Sankaran K, Fukuyama JA, McMurdie PJ, Holmes SP (2016) Bioconductor workflow for microbiome data analysis: from raw reads to community analyses. F1000Res. https://doi.org/10.12688/f1000research.8986.2
R Core Team (2017) R: a language and environment for statistical computing. R Foundation for Statistical Computing Vienna, Austria, Accessed at https://www.R-project.org/
McMurdie PJ, Holmes SP (2013) phyloseq: an R package fro reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8:e61217
Palarea-Albaladejo J, Martin-Fernandez JA (2015) R package for multivariate imputation of left-consored data under a compositional approach. Chemom Intell Lab Syst 143:85–96
Gloor GB, Wu JR, Pawlowsky-Glahn V, Egozcue JJ (2016) It’s all relative: analyzing microbiome data as compositions. Ann Epidemiol 26:322–329
Gloor GB, Reid G (2016) Compositional analysis: a valid approach to analyze microbiome high-throughput sequencing data. Can J Microbiol 62:692–703
Dedysh SN, Knief C, Dunfield PF (2005) Methylocella species are facultatively methanotrophic. J Bacteriol 187:4665–4670
Dunfield PF, Belova SE, Vorob’ev AV, Cornish SL, Dedysh SN (2010) Methylocapsa aurea sp. nov., a facultative methanotroph possessing a particulate methane monooxygenase, and emended description of the genus Methylocapsa. Int J Syst Evol Microbiol 60:2659–2664
Sizova M, Panikov NS, Spiridonova EM, Slobodova NV, Tourova TP (2007) Novel facultative anaerobic acidotolerant Telmatospirillum siberiense gen. nov. sp. nov. isolated from mesotrophic fen. Syst Appl Microbiol 30:213–220
Prewitt L, Kang Y, Kakumanu ML, Williams M (2014) Fungal and bacterial community succession differs for three wood types during decay in a forest soil. Microb Ecol 68:212–221
Weedon JT, Cornwell WK, Cornelissen JHC, Zanne AE, Wirth C, Coomes DA (2009) Global meta-analysis of wood decomposition rates: a role for trait variation among tree species? Ecol Lett 12:45–56
Levy JF (1987) The natural history of the degradation of wood. Philos Trans R Soc Lond Ser A 321:423–433
Rinta-Kanto JM, Sinkko H, Rajala T, Al-Soud WA, Sørensen SJ, Tamminen MV, Timonen S (2016) Natural decay process affects the abundance and community structure of Bacteria and Archaea in Picea abies logs. FEMS Microbiol Ecol 92(7):fiw087
Carper DL, Carrell AA, Kueppers LM, Frank AC (2018) Bacterial endophyte communities in Pinus flexilis are structured by host age, tissue type, and environmental factors. Plant Soil 428:335–352
Wang Z-P, Han S-J, Li H-L, Deng F-D, Zheng Y-H, Liu H-F, Han X-G (2017) Methane production explained largely by water content in the heartwood of living trees in upland forests. J Geophys Res Biogeosci. https://doi.org/10.1002/2017JG003991
Barrera-Bello ÁM, Lucas JM, Gora EM (2024) Suspended sections within downed deadwood are drier, have altered decomposer communities, and slower decomposition. Ecosystems 27:77–89
Carmichael MJ, Smith WK (2016) Standing dead trees: a conduit for the amospheric flux of greenhouse gases from wetlands? Wetlands 36:1183–1188
Barba J, Poyatos R, Capooci M, Vargas R (2021) Spatiotemporal variability and origin of CO2 and CH4 tree stem fluxes in an upland forest. Glob Change Biol 27:4879–4893
Machacova K, Bäck J, Vanhatalo A, Halmeenmäki E, Kolari P, Mammarella I, Pumpanen J, Acosta M, Urban O, Pihlatie M (2016) Pinus sylvestris as a missing source of nitrous oxide and methane in boreal forest. Sci Rep 6:23401
Pangala SR, Moore S, Horinbrook ERC, Gauci V (2012) Trees are major conduits for methane egress from tropical forested wetlands. New Phytol 197:524–531
Baskaran V, Patil PK, Antony ML, Avunje S, Nagaraju VT, Ghate SD, Nathamuni S, Dineshkumar N, Alavandi SV, Vijayan KK (2020) Microbial communiy profiling of ammonia nad nitrite oxidizing bacterial enrichments from braskishwater ecosystems for mitigating nitrogen species. Sci Rep 10:5201
