
Abstract
In ischemic stroke, the primary neuronal injury caused by the disruption of energy supply is further exacerbated by secondary sterile inflammation. The inflammatory cascade is largely initiated by the purine adenosine triphosphate (ATP) which is extensively released to the interstitial space during brain ischemia and functions as an extracellular danger signaling molecule. By engaging P2 receptors, extracellular ATP activates microglia leading to cytokine and chemokine production and subsequent immune cell recruitment from the periphery which further amplifies post-stroke inflammation. The ectonucleotidases CD39 and CD73 shape and balance the inflammatory environment by stepwise degrading extracellular ATP to adenosine which itself has neuroprotective and anti-inflammatory signaling properties. The neuroprotective effects of adenosine are mainly mediated through A1 receptors and inhibition of glutamatergic excitotoxicity, while the anti-inflammatory capacities of adenosine have been primarily attributed to A2A receptor activation on infiltrating immune cells in the subacute phase after stroke. In this review, we summarize the current state of knowledge on the ATP-adenosine axis in ischemic stroke, discuss contradictory results, and point out potential pitfalls towards translating therapeutic approaches from rodent stroke models to human patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Stroke ranks among the leading causes of death and disability worldwide. With 7.6 million new cases in 2019, ischemic stroke accounted for 62% of all strokes, and 3.3 million patients died from this widespread disease [1]. At present, treatment options of acute ischemic stroke are limited to intravenous (i.v.) thrombolysis and mechanical thrombectomy both aiming at reperfusion of the clotted vessel. Due to strict inclusion criteria, these therapies are only available for a subset of patients, and many patients present with persistent neurological deficits despite successful recanalization. Therefore, additional treatment strategies to combat ischemic brain injury are urgently needed. Research over the last three decades identified the substantial contribution of sterile inflammation to secondary neuronal injury following ischemic stroke. Consequently, targeting secondary infarct growth mediated by infiltrating immune cells emerged as a promising therapeutic strategy. The inflammatory cascade is initiated by the excessive release of danger-/damage-associated molecular patterns (DAMPs) which activate pattern recognition receptors on microglia and astrocytes with subsequent cytokine and chemokine production and immune cell attraction from the periphery [2]. Among these DAMPs, the purine adenosine triphosphate (ATP) plays a pivotal role. Geoffrey Burnstock pioneered the concept that ATP released by cells acts as an extracellular signaling molecule and thus exerts key functions beyond energy supply [3].
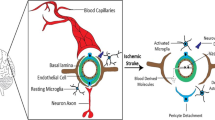
Under physiological conditions, cells within a tissue, including the brain, release ATP and nicotinamide adenine dinucleotide (NAD+) into the interstitial space in an activity-dependent and regulated manner. Release mechanisms include exocytosis, connexin or pannexin hemichannels, ATP-binding cassette (ABC) transporters, calcium homeostasis modulator (CALHM) channels, a macropore after P2X7 receptor overstimulation, maxi-anion channels (MACs), and volume-regulated ion channels (VRACs) as comprehensively reviewed by Giuliani et al. [4]. At sites of inflammation or ischemia, ATP is further liberated in an uncontrolled fashion via cell damage and death [4]. Indeed, an increase in extracellular ATP concentrations within the brain parenchyma during in vivo ischemia was measured by microdialysis [5, 6]. To improve the accuracy of extracellular ATP sensing and to circumvent methodological challenges of purine measurements within intact tissues, the group of Francesco Di Virgilio engineered a chimeric plasma membrane luciferase (pmeLUC) [7]. PmeLUC emits a measurable bioluminescent signal upon D-luciferin administration in the presence of extracellular ATP. Using this in vivo ATP biosensor, ATP release could be visualized as early as 30 min and up to 24 h after transient middle cerebral artery occlusion (tMCAO) [8]. After being released, ATP can either signal through its ionotropic P2X and metabotropic P2Y receptors or experience degradation to adenosine by ectoenzymes. The present review will give an overview about the different players of purinergic signaling, their expression in mice and humans (Fig. 1), and their contribution to tissue damage and regeneration in ischemic stroke. Details on the cited experimental stroke studies using mice with global genetic deficiency for the different players of purinergic signaling can be found in STab. 1.

Gene expression data heatmap of 26 enzymes and receptors (y axis) involved in purinergic signaling in 37 conserved cell types (x axis) between humans (left) and mice (right). All enzymes and receptors are listed with gene names. For human single-nucleus RNA-sequencing data (SMART-SEQ) of the middle temporal gyrus (MTG), 15,928 nuclei from 8 human tissue donors were analyzed [10] (Allen Institute for Brain Science. Allen Brain Map, Cell Types Database, RNA-Seq Data. Available from: https://celltypes.brain-map.org/rnaseq/human/mtg). For the mouse cortex single-cell RNA-sequencing dataset (SMART-SEQ) 14,249 cells from the primary visual cortex (V1) and 9573 cells from the anterior lateral motor cortex (ALM) were profiled [11] (Allen Institute for Brain Science. Allen Brain Map, Cell Types Database, RNA-Seq Data. Available from: https://celltypes.brain-map.org/rnaseq/mouse/v1-alm). Expression levels were quantified as log2-transformed counts per million (CPM) of intronic plus exonic reads. Homologous cell types between the two species [10] can be divided into 5 non-neuronal cells (astrocytes, microglia and perivascular macrophages (PVM), oligodendrocytes (Oligo), oligodendrocyte precursor cells (OPC), and endothelial cells), 12 excitatory neuron (Exc) types named after the originating brain lamina (L) and their projection targets (intratelencephalic (IT), extratelencephalic-pyramidal tract (ET), near-projecting (NP), corticothalamic (CT)) and 20 inhibitory neuron cell types. Inhibitory neurons are further subdivided into lysosomal associated membrane protein family member 5 (Lamp5), somatostatin (Sst), parvalbumin (Pvalb), vasoactive intestinal polypeptide (Vip), and paired box 6 (Pax6) expressing cells and chandelier neurons
Signaling of ATP via P2X receptors in stroke
Extracellular ATP serves as ligand for all purinergic receptors of the ionotropic P2X family and for several P2Y receptors, while its metabolite ADP binds exclusively to P2Y receptors. The P2X family comprises seven members (P2X1-P2X7). Three P2X receptor subunits form an ion channel that upon ATP-mediated gating conducts the rapid influx of calcium and sodium ions and efflux of potassium ions. The expression of P2X receptors has been ascribed to various brain-resident cells as well as to cells of the immune system [9] with P2X1, P2X4, and P2X7 being associated with the outcome of cerebral ischemic events. In the central nervous system (CNS) of both mice and humans, microglia show the highest expression of these three receptors [10,11,12,13] (Fig. 1).
Of importance in the context of ischemic stroke, P2X1 is expressed on platelets. ATP (10 μM) increases platelet aggregation in vitro, which can be prevented by the P2X1 antagonist NF449 [14]. P2X1 is also expressed by neutrophil granulocytes of the mouse, where its activation promotes neutrophil chemotaxis [15]. Pharmacological blockade of P2X1 and genetic deletion of P2rx1 in mice diminished the activation and recruitment of neutrophils to inflamed arteriolar endothelial cells, accompanied by impaired platelet aggregation and fibrin generation [16]. Therefore, targeting of P2X1 could be a promising new approach to ameliorate neutrophil driven post-stroke inflammation as well as stroke-associated thromboinflammation [17]. Apart from immune cells, P2X1 is also expressed on vascular smooth muscle cells (VSMCs) of rat middle cerebral arteries [18]. It has been shown that P2X1 and P2X4 form heterotrimers in VSMCs, which, in contrast to P2X1 homotrimers, contribute to ATP-mediated vasoconstriction. The impact of P2X1 signaling on VSMCs in the context of stroke still needs to be investigated.
ATP released during cerebral ischemia triggers P2X4 opening on brain innate immune cells such as microglia or infiltrating monocytes/macrophages and sustained P2X4 activation contributes to the ischemic injury [19, 20]. Among immune cells, P2X4 is expressed on T cells promoting early T cell activation in concert with P2X7 [21] as well as chemotaxis [22]. Indeed, P2rx4 deficiency in mice led to smaller infarcts after tMCAO [23]. Interestingly, when specifically deleting P2rx4 in myeloid cells using LysM-Cre mice, only female mice showed a reduction in infarct size, suggesting a sex difference in the P2X4 response after stroke [23]. Another experimental stroke study by the same group verified the detrimental role of P2X4 in brain ischemia by using the P2X4 antagonist 5-BDBD: When applied 4 hours (h) after tMCAO, 5-BDBD reduced infarct size, number of infiltrating pro-inflammatory myeloid cells, and blood-brain-barrier (BBB) disruption [24]. 5-BDBD was also shown to attenuate brain injury in a mouse model of intracerebral hemorrhage, significantly reducing brain edema, BBB disruption, neuronal cell death, and neurological deficits [25]. Another study demonstrated that P2X4 is also expressed by pyramidal neurons of the rat hippocampal CA1 region [26]. In line with tMCAO data, intracerebroventricular (i.c.v.) 5-BDBD treatment immediately after cerebral ischemia (four-vessel occlusion model) and once 24 h later was also protective by blocking neuronal apoptosis, thereby promoting neuronal survival. Similar results were obtained with the P2X4 antagonist PSB-12062 [26]. Given the central role of P2X4 in inflammation, cell migration, and also pain, new P2X4 antagonists are continuously developed. The group of Kenneth A. Jacobson recently described the generation and evaluation of MRS4719 and MRS4596, two potent new P2X4 antagonists (IC50 0.503 and 1.38 μM for human P2X4, respectively) with neuroprotective effects in murine ischemic stroke (tMCAO): MRS4719 dose dependently reduced infarct size and brain atrophy 3 and 35 days post-stroke [27]. First P2X4-specific antagonistic monoclonal antibodies have been developed by AstraZeneca [28]. The affinity-optimized clone IgG#151-LO showed high selectivity for human P2X4 and induced potent and complete blockage of P2X4 currents. Inhibition of spinal P2X4 either by intrathecal delivery of anti-P2X4 antibodies or by systemic delivery of an anti-P2X4 bispecific antibody with enhanced blood–spinal cord barrier permeability achieved analgesia for 7 days in a mouse model of neuropathic pain [28]. Further, single domain antibodies (nanobodies) against P2X4 have been developed, but did not show P2X4-antagonizing properties [29]. In principle, evaluation of new biological-based P2X4-targeting drugs in the setting of brain ischemia would be desirable, as these usually exhibit good pharmacological properties in vivo and are well suited for cell-specific targeting of P2X4. Both factors might be important for the successful translation into treating stroke patients. Two studies highlight the need for a differentiated approach regarding timing and cell tropism: Sustained absence of P2X4 signaling, as occurring in P2X4ko mice, reduced infarct size after tMCAO, but myeloid cell-specific P2X4 deficiency led to more depressive behavior 30 days after stroke onset compared to recovering wild-type (WT) mice [23]. As another example of potentially detrimental effects of P2X4 blockade, brain endothelial cells acquire ischemic tolerance in a P2X4-dependent fashion as 5-BDBD treatment 15 min before ischemic preconditioning prevents this tolerization [30]. Therefore, biologics-based spatiotemporal targeting of P2X4 could be the key to future therapies focusing on P2X4 in stroke. However, crossing the BBB still remains a challenge for most antibody-based constructs, an obstacle that needs to be overcome by modern antibody engineering technology [31].
P2X7 is most likely the most extensively studied P2X receptor in brain ischemia. P2X7 activation is known as a potent trigger for the NLRP3 inflammasome formation in macrophages and microglia, leading to the release of pro-inflammatory interleukin-1β (IL-1β), a powerful driver of post-stroke inflammation [32,33,34]. Yet, deciphering the complex role of P2X7 during the interlinked processes of post-stroke inflammation, BBB breakdown and neuronal recovery remains challenging.
First studies by Nancy Rothwell’s group did not show a difference in stroke size when comparing P2rx7-deficient and WT mice [35], and i.c.v. treatment with P2X antagonists oxidized ATP and PPADS immediately before and again 30 minutes (min) after tMCAO did not affect lesion size after tMCAO either [36]. However, other studies demonstrated that intraperitoneal treatment with the broad P2 antagonist Reactive Blue 2 5 min after intraluminal permanent MCAO (pMCAO) in rats reduced brain damage and that MCAO led to an upregulation of P2X7 on microglia [37]. P2X7 upregulation had already been observed by earlier studies with hypertensive rats subjected to pMCAO by electrocoagulation [38]. Other studies suggested a protective role of P2X7 in stroke: I.c.v. injection of the P2X7-specific agonist BzATP into rats 1 h after tMCAO improved their motor functions when compared to mock-injected controls [39]. Studies using Brilliant Blue G (BBG) as P2X7 antagonist reported different outcomes regarding the role of P2X7 in rodent stroke models: The group of Carlos Matute reported that intraperitoneal BBG treatment 30 min after ischemia onset reduced the lesion size in rats subjected to tMCAO [40] and later reproduced their findings in mice [41]. The study by Caglayan and colleagues in 2017 again confirmed that i.c.v. injection of BBG reduced lesion size in mice when injected 30 min before tMCAO [42]. Similarly, intraperitoneal BBG administration also turned out to be protective in murine photothrombotic stroke when applied 1, 3, and 5 days after ischemia [43]. In contrast, a study, in which BBG was injected intraperitoneally (i.p.) daily for 3 days immediately after a short (15 min) tMCAO in mice, did not demonstrate a difference in lesion size between treatment and control group [44].
As for P2X7 targeting compounds, contrasting results have also been reported in studies using P2rx7-deficient mice. As mentioned above, first stroke experiments with P2X7ko mice did not reveal differences in stroke size [35]. Later in 2015, the group of Carlos Matute reported smaller stroke lesions (tMCAO) in P2X7ko mice when compared to WT [41]. In contrast, the group of Michael Schäfer reported reduced edema formation in P2X7ko mice 3 days after tMCAO, while stroke lesion size did not differ between P2X7ko and WT mice [45]. A recent study published by our group again suggested a detrimental role of P2X7 in stroke: Mice overexpressing P2X7 in cells that naturally express P2X7 [46] exhibited larger infarcts compared to WT controls. Further, i.c.v. injection of P2X7-blocking nanobodies [47] in mice directly before tMCAO significantly reduced lesion size compared to the control group [8]. This study represents the first approach to use highly specific biologicals to block P2X7 in the context of stroke. As mentioned above, the BBB constitutes a major obstacle for i.v. application of antibody-based drugs targeting brain-resident cells like microglia. Even for the small P2X7-specific nanobodies only high doses beyond 1 mg lead to detectable amounts of the nanobodies on brain microglia [48]. Interestingly, an alternative approach based on adeno-associated virus (AAV)-mediated muscle cell transduction followed by in vivo production of P2X7 antagonistic nanobodies led to the successful diffusion of the nanobodies into the brain where they could be detected on microglia for up to 120 days after transduction [48]. Since the role of P2X7 in long-term recovery after stroke still remains elusive, the AAV approach could be utilized to investigate this technically challenging question of high translational relevance.
When using antibody or nanobody-based therapeutics, one has to monitor for potential immune-related adverse events (irAE). Antibody-induced inflammation via complement activation or activation of natural killer (NK) cells and macrophages can be prevented by modifying the Fc-region of antibodies. Nanobody monomers lack Fc regions in principle but can be tailored as Fc-fusion proteins with desired or no Fc-related functions [49, 50]. To date, no adverse effects of P2X7 blockade by antibodies or nanobodies have been described. However, one has to keep in mind that P2X7 blockade can also potentially alter the susceptibility towards infections. Indeed, the P2X7 receptor antagonism can have both beneficial and deleterious effects depending on the type of pathogen, its virulence, and the severity of infection as reviewed in [51].
In general, though various studies suggest a role of P2X7 in stroke, many P2X7-related pathways and mechanisms that participate in post-stroke inflammation, thromboinflammation, BBB breakdown, neuroprotection and recovery remain to be revealed and connected. Contrasting results may be the consequence of (1) variations in the specificity of applied P2X7 antagonists, (2) different stroke models that do or do not allow reperfusion or that are based on photothrombosis, (3) differences related to administration routes or pre- versus post-treatment, or (4) the use of P2rx7-deficient mice of different origin. For the latter aspect, differences between the GlaxoSmithKline (GSK) and the Pfizer P2X7ko mice have already been demonstrated [52]: GSK P2X7ko mice lack P2X7 expression on innate immune cells but exhibit robust expression of P2X7 on T cells due to an escape splice variant. In contrast, the Pfizer P2X7ko mice lack P2X7 in both immune cell compartments. These differences might explain the opposite outcome of experimental autoimmune encephalomyelitis (EAE) studies: GSK P2X7ko mice showed suppressed EAE development compared to WT mice [53], whereas Pfizer P2X7ko mice exhibited an exacerbated disease course in EAE when compared to WT mice [54]. GSK P2X7ko mice have not been investigated in murine stroke models yet. Additionally, one has to keep in mind that the Pfizer P2X7ko mice are congenic and therefore might contain passenger mutations from 129 embryonic stem cells used for P2rx7 gene targeting. Indeed, our group could identify a P2rx7 passenger mutation in the commonly used P2X4ko mouse [55, 56]. Since P2rx4 and P2rx7 are direct neighboring genes, differentially expressed genes in congenic P2X4ko might also occur in congenic P2X7ko mice. If such passenger mutations also exist in the widely used Pfizer P2X7ko mouse [57] still needs to be investigated, in particular if they affect the outcome of stroke.