Chan CS, Chan K-G, Ee R, Hong K-W, Urbieta MS, Donati ER, Shamsir MS, Goh KM (2017) Effects of physiochemical factors on prokaryotic biodiersity in Malaysian circumneutral hot springs. Front Microbiol 8:1252
Costa PS, Reis MP, Ávila MP, Leite LR, de Araújo FMG, Salim ACM, Oliveira G, Barbosa F, Chartone-Souza E, Nascimento AMA (2015) Metagenome of a microbial community inhabiting a metal-rich tropical stream sediment. PLoS ONE 10:e0119465
Gwak J-H, Awala SI, Nguyen N-L, Yu W-J, Yang H-Y, von Bergen M, Jehmlich N, Kits KD, Lloy A, Dunfield PF, Dhal C, Hyun J-H, Rhee S-K (2022) Sulfur and methane oxidation by a single microorganism. Proceed Natl Acad Sci 119:e2114799119
Dedysh SN, Beretovskaya YY, Vasylieva LV, Belova SE, Khmelenia VN, Suzina NE, Trotsenko YA, Liesack W, Zavarzin GA (2004) Methylocella tundrae sp. nov., a novel methanotrophic bacterium from acidic tundra peatlands. Int J Syst Evol Microbiol 54:151–156
Dedysh SN, Liesack W, Khmelenia VN, Suzina NE, Trotsenko YA, Semrau JD, Bares AM, Panikov NS, Tiedje JM (2000) Methylocella palustris gen. nov., sp. nov., a new methane-oxidizing acidophilic bacterium from peat bogs, representing a novel subtype of serine-pathway methanotrophs. Int J Syst Evol Microbiol 50:955–969
Dunfield PF, Khmelenia VN, Suzina NE, Trotsenko YA, Dedysh SN (2004) Methylocella silvestris sp. nov., a novel methanotroph isolated from an acidic forest cambisol. Int J Syst Evol Microbiol 53:1231–1239
Tveit A, Hestensa AG, Robinsona SL, Schintlmeister A, Dedysh SN, Jehmlich N, von Bergen M, Herbold C, Wagner M, Richterf A, Svenning MM (2019) Widespread soil bacterium that oxidizes atmospheric methane. Proceed Natl Acad Sci 116:8515–8524
Tveit A, Schmider T, Hestnes AG, Lindgren M, Didriksen A, Svenning MM (2021) Simultaneous oxidation of atmospheric methane, carbon monoxide and hydrogen for bacterial growth. Microorganisms 9:153
Kaupper T, Mendes LW, Harnisz M, Krause SMB, Horn MA, Ho A (2021) Recovery of methanotrophic activity Is not reflected in the methane-driven interaction network after peat mining. Appl Environ Microbiol 87:e02355-e12320
Martins PD, de Jong A, Lenstra WK, van Helmond NAGM, Slomp CP, Jetten MSM, Welte CU, Rasigraf O (2021) Enrichment of novel Verrucomicrobia, Bacteroidetes, and Krumholzibacteria in an oxygen-limited methane-and iron-fed bioreactor inoculated with Bothnian Sea sediments. MicrobiologyOpen 10:e1175
Gogarten JP, Doolittle WF, Lawrence JG (2002) Prokaryotic evolution in light of gene transfer. Mol Biol Evol 19:2226–2238
Kang CS, Dunfield PF, Semrau JD (2019) The origin of aerobic methanotrophy within the proteobacteria. FEMS Microbiol Lett. https://doi.org/10.1093/femsle/fnz096
Osborne CD, Haritos VS (2018) Horizontal gene transfer of three co-inherited methane monooxygenase systems gave rise to methanotrophy in the Proteobacteria. Mol Phylogenet Evol 129:171–181
Al-Shayeb B, Schoelmerich MC, West-Roberts J, Valentin-Alvarado L, Sachdeva R, Mullen S, Crits-Christoph A, Wilkins MJ, Williams KH, Doudna JA, Banfield JF (2022) Borgs are giant genetic elements with potential to expand metabolic capacity. Nature 610:731–736
Okamura K, Kawai A, Yamada T, Hiraishi A (2011) Acidipila rosea gen. nov., sp. nov., an acidophilic chemoorganotrophic bacterium belonging to the phylum Acidobacteria. FEMS Microbiol Lett 317:138–142
Belova SE, Pankratov TA, Detkova EN, Kaparullina EN, Dedysh SN (2009) Acidisoma tundrae gen. nov., sp. nov. and Acidisoma sibiricum sp. nov., two acidophilic, psychrotolerant members of the Alphaproteobacteria from acidic northern wetlands. Int J Syst Evol Microbiol 59:2283–2290