Of note, the ATP-gated P2X receptors expressed in the brain differ greatly in their sensitivity to ATP. As measured in transfected X. laevis oocytes or HEK293 cells by electrophysiology, the ATP concentration to induce the half-maximal response (K1/2) is about ten times lower for rat P2X1 (0.82 μM) than for rat P2X4 (11 μM). Rat P2X7, in turn, has a K1/2 of 130 μM and is accordingly about ten times less sensitive to ATP than P2X4 [58]. Thus, the activation of P2X receptors in the brain does not only depend on the expression pattern of the receptors, but also on the availability of extracellular ATP.
Role of P2Y receptors in stroke
P2Y receptors are G-protein-coupled receptors activated by ATP, ADP, and other naturally occurring nucleoside phosphates. The most abundant P2Y receptors in the brain are P2Y12 and P2Y13, expressed at high levels in microglial cells (Fig. 1). The natural ligand for these two receptors is ADP, which upon engagement inhibits adenylate cyclase leading to a decrease in cyclic AMP (cAMP) and subsequent activation of microglia. P2Y12 receptor inhibitors, such as clopidogrel and ticagrelor, are commonly used for the secondary prevention of atherosclerotic ischemic stroke because of their effective reduction of P2Y12-mediated platelet aggregation. However, the beneficial effects of these inhibitors are not limited to this anti-thrombotic effect, since they are also neuroprotective in murine tMCAO (oral administration from 24 h before to 8 h after occlusion) [59], anti-atherosclerotic [60], and can reduce microglial activation and chemotaxis after pMCAO in rats (oral treatment 10 min, 22 and 36 h after occlusion) [61]. Interestingly, physiological concentrations of ADP facilitate the contact of microglia processes with neuronal cell bodies. After neuronal injury, higher concentrations of ADP triggered microglial-mediated neuroprotection in a P2Y12 receptor-dependent manner, sparing viable neurons from cell death and maintaining functional connectivity [62]. These data pose a warning to the treatment with P2Y12 inhibitors for stroke prevention and prompt the analysis of neuronal function in MCAO models under P2Y12 inhibitor treatment.
ADP can also activate P2Y1 receptors, expressed in astrocytes, and is a partial agonist for the P2Y6 receptor, expressed by microglial cells. While pre- and post-stroke blockade of P2Y1 by MRS2500 i.c.v. proved beneficial for stroke outcome in transient and permanent MCAO models [63, 64], the role of P2Y6 in stroke is still controversial: Pharmacological inhibition of P2Y6-mediated phagocytosis with MRS2578 on 3 consecutive days after tMCAO aggravated infarct size, brain atrophy, and neurological deficits [65]. However, in a model of brain ischemia induced by endothelin-1 injection, knockdown of the P2Y6 receptor had a beneficial effect by delaying phagocytosis of stressed neurons in the peri-infarct region [66]. P2Y2 and P2Y11 receptors are not expressed on brain-resident cells but are present on immune cells that can be attracted to the brain after ischemia-reperfusion injury. Indeed, ATP signaling through P2Y2 and P2Y11 promotes neutrophil and monocyte migration [67,68,69]. Therefore, a transient increase of systemic ATP after stroke may contribute to brain immune cell infiltration. Of note, ATP has antagonistic effects on P2Y12 [70], which may constitute an intrinsic regulatory mechanism to prevent extended inflammation.
Ectoenzymes convert extracellular ATP via ADP and AMP to adenosine
Extracellular ATP has a half-life of less than 5 min. Ectonucleotidases sequentially degrade extracellular ATP to dampen the pro-inflammatory effects of ATP signaling and to generate adenosine as a metabolite with signaling properties itself [4]. Once produced, adenosine signals through four different G-protein-coupled P1 receptors and has neuroprotective [71] and anti-inflammatory [72] properties. Modulation of ischemic brain injury by adenosine signaling will be elucidated in the next section.
On the canonical pathway of adenosine production (Fig. 2), ecto-nucleoside triphosphate diphosphohydrolase 1 (NTPDase1/CD39) hydrolyzes ATP to adenosine diphosphate (ADP) and then to adenosine monophosphate (AMP). AMP is metabolized to adenosine by ecto-5’-nucleotidase (NT5E/CD73) which is the dominant adenosine-generating enzyme. CD73 activity can be inhibited by high concentrations of ATP and ADP [73]. The alternative non-canonical pathway to generate AMP for subsequent adenosine formation independently of CD39 involves NAD glycohydrolase/CD38 and ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1/CD203a): CD38 hydrolyzes extracellular NAD+ to ADP ribose (ADPR) which is further converted to AMP by ENPP1 [74]. On top of that, tissue non-specific alkaline phosphatase (TNAP) mainly expressed at the vasculature contributes to adenosine formation in the rodent and human brain by catalyzing all steps in the degradation of nucleoside 5’-tri-, -di-, and -monophosphates to adenosine [75, 76].

Nucleotide-metabolizing enzymes. Extracellular ATP serves as ligand for ionotropic P2X receptors, e.g., P2X7. On the canonical pathway, ATP is degraded to ADP and AMP by the ectonucleotidase CD39. ADP is a natural ligand for P2Y12. CD73 finally dephosphorylates AMP to generate adenosine (ADO). On the alternative non-canonical pathway, AMP is generated from NAD+: CD38 hydrolyzes NAD+ to generate ADPR, which is further converted to AMP by ENPP1. Extracellular adenosine serves as ligand for adenosine receptors, e.g., A2AR
In mouse and human brain, CD39 expression is highest on microglia followed by endothelial cells [10,11,12,13, 76] (Fig. 1). In the immune cell compartment it is expressed by neutrophils, monocytes, dendritic cells (DCs), B cells, regulatory T cells (Tregs), and NK cells (Immunological Genome Project). In the mouse brain, CD73 expression is highest on oligodendrocytes whereas both astrocytes and oligodendrocytes show high expression in humans [10,11,12,13] (Fig. 1). It has to be noted that, in contrast to human brain endothelial cells [77], CD73 could not be detected on mouse brain endothelial cells in vivo but shows high expression in the olfactory bulb and in the caudoputamen [76, 78, 79]. In the immune cell compartment CD73 is expressed by T and B cells, neutrophils, and macrophages (Immunological Genome Project). CD38 expression is highest on astrocytes followed by endothelial cells in both mice and humans, whereas ENPP1 is mainly expressed by microglia in both species and by oligodendrocytes in mice [10,11,12,13] (Fig. 1). The enzyme adenosine deaminase (ADA) completes the purine-inactivating machinery and catalyzes the deamination of adenosine to inosine. The highest tissue ADA enzyme activity in humans was found in lymphoid tissues, particularly the thymus, the brain, and the gastrointestinal tract [80]. In the rodent brain, immunohistochemistry revealed an extensive plexus of ADA-containing neurons in the hypothalamus [81]. Genetic deficiency of ADA2 results in an autoimmune syndrome characterized by systemic vasculitis and inflammation. Ischemic strokes occur early in childhood and are often a consequence of systemic vasculitis affecting the CNS, but stroke can also represent the initial clinical symptom without any signs of cerebral vasculitis [82, 83].
Taken together, ectonucleotidases shape the inflammatory microenvironment by tightly controlling and balancing the extracellular concentration of pro-inflammatory ATP and anti-inflammatory adenosine.
In the late 1990s, CD39 was identified as a potent thromboregulator by rapidly degrading ADP released from activated platelets at sites of vascular injury, thus inhibiting ADP-induced platelet activation, recruitment, and aggregation [84,85,86]. Consistent with the notion that platelet and fibrin accumulation occurs distal to the occluded vessel leading to microvascular thrombosis [87], CD39 knockout mice exhibited increased cerebral infarct volumes and reduced post-ischemic reperfusion after tMCAO [88], whereas treatment of WT mice with recombinant soluble human CD39 up to 3 h after induction of stroke reduced infarct sizes at 24 h and promoted an increase in post-ischemic blood flow [88]. Global transgenic overexpression of CD39 in mice was likewise protective in photothrombotic stroke [89]. Selective CD39 overexpression in myeloid lineage cells also conferred a reduction in infarct volume indicating a contribution of the ATP-degrading machinery on infiltrating immune cells to the protective phenotype [89]. However, these experiments were only conducted in a small number of animals.
Regarding the role of CD73 in ischemic stroke, contradictory results can be found in the literature. While mice deficient for CD73 exhibited larger infarcts and increased immune cell infiltration into the ischemic hemisphere than wild-type mice after photothrombotic stroke [79], global CD73 deficiency did not affect infarct volume, immune cell infiltration, and glia cell activation profiles following ischemia-reperfusion injury induced by tMCAO in our laboratory [90]. It seems most plausible that these conflicting findings can be attributed to the different experimental stroke models used: In the photothrombotic stroke model, focal activation of the photosensitive dye induces severe endothelial cell injury, local thrombus formation, and rapid blood-brain barrier breakdown leading to pronounced vascular (extracellular) edema. The irreversibly injured infarct core is not surrounded by functionally impaired tissue predisposed for infarction but capable of surviving, the so-called penumbra, and the vessel is permanently occluded not allowing reperfusion. On the contrary, ischemic stroke induced by tMCAO is characterized by a leading cytotoxic (intracellular) edema, a penumbra with delayed neuronal cell death due to secondary mechanisms such as inflammation and reperfusion [91].
When interpreting and extrapolating results on CD73 from experimental stroke studies conducted in mice, one has to keep in mind that CD73 is absent on murine brain endothelial cells but highly expressed on peripheral endothelial cells and human brain endothelial cells [76, 77, 92] (Fig. 1). These expression differences account for the finding that CD73 knockout mice exhibited increased vascular leakage in peripheral organs where CD73-derived adenosine signaling is implicated in endothelial barrier function via A2A and A2B adenosine receptors (AR), whereas vascular permeability in the brain was not affected [93].
Beyond brain-resident cells, infiltrating immune cells play a central role in secondary neuronal injury, and their CD73 expression can also influence stroke outcome. Murine CD4+CD25+Foxp3+ Tregs co-express CD39 and CD73, and adenosine generated by their coordinated activity mediates both their immunosuppressive effects on activated T effector cells and their own expansion and functionality through A2AR signaling [94, 95]. In vivo expansion of Tregs with interleukin-2/interleukin-2 antibody (IL-2/IL-2Ab) complex reduced infarct volume and infiltration of T cells and macrophages into the ischemic hemisphere after tMCAO. Mechanistically, CD73 expression on Tregs played a crucial role in IL-2/IL-2Ab-afforded protection as Tregs prepared from CD73 knockout mice with or without IL-2/IL-2Ab treatment failed to protect WT-recipient mice in an adoptive transfer experiment [96]. Analogous to Treg functions, Th17 responses are also shaped by CD73 activity [4]. On γδ T cells, low CD73 expression correlated with enhanced Th17-response-promoting activity [97]. Given that IL-17A-producing γδ T cells promote early detrimental neutrophil infiltration after stroke [98], modulating their CD73 expression may attenuate post-stroke inflammation.
The ectoenzyme CD38 catalyzes, inter alia, the conversion of NAD+ to ADPR on the alternative pathway of adenosine formation and controls NAD+ bioavailability and activity of NAD-dependent enzymes in the brain as reviewed by Guerreiro et al. [99]. Besides, chemotaxis of myeloid cells in response to different chemoattractants depends on CD38 and its products ADPR and cyclic ADPR [100, 101]. Several studies demonstrated protective effects of CD38 deficiency in animal models of neurodegeneration and neuroinflammation [99]. Regarding ischemic stroke, CD38 knockout mice showed smaller infarcts after tMCAO [102] and reduced hippocampal neuronal cell death after transient forebrain ischemia due to occlusion of both common carotid arteries and concomitant reduction of mean arterial blood pressure [103]. CD38 knockout mice also showed a drastic reduction of macrophage recruitment to the ischemic hemisphere which probably contributed to the protective phenotype [102]. Beneficial effects of CD38 deletion in brain pathologies were additionally attributed to increased NAD+ levels itself [99, 104] as it has strong neuroprotective and anti-inflammatory properties [105]. NAD+ depletion, on the other hand, leads to mitochondrial dysfunction and oxidative stress and is a common finding in neurodegenerative disorders [99, 106]. CD38 expression and enzymatic activity were recently shown to increase with age in the rat brain [107] complementing previous findings on an age-dependent decrease in brain NAD+ levels in healthy humans [108]. Spontaneously hypertensive stroke-prone rats, the most frequently used animal model for cerebral small vessel disease leading to lacunar infarction and cognitive decline, also exhibited higher CD38 expression and activity in the brain than age-matched normotensive controls [107]. Taken together, there is growing indirect evidence that CD38 might link aging with neuroinflammation and neurodegeneration and thus remains an interesting target in stroke.
Role of adenosine receptors in cerebral ischemia
The concentration of extracellular adenosine increases substantially during in vivo ischemia. By microdialysis technique, Melani et al. measured a basal concentration of 130 nmol/l in the rat striatum which increased 10-fold within the first hour after intraluminal pMCAO and then decreased again but remained elevated until the end of the observation period 4 h after stroke induction [6, 109]. The half-life of extracellular adenosine is very short (4–10 s), as it is swiftly degraded by adenosine deaminase to its metabolite inosine. Therefore, to maintain an increased level of adenosine implies a continuous production. The increase of extracellular adenosine during hypoxia mainly relies on two mechanisms: Within the first 20 min of ischemia, adenosine derives mostly from hydrolysis of extracellular ATP involving CD73 leading to a higher extracellular than intracellular adenosine concentration [6]. Thereafter, extracellular adenosine originates mainly from parenchymal cells where it accumulates in a pO2-dependent manner [110]. Consequently, the gradient between extracellular and intracellular adenosine concentrations is reversed, and adenosine is released from the cells to the extracellular space [6, 111]. Under hypoxic conditions, mitochondria consume ATP, and accumulating AMP cannot be reconverted to ATP due to the lack of oxygen and glucose. Hence, ATP and ADP levels decrease below the threshold to inhibit cytosolic 5’-nucleotidase [112], and adenosine is excessively formed from AMP. Intracellular adenosine concentrations further rise due to the hypoxia-induced inhibition of adenosine kinase which removes adenosine by phosphorylation into AMP and represents another key enzyme for the regulation of ambient adenosine levels [113,114,115]. Given that all these cellular and enzymatic processes determine adenosine concentration, caution should be taken in extrapolating tissue adenosine levels from measurements of CD73 mRNA or protein expression only [111].
Adenosine release during ischemia is considered an endogenous neuroprotective response as adenosine infusion through a microdialysis probe into the rat striatum reduced neurological deficits after tMCAO [116]. Pre- and post-ischemic (2 h) i.v. delivery of adenosine-containing nanoparticles allowing prolonged circulation of the nucleoside also resulted in a significant improvement of infarct volume and neurologic deficit score after transient and permanent intraluminal MCAO [117]. Administration of an adenosine kinase inhibitor likewise attenuated ischemic damage in rats after tMCAO [118], whereas transgenic overexpression of adenosine kinase in the murine brain leads to increased infarct volumes after tMCAO [119]. Given that targeted delivery of adenosine to the CNS is hampered by a very short half-life in plasma, low blood-brain barrier permeability [120], and major systemic side effects such as bradycardia via cardiac A1R [121], therapeutic approaches focused on the modulation of adenosine receptor signaling. The A1, A2A, A2B, and A3 receptors are the four known AR subtypes, which all belong to the superfamily of G-protein-coupled receptors and differ regarding their affinity for adenosine and tissue distribution. Apart from inhibition or stimulation of adenylate cyclase, signaling occurs through phospholipase C (PLC), calcium, and mitogen-activated protein kinases (MAPK) [122] (Fig. 3). At the concentration range during physiologic conditions, adenosine can stimulate the high affinity adenosine receptors A1 and A2A, but during ischemia, adenosine reaches concentrations in the low micromolar range sufficient to also activate low affinity A2B and A3R [123]. A1R shows an overall higher expression in mice than in humans with a quite even distribution across neurons in cortex, hippocampus, and cerebellum, oligodendrocytes, and astrocytes. At the cellular level, A2AR expression is highest on endothelial cells followed by astrocytes in both species, and at the spatial level, A2AR density is highest at post-synaptic neurons in the striatum. In the striatum, A2ARs mainly signal through activation of stimulatory Golf [124]. While astrocytes followed by murine OPCs show the highest expression of A2BR in the CNS, A3R expression at the cellular level is highest on microglia in both mice and humans and at the spatial level most dense on hippocampal neurons [10,11,12,13, 125] (Fig. 1). The main approach in the development of AR agonists has been the chemical modification of adenosine itself, whereas AR antagonists are mainly derived from xanthines such as caffeine and theophylline [122]. A review by Pedata et al. comprehensively summarized our current knowledge on the role of all four adenosine receptors in brain ischemia both in vitro and in vivo [126]. In the present review, we will therefore only give a condensed overview with focus on data from in vivo stroke studies. STab. 2 provides detailed information for all the cited studies using pharmacological modulation of AR signaling with focus on common quantitative outcome measures of translational relevance, e.g., infarct volume and neurological outcome.