Mieszkin S, Pouder E, Uroz S, Simon-Colin C, Alain K (2021) Acidisoma silvae sp. nov. and Acidisoma cellulosilytica sp. nov., two acidophilic bacteria isolated from decaying wood, hydrolyzing cellulose and producing poly-3-hydroxybutyrate. Microorganisms. https://doi.org/10.3390/microorganisms9102053
Hiraishi A, Matsuzawa Y, Kanbe T, Wakao N (2000) Acidisphaera rubrifaciens gen. nov., sp. nov., an aerobic bacteriochlorophyll-containing bacterium isolated from acidic environments. Int J Syst Evol Microbiol 50:1539–1546
Jones RM, Hedrich S, Johnson DB (2013) Acidocella aromatica sp. nov.: an acidophilic heterotrophic alphaproteobacterium with unusual phenotypic traits. Extremophiles 17:841–850
Kimoto K, Aizawa T, Urai M, Ve NB, Suzuki K, Nakajima M, Sunairi M (2010) Acidocella aluminiidurans sp. nov., an aluminiumtolerant bacterium isolated from Panicum repens grown in a highly acidic swamp in actual acid sulfate soil area of Vietnam. Int J Syst Evol Microbiol 60:764–768
Okamoto R, Kohirna H, Fukui M (2017) Acidocella aquatica sp. nov., a novel acidophilic heterotrophic bacterium isolated from a freshwater lake. Int J Syst Evol Microbiol 67:4773–4776
Mohagheghi A, Grohmann K, Himmel M, Leighton L, Updegraff DM (1986) Isolation and characterization of Acidothermus cellulolyticus gen. nov., sp. nov., a new genus of thermophilic, acidophilic, cellulolytic bacteria. Int J Syst Bacteriol 36:435–443
Kulichevskaya IS, Suzina NE, Liesack W, Dedysh SN (2010) Bryobacter aggregatus gen. nov., sp. nov., a peat-inhabiting, aerobic chemo-organotroph from subdivision 3 of the Acidobacteria. Int J Syst Evol Microbiol 60:301–306
Dedysh SN, Kulichevskaya IS, Serkebaeva YM, Mityaeva MA, Sorokin VV, Suzina NE, Rijpstra WI, Damsté JSS (2012) Bryocella elongata gen. nov., sp. nov., a member of subdivision 1 of the Acidobacteria isolated from a methanotrophic enrichment culture, and emended description of Edaphobacter aggregans Koch et al. 2008. Int J Syst Evol Microbiol 62:654–664
Belova SE, Suzina NE, Rijpstra WIC, Damsté JSS, Dedysh SN (2016) Edaphobacter lichenicola sp. nov., a member of the family Acidobacteriaceae from lichen-dominated forested tundra. Int J Syst Evol Microbiol 68:1265–1270
Koch IH, Gich F, Dunfield PF, Overmann J (2008) Edaphobacter modestus gen. nov., sp. nov., and Edaphobacter aggregans sp. nov., acidobacteria isolated from alpine and forest soils. Int J Syst Evol Microbiol 58:1114–1122
Wang J, Chen M-h, Lv Y-y, Jiang Y-w, Qiu L-h (2016) Edaphobacter dinghuensis sp. nov., an acidobacterium isolated from lower subtropical forest soil. Int J Syst Evol Microbiol 66:276–282
Xia F, Ou-yang T, Gao Z-h, Qiu L-h (2018) Edaphobacter flagellatus sp. nov. and Edaphobacter bradus sp. nov., two acidobacteria isolated from forest soil. Int J Syst Evol Microbiol 68:2530–2537
Pankratov TA, Dedysh SN (2010) Granulicella paludicola gen. nov., sp. nov., Granulicella pectinivorans sp. nov., Granulicella aggregans sp. nov. and Granulicella rosea sp. nov., acidophilic, polymer-degrading acidobacteria from Sphagnum peat bogs. Int J Syst Evol Microbiol 60:2951–2959
Dunfield PF, Yuryev A, Senin P, Smirnova AV, Stott MB, Hou S, Ly B, Saw JH, Zhou Z, Ren Y, Wang J, Mountain BW, Crowe MA, Weatherby TM, Bodelier PLE, Liesack W, Feng L, Wang L, Alam M (2007) Methane oxidation by an extremely acidophilic bacterium of the phylum Verrucomicrobia. Nature 450:879–882
Hou S, Makarova KS, Saw JHW, Senin P, Ly BV, Zhou Z, Ren Y, Wang J, Galperin MY, Omelchenko MV, Wolf YI, Yutin N, Koonin EV, Stott MB, Mountain BW, Crowe MA, Smirnova AV, Dunfield PF, Feng L, Wang L, Alam M (2008) Complete genome sequence of the extremely acidophilic methanotroph isolate V4, Methylacidiphilum infernorum, a representative of the bacterial phylum Verrucomicrobia. Biol Direct 3:26