Overview on the signaling pathways and affinity of the four known subtypes of adenosine receptors (ARs), stimulatory (+) and inhibitory (–) compounds used in the cited studies on OGD or in vivo ischemic stroke and their role in brain ischemia. For details, see Supplementary Table 2. Images from BioRender.com
A1R signaling in response to ischemia exerts protective effects by inhibiting excitatory synaptic transmission [127,128,129], in particular glutamate, which is crucial for the functional recovery of hippocampal circuits upon reoxygenation [130,131,132]. Acute systemic administration of different A1R agonists (CHA, CPA, ADAC) was protective in a model of global cerebral ischemia in gerbils [133,134,135], whereas acute treatment with the antagonist DPCPX exacerbated ischemic brain injury [134]. Of note, the effects of chronic administration, i.e., pre-treatment for several days before the induction of stroke, are opposite [134]. It has been suggested that ischemic protection by chronic administration of adenosine antagonists may be a consequence of A1R upregulation and that detrimental effects of chronic A1R agonism are due to the phenomenon of A1R desensitization [136] which has been described for rat hippocampal slices upon hypoxia [137]. A1R signaling also plays a central role in mediating the beneficial effects of ischemic preconditioning in the brain [138, 139], a phenomenon in which the previous exposure to a short period of ischemia confers resistance to a following prolonged ischemic insult. To overcome the limitations of full A1R agonists for clinical application, mainly receptor desensitization and cardiovascular side effects such as bradycardia and hypotension, the neuroprotective potential of partial agonists (2′-dCCPA, 3′-dCCPA) was recently investigated and proven in oxygen-glucose deprivation (OGD) as the most frequently used in vitro model of cerebral ischemia [140].
Protective effects of A2AR antagonism (CGS 15943) on ischemic injury were first demonstrated in 1994 by Gao and Phillis after global forebrain ischemia in gerbils [141]. In the following years, many studies using different models of rodent experimental stroke (global forebrain ischemia, four-vessel occlusion, intraluminal pMCAO, pMCAO by electrocoagulation) confirmed the beneficial role of various A2AR antagonists—namely, CSC, CP 66,713, ZM 241385, SCH 58261—when being administered before ischemia [142, 143] or acutely after stroke [144,145,146]. The latter three studies on SCH 58261 evaluated infarct volume at 24 h after stroke. Later, Melani et al. demonstrated that ischemic protection by SCH 58261 was limited to outcome parameters on day 1, whereas prolonged treatment for 7 days did not confer sustained reduction in infarct volume [147]. In line with the findings on drug-mediated antagonism, mice genetically deficient for A2AR exhibited smaller infarcts 2 h, 24 h, and 48 h after tMCAO [148, 149] and 24 h after embolic MCAO [150]. Interestingly, RNA-sequencing analysis of the ipsilesional and contralesional primary motor cortices (iM1 and cM1) 15 days after tMCAO revealed that downregulation of Adora2a encoding A2AR in cM1 correlated with improved recovery [151]. Detrimental effects of A2AR signaling are largely attributed to an increase of extracellular glutamate concentrations leading to excitotoxicity, thus counteracting the depression of excitatory transmission mediated by A1R [126]. Several mechanisms contribute to glutamatergic excitatory transmission upon A2AR stimulation: First, it increases neuronal glutamate release during ischemia in vitro [152, 153], and genetic A2AR deficiency was shown to reduce striatal glutamate outflow measured by microdialysis during tMCAO and reperfusion [148]. Second, A2AR activation on astrocytes inhibits glutamate uptake [154,155,156,157]. Third, A1 and A2AR can form heteromers on striatal glutamatergic nerve terminals in which A2AR activation reduces the affinity of the A1R for agonists shifting the balance towards A2AR effects in an adenosine-rich milieu such as ischemia [158].
Beyond the control of glutamatergic excitotoxicity, A2AR antagonists exert their protective effects through inhibition of phospho-p38 MAPK [145] and by reducing JNK MAPK activation in oligodendrocytes [159]. Moreover, A2AR activation was shown to drive microglia process retraction into an amoeboid shape upon activation which represents a hallmark of neuroinflammation and modulates their phagocytotic capacities [160]. A2AR signaling on brain endothelial cells contributes to ischemic brain injury as endothelium-specific A2AR-deficient mice also showed reduced infarct volumes 24 h after stroke [150].
When given systemically and chronically 13 days before global brain ischemia or until day 7 after tMCAO, A2AR agonists (APEC, CGS 21680) also conferred protection [161, 162]. These findings seem paradoxically but rely on different mechanisms than protection by antagonists. While central effects like regulation of cerebral blood flow [161] and control of neurotrophic factors [163] might account in part for this phenomenon, protection is mainly attributed to immunomodulatory effects of A2AR signaling. The substantial contribution of sterile inflammation to secondary neuronal injury is nowadays widely accepted and summarized elsewhere [2, 164, 165]. A2AR is abundantly expressed on innate and adaptive immune cells, and its activation serves as a strong anti-inflammatory signal [166, 167]. A2ARs even increase in density and affinity on lymphocytes from ischemic stroke patients indicating the translational relevance of this pathway [168]. It has to be noted that the A2AR agonist CGS 21680 only conferred protection when being chronically administered, whereas delivery limited to the first 24 h after stroke did not reduce infarct volume [169] as it probably missed the peak of immune cell infiltration at day 3 after tMCAO [170].
Taken together, research over the last decades demonstrated a dual role of A2AR signaling in brain ischemia depending on the timepoint and the leading mechanism of injury: A2AR antagonists reduce ischemic injury early after stroke by reducing excitotoxicity, whereas A2AR agonists confer protection when being administered for several days post-ischemia by attenuating secondary sterile inflammation. From a translational perspective, therapeutic strategies with antagonists or agonists at the A2AR should therefore be carefully evaluated in terms of timing.
Compared to A2AR signaling, far less literature is available on the role of A2BR signaling in ischemic stroke. Though ubiquitously expressed [171], the low affinity of A2BR for its endogenous ligand adenosine and the lack of potent and selective agonists and antagonists has hampered investigations into its specific function in health and disease for a long time. Moreover, the role of A2BR is often masked by co-expression of A2AR, which especially holds true for monocytes and macrophages, where A2AR remains the predominant adenosine receptor. Due to recent innovations regarding pharmacological tools and molecular approaches, A2BRs are increasingly recognized as orchestrators of immunity and inflammation as reviewed by Haskó and colleagues [172]. In the immune cell compartment, A2BR activation increases pro-inflammatory cytokine production by mast cells, whereas it shapes the function and cytokine profile of antigen-presenting cells such as DCs and macrophages towards an anti-inflammatory and tolerance-inducing phenotype [172]. A2BR knockout mice displayed a mild but significant increase in pro-inflammatory cytokines under basal conditions [171].
On endothelial cells, A2BR signaling plays a central role in controlling barrier function. The protective effect is even potentiated under hypoxic conditions because of adenosine accumulation and A2BR upregulation [173, 174]. Consequently, A2BR knockout mice exhibited increased vascular leakage measured by Evans’s blue extravasation in multiple organs including the brain during hypoxia, whereas A1R, A2AR, and A3R deficiency did not accentuate hypoxia-induced vascular leakage compared to wild-type littermate controls [175]. A2BR knockout mice expressed higher levels of adhesion molecules at the peripheral vasculature resulting in higher numbers of leukocytes adhering at the endothelium under basal conditions [171] and increased infiltration of polymorphonuclear leukocytes in various organs including the brain after hypoxia [175]. Bone marrow transplantation experiments demonstrated that A2BR signaling on hematopoietic cells mainly accounted for A2BR-mediated attenuation of hypoxia-associated neutrophil transmigration, while vascular A2BR conferred protection to fluid leakage [171, 175].
A recent in vivo study in rats demonstrated that systemic and chronic treatment with the A2BR agonist BAY60-6583 after tMCAO diminished immune cell infiltration at day 2 and reduced infarct volume at day 7 after stroke [176]. These protective effects of A2BR signaling are contrasted by a study indicating a rather detrimental role of central A2BR signaling: In transient global brain ischemia induced by four-vessel occlusion in rats, the selective A2BR antagonist MRS1754 reduced the phosphorylation of p38 MAPK and subsequent activation of neutral sphingomyelinase 2 which otherwise leads to pro-inflammatory ceramide accumulation in hippocampal astrocytes [177]. Taken together, central A2BR signaling on astrocytes seems to exert a pro-inflammatory role, while A2BR activation on immune cells and endothelial cells dampens inflammation and vascular leakage.
Several in vivo stroke studies demonstrated a protective effect of A3R agonists: Pre-treatment with the A3R agonist CI-IB-MECA either once i.c.v. 15 min or twice i.v. 165 min and 15 min before transient MCA ligation reduced infarct volume at day 2 in rats and mice [178]. In the same study, A3R knockout mice exhibited larger infarcts after tMCAO than WT controls [178]. With higher relevance from a translational perspective, post-treatment with agonists also proved effective: Administration of the A3R agonist LJ529 twice within the first seven hours after tMCAO reduced infarct volume at day 1 in rats. This effect was only reversed by application of the A3R antagonist MRS1523 but not by co-administration of an A2AR antagonist (SCH 58261) indicating that ischemic protection was indeed mediated by A3R [179]. Recently, delivery of the A1/A3R agonist AST-004 via bolus and continuous infusion starting 2 h after tMCAO limited the lesion growth rate measured by repeated magnetic resonance imaging (MRI) scans and overall stroke volume at day 5 in macaques as an example of non-human primates [180]. In vitro data from various OGD experiments seemed to be contradictory regarding the effects of A3R signaling on synaptic transmission and excitotoxicity during ischemia with both agonists and antagonists exerting ischemic protection under different experimental conditions [126]. In 2007, Pugliese and colleagues deciphered the divergent contribution of A3R signaling depending on the severity of OGD in rat hippocampal slices: While A3R agonists had an acute A1R-like inhibitory effect on synaptic transmission during a short period of OGD (2–5 min), both antagonists (LJ1251, MRS1523) and prolonged exposure to agonists (CI-IB-MECA, VT72, VT158, VT160, VT163, AR132) prevented or delayed anoxic depolarization and were protective during severe OGD (7 min) [181]. It is assumed that severe ischemia switches A3R-mediated effects from beneficial to detrimental via sustained activation of PLC and protein kinase C through Gq proteins and subsequent calcium mobilization. The yet protective effects of chronic A3R agonist treatment are a consequence of receptor desensitization in an per se adenosine-rich microenvironment [181], which has been shown for both rat and human A3R [182, 183].
This noxious role of A3R signaling in conditions of severe ischemia explains early findings by von Lubitz and colleagues, who showed that acute treatment with the A3R agonist IB-MECA right before global forebrain ischemia increased mortality until day 7 after ischemia in gerbils, whereas chronic pre-treatment over 10 days, probably leading to A3R desensitization, enhanced neuronal survival in the hippocampus [184].
Beyond receptor desensitization, the protective effects of A3R agonists are also mediated by immune cells as demonstrated by Choi et al. [179]. The A3R agonist LJ529 when given twice at 2 h and 7 h after tMCAO reduced the migration of activated microglia and infiltration of monocytes into ischemic cortical and striatal lesions. In a microglial cell culture, LJ529 inhibited chemotaxis of microglia and monocytes, and co-treatment with A3 and A2AR antagonists indicated that this effect was selectively mediated by A3R [179]. A3R was found to be overexpressed in inflammatory tissue and on peripheral blood mononuclear cells from patients with different autoimmune inflammatory diseases such as rheumatoid arthritis, psoriasis, and Crohn’s disease [185, 186]. Various A3R agonists exerted anti-inflammatory effects across different experimental models of inflammation [187] which is mediated by de-regulation of the NF-kB signaling pathway inhibiting cytokine production and enhancing apoptosis of inflammatory cells [186]. The highly selective A3R agonist CF101 even proved effective and safe in a phase II clinical trial in rheumatoid arthritis [188]. If the evidence for a protective role of A3R agonists in ischemic stroke mounts, CF101 might indeed represent an interesting therapeutic perspective.
Taken together, A3R signaling takes on a noxious role in conditions of severe ischemia such as long OGD and in vivo stroke, and the yet protective effects of chronic A3R agonist treatment are mainly attributed to receptor desensitization.
Differences between murine and human purinergic signaling pathways
Mice and humans differ in their expression and regulation of P2 receptors, particularly with respect to P2X7. P2X7 is expressed at lower levels in humans (at least on circulating T cells) than in mice, as shown, for example, for Tregs, which have a very high P2X7 expression in mice, but low expression in humans [189, 190]. Compared to rodent P2X7, human P2X7 is around ten times less sensitive to ATP [58] and requires ATP concentrations in the high micromolar to low millimolar range for activation [191]. Additionally, only murine P2X7 is activated by low NAD+ concentrations through ARTC2.2-mediated ADP ribosylation, as there is no human orthologue of ARTC2.2 [192, 193]. P2Y11, an ATP-gated G-protein-coupled receptor, is expressed in humans, but not in mice. P2Y11 interacts with P2X7 and inhibits P2X7-mediated pore formation and cell death [194]. Overall, the hurdle to activate P2X7 appears to be significantly higher in humans than in mice.
Major differences between mice and humans also exist in the ectonucleotidases degrading ATP to adenosine. As mentioned earlier, CD73 is highly expressed on human, but not on murine brain endothelial cells [76, 77, 92]. This could strongly influence brain adenosine concentrations at the site of ischemic stroke. Unlike hypoxia-induced upregulation of CD73 on human microvascular endothelial cells in vitro [173], our group did not detect an upregulation of CD73 on murine cerebral microvessels after ischemic stroke [90]. The expression of CD73 on infiltrating immune cells also differs between the two species. Murine Tregs express both CD39 and CD73 on the cell surface, allowing the generation of adenosine from ATP by the concerted action of both ectonucleotidases [94]. In contrast, human Tregs exhibit very low CD73 expression [195]. Similarly, almost all conventional T cells in mice express CD73, whereas human CD4 and CD8 cells (except for naïve CD8 T cells and a subset of memory cells) have low CD73 expression [195, 196]. A reason for the differential expression of CD73 in mice and humans may be that adenosine receptor signaling has different effects in the two species. In contrast to mice, where adenosine promotes Treg expansion, adenosine signaling in humans inhibits the activation of Tregs [95, 197,198,199]. The absence of CD73 on Tregs could prevent adenosine-mediated auto-inhibition. Adenosine is degraded by ADA, which in humans, but not in mice, is “anchored” to the plasma membrane by the ectonucleotidase CD26. ADA in close proximity to the cell surface could reduce the autocrine effect of adenosine in humans.
Due to these differences in purinergic signaling between mice and humans, results from mouse models need to be carefully evaluated and are unlikely to be directly applicable to humans.
Concluding remark
A large number of preclinical studies clearly demonstrated the involvement of the ATP-adenosine axis in stroke pathophysiology reaching from excitotoxicity over post-stroke inflammation to recovery. From today’s perspective, the translation of drugs targeting key players of purinergic signaling from experimental stroke research into clinical routine is mainly hampered by (i) the BBB that constitutes a major obstacle for the i.v. application of antibody-based therapeutic constructs, (ii) a potentially dual role of certain targets depending on the timepoint and leading mechanism of injury (e.g., A2AR), (iii) major cardiovascular side effects (e.g., A1R agonists), (iv) gene expression differences between mice and humans (e.g., CD73), and (v) futility of pre-treatment strategies in human stroke. Especially when aiming at reducing post-stroke inflammation, e.g., through P2X7 blockade, the rapid onset of P2X7 signaling after stroke will probably leave a narrow time window for treatment. To our opinion, the engineering of P2X7-blocking nanobodies with improved ability to cross the BBB after i.v. injection could be a huge step towards translation. Further, the application of nanobodies via the intranasal route could be a promising therapeutic approach. A high dose of P2X7-blocking nanobodies could be given as nasal spray at the first signs of stroke. The successful distribution of nanobodies via the nasal route targeting, e.g., transthyretin in the CNS has already been demonstrated in mice [200]. Apart from targeting P2X7, the use of partial AR agonists offers the greatest therapeutic potential. Nevertheless, any therapeutic approach must be carefully evaluated in terms of timing, efficiency irrespective of age and gender, specificity, and effects on long-term outcome.