Islam T, Jensen S, Reigstad LJ, Larsen Ø, Birkeland N-K (2008) Methane oxidation at 55 °C and pH 2 by a thermoacidophilic bacterium belonging to the Verrucomicrobia phylum. Proceed Natl Acad Sci 105:300–304
Mohammadi S, Pol A, van Alen TA, Jetten MSM, Op den Camp HJM (2017) Methylacidiphilum fumariolicum SolV, a thermoacidophilic ‘Knallgas’ methanotroph with both an oxygen-sensitive and-insensitive hydrogenase. ISME J 11:945–958
Picone N, Blom P, Hogendoom C, Frank J, van Alen T, Pol A, Gagliano AL, Jetten MSM, D’Alessandro W, Quatrini P, Op den Camp HJM (2021) Metagenome assembled genome of a novel Verrocomicrobial methanotroph from Pantelleria Island. Front Microbiol 12:666929
Foesel BU, Mayer S, Luckner M, Wanner G, Rohde M, Overmann J (2016) Occallatibacter riparius gen. nov., sp. nov. and Occallatibacter savannae sp. nov., acidobacteria isolated from Namibian soils, and emended description of the family Acidobacteriaceae. Int J Syst Evol Microbiol 66:219–229
Seki T, Matsumoto A, Shimada R, Inahashi Y, Õmura S, Takahashi Y (2012) Conexibacter arvalis sp. nov., isolated from a cultivated field soil sample. Int J Syst Evol Microbiol 62:2400–2404
Choi G-M, Lee Y-W, Choe H, Kim S-Y, Im W-T (2022) Humibacter ginsenosidimutans sp. nov., with ginsenoside-converting activity isolated from activated sludge. Int J Systematic Evolut Microbiol. https://doi.org/10.1099/ijsem.1090.005442
Garcia-Fraile P, Benada O, Cajthaml T, Baldrian P, Lladó S (2016) Terracidiphilus gabretensis gen. nov., sp. nov., an abundant and active forest soil Acidobacterium important in organic matter transformation. Appl Environ Microbiol 82:560–569
Ohshima S, Sato Y, Fujimura R, Takashima Y, Hamada M, Nishizawa T, Narisawa K, Ohta H (2016) Mycoavidus cysteinexigens gen. nov., sp. nov., an endohyphal bacterium isolated from a soil isolate of the fungus Mortierella elongata. Int J Syst Evol Microbiol 66:2052–2057
Zhang L, Chen X-L, Hu Q, Chen K, Yan X, Li S-P, Freilich S, Jiang J-D (2016) Haoranjiania flava gen. nov., sp. nov., a new member of the family Chitinophagaceae, isolated from activated sludge. Int J Syst Evolut Microbiol 66:4686–4691
Martineau C, Villeneuve C, Mauffrey F, Villemur R (2013) Hyphomicrobium nitrativorans sp. nov., isolated from the biofilm of a methanol-fed denitrification system treating seawater at the Montreal Biodome. Int J Syst Evol Microbiol 63:3777–3781
Xu Q, Zhang Y, Wang X, Wang G (2021) Hyphomicrobium album sp. nov., isolated from mountain soil and emended description of genus Hyphomicrobium. Arch Microbiol 203:5931–5936
Kim Y-J, Nguyen N-L, Weon H-Y, Yang D-C (2013) Sediminibacterium ginsengisoli sp. nov., isolated from soil of a ginseng field, and emended descriptions of the genus Sediminibacterium and of Sediminibacterium salmoneum. Int J Syst Evol Microbiol 63:905–912
Wu S, Zhong L, Liao S, Li T, Zhou Z, Wang G (2021) Sediminibacterium soli sp. nov., isolated from soil. Arch Microbiol 203:967–973
Izumi Y, Takizawa M, Tani Y, Yamada H (1982) An obligate methylotrophic Hyphomicrobium strain identification, growth characteristics and cell composition. J Ferment Technol 60:371–375
McDonald IR, Doronina NV, Trotsenko YA, McAnulla C, Murrell JC (2001) Hyphomicrobium chloromethanicum sp. nov. and Methylobacterium chloromethanicum sp. nov., chloromethane-utilizing bacteria isolated from a polluted environment. Int J Syst Evol Microbiol 51:119–122
Urakami T, Sasaki J, Suzuki K-I, Komagata K (1995) Characterization and description of Hyphomicrobium denitrificans sp. nov. Int J Syst Bacteriol 45:528–532
Ramírez-Bahena M, Tejedor C, Martín I, Velázquez E, Peiz A (2013) Endobacter medicaginis gen. nov., sp. nov., isolated from alfalfa nodules in an acidic soil. Int J Syst Evol Microbiol 63:1760–1765
Kim E-K, Hoang V-A, Kim Y-J, Nguyen N-L, Sukweenadhi J, Kang J-P, Yang D-C (2015) Humibacter ginsengiterrae sp. nov., and Humibacter ginsengisoli sp. nov., isolated from soil of a ginseng field. Int J Syst Evol Microbiol 65:2734–2740