Abbreviations
- AAV:
-
Adeno-associated virus
- ADA:
-
Adenosine deaminase
- ADAC:
-
Adenosine amine congener
- ADP:
-
Adenosine diphosphate
- ADPR:
-
ADP ribose
- AMP:
-
Adenosine monophosphate
- APEC:
-
2-[(2-Aminoethylamino)-carbonylethylphenylethylamino]-5′-N-ethylcarboxoamidoadenosine
- AR:
-
Adenosine receptor
- AR132:
-
N6-Methyl-2-phenylethynyladenosine
- ATP:
-
Adenosine triphosphate
- BAY60-6583:
-
2-[[6-Amino-3,5-dicyano-4-[4-(cyclopropylmethoxy) phenyl]-2-pyridinyl] thio]-acetamide
- BBB:
-
Blood-brain-barrier
- BBG:
-
Brilliant Blue G
- 5-BDBD:
-
5-(3-Bromophenyl)-1,3-dihydro-2H-benzofuro[3,2-e]-1,4-diazepin-2-one
- BzATP:
-
2'-3'-O-(4-benzoylbenzoyl) adenosine 5'-triphosphate
- cAMP:
-
Cyclic AMP
- CGS 15943:
-
9-Chloro-2-(2-furanyl)-[1,2,4]triazolo[1,5-c]quinazolin-5-amine
- CGS 21680:
-
2-p-(2-Carboxyethyl)-phenethylamino-5′-N-ethylcarboxamidoadenosine
- CHA:
-
Cyclohexyladenosine
- CI-IB-MECA:
-
Chloro-N6-(3-iodo-benzyl)-adenosine-5′-N-methyluronamide
- cM1:
-
Contralesional primary motor cortex
- CNS:
-
Central nervous system
- CPA:
-
N6-cyclopentyladenosine
- CP 66,713:
-
4-Amino-l-phenyl[1,2,4]-triazolo[4,3-a] quinoxaline
- CSC:
-
8-(3-Chlorostyryl) caffeine
- DAMPs:
-
Danger-/damage-associated molecular patterns
- DCs :
-
Dendritic cells
- 2′-dCCPA:
-
2-Chloro-2′-deoxy-N6-cyclopentyladenosine
- 3′-dCCPA :
-
2-Chloro-3′-deoxy-N6-cyclopentyladenosine
- DPCPX:
-
1,3-Dipropyl-8-cyclopentylxanthine
- EAE:
-
Experimental autoimmune encephalomyelitis
- ENPP1/CD203a:
-
Ectonucleotide pyrophosphatase/phosphodiesterase 1
- GSK:
-
GlaxoSmithKline
- h:
-
Hours
- i.c.v.:
-
Intracerebroventricular
- IL-2/IL-2Ab:
-
Interleukin-2/interleukin-2 antibody
- iM1:
-
Ispilesional primary motor cortex
- i.p.:
-
Intraperitoneal
- irAE:
-
Immune-related adverse event
- i.v.:
-
Intravenous
- LJ529:
-
2-Chloro-N6-(3-iodobenzyl)-5′-N-methylcarbamoyl-4′-thioadenosine
- LJ1251:
-
(2R,3R,4S)-2-(2-chloro-6-(3-iodobenzylamino)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol
- MAPK:
-
Mitogen-activated protein kinase
- K 1/2 :
-
Half-maximal response
- min:
-
Minutes
- MRI:
-
Magnetic resonance imaging
- MRS1523:
-
3-Propyl-6-ethyl-5-[(ethylthio)carbonyl]-2-phenyl-4-propyl-3-pyridine carboxylate
- MRS1754:
-
8-[4-[((4-Cyanophenyl)carbamoylmethyl)oxy]phenyl]-1,3-di(n-propyl) xanthine hydrate
- MRS2500:
-
(1R*,2S*)-4-[2-iodo-6-(methylamino)-9H-purin-9-yl]-2-(phosphono-oxy)bicycle[3.1.0] hexane-1-methanol dihydrogen phosphate ester
- MRS2578:
-
1,4-Di[3-(3-isothiocyanatophenyl)thioureido]butane
- MRS4719:
-
5-(2-(5-Thioxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)pyridin-4-yl)-1,5-dihydro-2H-naphtho[1,2-b][1,4]diazepine-2,4(3H)-dione triethylamine salt
- NAD+ :
-
Nicotinamide adenine dinucleotide
- NF449:
-
4,4',4",4"'-(Carbonylbis (imino-5,1,3-benzenetriylbis (carbonylimino)))tetrakis-benzene-1,3-disulfonic acid
- NK cells:
-
Natural killer cells
- NTPDase1/CD39:
-
Ecto-nucleoside triphosphate diphosphohydrolase 1
- NT5E/CD73:
-
Ecto-5’-nucleotidase
- OGD:
-
Oxygen-glucose deprivation
- PLC:
-
Phospholipase C
- pMCAO:
-
Permanent middle cerebral artery occlusion
- pmeLUC:
-
Plasma membrane luciferase
- PPADS:
-
Pyridoxalphosphate-6-azophenyl-2′,4′-disulphonic acid
- SCH 58261:
-
7-(2-Phenylethyl)-5-amino-2-(2-furyl)-pyrazolo-[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine
- tMCAO:
-
Transient middle cerebral artery occlusion
- TNAP:
-
Tissue non-specific alkaline phosphatase
- Tregs:
-
Regulatory T cells
- WT:
-
Wild type
- VSMCs:
-
Vascular smooth muscle cells
- VT72:
-
N6-Methoxy-2-phenylethynyl
- VT158:
-
N6-Methoxy-2-phenylethynyl
- VT160:
-
N6-Methoxy-2-(2-pyridinyl)-ethynyl
- VT163:
-
N6-Methoxy-2-p-acetylphenylethynyl
- ZM 241385:
-
4-(-2-[7-Amino-2-{2-furyl}{1,2,4}triazolo{2,3-a}{1,3,5}triazin-5-yl-amino]ethyl)phenol
References
Feigin VL, Stark BA, Johnson CO et al (2021) Global, regional, and national burden of stroke and its risk factors, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol 20:795–820. https://doi.org/10.1016/S1474-4422(21)00252-0
Anrather J, Iadecola C (2016) Inflammation and stroke: an overview. Neurotherapeutics 13:661–670. https://doi.org/10.1007/s13311-016-0483-x
Burnstock G (1972) Purinergic Nerves. Pharmacol Rev 24:509–581
Giuliani AL, Sarti AC, Di Virgilio F (2021) Ectonucleotidases in acute and chronic inflammation. Front Pharmacol 11:1–20. https://doi.org/10.3389/fphar.2020.619458
Melani A, Turchi D, Giuliana M et al (2005) ATP extracellular concentrations are increased in the rat striatum during in vivo ischemia. Neurochem Int 47:442–448. https://doi.org/10.1016/j.neuint.2005.05.014
Melani A, Corti F, Stephan H et al (2012) Ecto-ATPase inhibition: ATP and adenosine release under physiological and ischemic in vivo conditions in the rat striatum. Exp Neurol 233:193–204. https://doi.org/10.1016/j.expneurol.2011.09.036
Pellegatti P, Falzoni S, Pinton P et al (2005) A novel recombinant plasma membrane-targeted luciferase reveals a new pathway for ATP secretion. Mol Biol Cell 16:3659–3665. https://doi.org/10.1091/mbc.e05-03-0222
Wilmes M, Espinoza CP, Ludewig P et al (2022) Blocking P2X7 by intracerebroventricular injection of P2X7-specific nanobodies reduces stroke lesions. J Neuroinflammation 19:256. https://doi.org/10.1186/s12974-022-02601-z
Burnstock G (2016) P2X ion channel receptors and inflammation. Purinergic Signal 12:59–67. https://doi.org/10.1007/s11302-015-9493-0
Hodge RD, Bakken TE, Miller JA et al (2019) Conserved cell types with divergent features in human versus mouse cortex. Nature 573:61–68. https://doi.org/10.1038/s41586-019-1506-7
Lein ES, Hawrylycz MJ, Ao N et al (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445:168–176. https://doi.org/10.1038/nature05453
Zhang Y, Chen K, Sloan SA et al (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. https://doi.org/10.1523/JNEUROSCI.1860-14.2014
Zhang Y, Sloan SA, Clarke LE et al (2016) Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89:37–53. https://doi.org/10.1016/j.neuron.2015.11.013
Karunarathne W, Ku CJ, Spence DM (2009) The dual nature of extracellular ATP as a concentration-dependent platelet P2X1 agonist and antagonist. Integr Biol 1:655–663. https://doi.org/10.1039/b909873a
Lecut C, Frederix K, Johnson DM et al (2009) P2X 1 ion channels promote neutrophil chemotaxis through rho kinase activation. J Immunol 183:2801–2809. https://doi.org/10.4049/jimmunol.0804007
Darbousset R, Delierneux C, Mezouar S et al (2014) P2X1 expressed on polymorphonuclear neutrophils and platelets is required for thrombosis in mice. Blood 124:2575–2585. https://doi.org/10.1182/blood-2014-04-571679
Rawish E, Langer HF (2022) Platelets and the role of P2X receptors in nociception, pain, neuronal toxicity and thromboinflammation. Int J Mol Sci 23:6585. https://doi.org/10.3390/ijms23126585
Harhun MI, Povstyan OV, Albert AP, Nichols CM (2014) ATP-evoked sustained vasoconstrictions mediated by heteromeric P2X1/4 receptors in cerebral arteries. Stroke 45:2444–2450. https://doi.org/10.1161/STROKEAHA.114.005544
Cheng RD, Ren JJ, Zhang YY, Ye XM (2014) P2X4 receptors expressed on microglial cells in post-ischemic inflammation of brain ischemic injury. Neurochem Int 67:9–13. https://doi.org/10.1016/j.neuint.2014.01.011