Lin S-Y, Hameed A, Liu Y-C, Hsu Y-H, Hsieh Y-T, Lai W-A, Young C-C (2017) Humibacter aquilariae sp. nov., an actinobacterium isolated from an agarwood chip. Int J Syst Evol Microbiol 67:1468–1472
Bouam A, Armstrong N, Levasseur A, Drancourt M (2018) Mycobacterium terramassiliense, Mycobacterium rhizamassiliense and Mycobacterium numidiamassiliense sp. nov., three new Mycobacterium simiae complex species cultured from plant roots. Sci Rep. https://doi.org/10.1038/s41598-018-27629-1
Huang H-Y, Li J, Zhao G-Z, Zhu W-Y, Yang L-L, Tang H-Y, Xu L-H, Li W-J (2012) Sphingomonas endophytica sp. nov., isolated from Artemisia annua L. Int J Syst Evol Microbiol 62:1576–1580
Rivas R, Abril A, Trujillo ME, Velázquez E (2004) Sphingomonas phyllosphaerae sp. nov., from the phyllosphere of Acacia caven in Argentina. Int J Syst Evol Microbiol 54:2147–2150
Takeuchi M, Sakane T, Yangai M, Yamasoto K, Hamana K, Yokota A (1995) Taxonomic study of bacteria isolated from plants: proposal of Sphingomonas rosa sp. nov., Sphingomonas pruni sp. nov., Sphingomonas asaccharolytica sp. nov., and Sphingomonas mali sp. nov. Int J Syst Bacteriol 45:334–341
Yan Z-F, Lin P, Won K-H, Li C-T, Park G, Chin B, Kook M, Wang Q-J, Yi T-H (2018) Sphingomonas rhizophila sp. nov., isolated from rhizosphere of Hibiscus syriacus. Int J Syst Evol Microbiol 68:681–686
Whang K-S, Lee J-C, Lee H-R, Han S-I, Chung S-H (2014) Terriglobus tenax sp. nov., an exopolysaccharideproducing acidobacterium isolated from rhizosphere soil of a medicinal plant. Int J Syst Evol Microbiol 64:431–437
Acknowledgements
The authors would like to thank Meredith Keppel and Destinee Parson for field and lab assistance, and the members of the Fall 2023 Hollins University Microbial Ecology class for thoughtful feedback on the manuscript. We are grateful for the assistance of North Carolina State University’s Genomic Sciences Laboratory for library preparation and Illumina sequencing and for the assistance of Allison Dickey at North Carolina State University’s Bioinformatics Consulting and Service Corps at North Carolina State University for help with bioinformatic analyses.
Funding
This work was supported by the National Science Foundation [DEB1713592] to Marcelo Ardón, by a Hollins University Summer Collaborative Research Award to Mary Jane Carmichael, and by a 2019 North Carolina Sea Grant/Space Grant Fellowship to Melinda Martinez. Melinda Martinez was also supported by a USGS Mendenhall Postdoctoral Research Fellowship funded by the USGS Ecosystems Mission Area. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
Author information
Authors and Affiliations
Contributions
Conceptualization and methodology: Marcelo Ardón, Mary Jane Carmichael, and Melinda Martinez; Formal analysis and investigation: Suzanna Bräuer, Mary Jane Carmichael, and Melinda Martinez; Writing- original draft preparation: Mary Jane Carmichael; Writing- review, editing, and final approval: Marcelo Ardón, Mary Jane Carmichael, Suzanna Bräuer, Melinda Martinez; Funding acquisition: Marcelo Ardón, Mary Jane Carmichael, and Melinda Martinez; Resources and supervision: Marcelo Ardón, Mary Jane Carmichael.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests or conflicts of interest.
Ethics Approval
There were no human or animal subjects involved in this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Carmichael, M.J., Martinez, M., Bräuer, S.L. et al. Microbial Communities in Standing Dead Trees in Ghost Forests are Largely Aerobic, Saprophytic, and Methanotrophic. Curr Microbiol 81, 229 (2024). https://doi.org/10.1007/s00284-024-03767-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00284-024-03767-w