Rivera A, Vanzulli I, Butt AM (2016) A central role for ATP signalling in glial interactions in the CNS. Curr Drug Targets 17:1829–1833. https://doi.org/10.2174/1389450117666160711154529
Brock VJ, Wolf IMA, Er-Lukowiak M et al (2022) P2X4 and P2X7 are essential players in basal T cell activity and Ca2+ signaling milliseconds after T cell activation. Sci Adv 8:eabl9770. https://doi.org/10.1126/sciadv.abl9770
Ledderose C, Liu K, Kondo Y et al (2018) Purinergic P2X4 receptors and mitochondrial ATP production regulate T cell migration. J Clin Invest 128:3583–3594. https://doi.org/10.1172/JCI120972
Verma R, Cronin CG, Hudobenko J et al (2017) Deletion of the P2X4 receptor is neuroprotective acutely, but induces a depressive phenotype during recovery from ischemic stroke. Brain Behav Immun 66:302–312. https://doi.org/10.1016/j.bbi.2017.07.155
Srivastava P, Cronin CG, Scranton VL et al (2020) Neuroprotective and neuro-rehabilitative effects of acute purinergic receptor P2X4 (P2X4R) blockade after ischemic stroke. Exp Neurol 329:113308. https://doi.org/10.1016/j.expneurol.2020.113308
Wu ST, Han JR, Yao N et al (2022) Activation of P2X4 receptor exacerbates acute brain injury after intracerebral hemorrhage. CNS Neurosci Ther 28:1008–1018. https://doi.org/10.1111/cns.13831
Xiang Z, Jiang X, Ji R, Yuan H (2021) Enhanced expression of P2X4 purinoceptors in pyramidal neurons of the rat hippocampal CA1 region may be involved ischemia-reperfusion injury. Purinergic Signal 17:425–438. https://doi.org/10.1007/s11302-021-09780-z
Toti KS, Verma R, McGonnigle MJ et al (2022) Structure–activity relationship and neuroprotective activity of 1,5-dihydro-2 h -naphtho[1,2- b ][1,4]diazepine-2,4(3 H )-diones as P2X4 receptor antagonists. J Med Chem 65:13967–13987. https://doi.org/10.1021/acs.jmedchem.2c01197
Williams WA, Linley JE, Jones CA et al (2019) Antibodies binding the head domain of P2X4 inhibit channel function and reverse neuropathic pain. Pain 160:1989–2003. https://doi.org/10.1097/j.pain.0000000000001587
Bergmann P, Garcia de Paco E, Rissiek B et al (2019) Generation and characterization of specific monoclonal antibodies and nanobodies directed against the ATP-gated channel P2X4. Front Cell Neurosci 13:498. https://doi.org/10.3389/fncel.2019.00498
Ozaki T, Muramatsu R, Sasai M et al (2016) The P2X4 receptor is required for neuroprotection via ischemic preconditioning. Sci Rep 6:25893. https://doi.org/10.1038/srep25893
Neves V, Aires-da-silva F, Corte-real S, Castanho MARB (2016) Antibody approaches to treat brain diseases. Trends Biotechnol 34:36–48. https://doi.org/10.1016/j.tibtech.2015.10.005
Murray KN, Parry-Jones AR, Allan SM (2015) Interleukin-1 and acute brain injury. Front Cell Neurosci 9:18. https://doi.org/10.3389/fncel.2015.00018
Schädlich IS, Vienhues JH, Jander A et al (2022) Interleukin-1 mediates ischemic brain injury via induction of IL-17A in γδ T cells and CXCL1 in astrocytes. NeuroMolecular Med. 24:437–451. https://doi.org/10.1007/s12017-022-08709-y
Stroemer RP, Rothwell NJ (1998) Exacerbation of ischemic brain damage by localized striatal injection of interleukin-1β in the rat. J Cereb Blood Flow Metab 18:833–839. https://doi.org/10.1097/00004647-199808000-00003
Le Feuvre RA, Brough D, Rothwell N (2002) Extracellular ATP and P2X7 receptors in neurodegeneration. Eur J Pharmacol 447:261–269. https://doi.org/10.1016/s0014-2999(02)01848-4
Le Feuvre RA, Brough D, Touzani O, Rothwell NJ (2003) Role of P2X7 receptors in ischemic and excitotoxic brain injury in vivo. J Cereb Blood Flow Metab 23:381–384. https://doi.org/10.1097/01.WCB.0000048519.34839.97
Melani A, Amadio S, Gianfriddo M et al (2006) P2X7 receptor modulation on microglial cells and reduction of brain infarct caused by middle cerebral artery occlusion in rat. J Cereb Blood Flow Metab 26:974–982. https://doi.org/10.1038/sj.jcbfm.9600250
Franke H, Al G, Grosche J et al (2004) P2X7 receptor expression after ischemia in the cerebral cortex of rats. J Neuropathol Epxperimental Neurol 63:686–699. https://doi.org/10.1093/jnen/63.7.686
Yanagisawa D, Kitamura Y, Takata K et al (2008) Possible involvement of P2X7 receptor activation in microglial neuroprotection against focal cerebral ischemia in rats. Biol Pharm Bull 31:1121–1130. https://doi.org/10.1248/bpb.31.1121
Arbeloa J, Pérez-Samartín A, Gottlieb M, Matute C (2012) P2X7 receptor blockade prevents ATP excitotoxicity in neurons and reduces brain damage after ischemia. Neurobiol Dis 45:954–961. https://doi.org/10.1016/j.nbd.2011.12.014
Cisneros-Mejorado A, Gottlieb M, Cavaliere F et al (2015) Blockade of P2X7 receptors or pannexin-1 channels similarly attenuates postischemic damage. J Cereb Blood Flow Metab 35:843–850. https://doi.org/10.1038/jcbfm.2014.262
Caglayan B, Caglayan AB, Beker MC et al (2017) Evidence that activation of P2X7R does not exacerbate neuronal death after optic nerve transection and focal cerebral ischemia in mice. Exp Neurol 296:23–31. https://doi.org/10.1016/j.expneurol.2017.06.024
Ye X, Shen T, Hu J et al (2017) Purinergic 2X7 receptor / NLRP3 pathway triggers neuronal apoptosis after ischemic stroke in the mouse. Exp Neurol 292:46–55. https://doi.org/10.1016/j.expneurol.2017.03.002
Kang SS, Keasey MP, Hagg T (2013) P2X7 receptor inhibition increases CNTF in the subventricular zone , but not neurogenesis or neuroprotection after stroke in adult mice. Transl Stroke Res 4:533–545. https://doi.org/10.1007/s12975-013-0265-2
Kaiser M, Penk A, Franke H et al (2016) Lack of functional P2X7 receptor aggravates brain edema development after middle cerebral artery occlusion. Purinergic Signal 12:453–463. https://doi.org/10.1007/s11302-016-9511-x
Kaczmarek-Hajek K, Zhang J, Kopp R et al (2018) Re-evaluation of neuronal P2X7 expression using novel mouse models and a P2X7-specific nanobody. Elife 7:e36217. https://doi.org/10.7554/elife.36217
Danquah W, Meyer-Schwesinger C, Rissiek B et al (2016) Nanobodies that block gating of the P2X7 ion channel ameliorate inflammation. Sci Transl Med 8:366ra162. https://doi.org/10.1126/scitranslmed.aaf8463
Pinto-Espinoza C, Guillou C, Rissiek B et al (2022) Effective targeting of microglial P2X7 following intracerebroventricular delivery of nanobodies and nanobody-encoding AAVs. Front Pharmacol 13:1029236. https://doi.org/10.3389/fphar.2022.1029236
Wesolowski J, Alzogaray V, Reyelt J et al (2009) Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med Microbiol Immunol 198:157–174. https://doi.org/10.1007/s00430-009-0116-7
Muyldermans S (2021) A guide to: generation and design of nanobodies. FEBS J 288:2084–2102. https://doi.org/10.1111/febs.15515
Savio LEB, de Andrade Mello P, da Silva CG, Coutinho-Silva R (2018) The P2X7 receptor in inflammatory diseases: angel or demon? Front Pharmacol 9:52. https://doi.org/10.3389/fphar.2018.00052
Taylor SRJ, Gonzalez-Begne M, Sojka DK et al (2009) Lymphocytes from P2X7-deficient mice exhibit enhanced P2X7 responses. J Leukoc Biol 85:978–986. https://doi.org/10.1189/jlb.0408251
Sharp AJ, Polak PE, Simonini V et al (2008) P2x7 deficiency suppresses development of experimental autoimmune encephalomyelitis. J Neuroinflammation 5:33. https://doi.org/10.1186/1742-2094-5-33
Chen L, Brosnan CF (2006) Exacerbation of experimental autoimmune encephalomyelitis in P2X7R−/− mice: evidence for loss of apoptotic activity in lymphocytes. J Immunol 176:3115–3126. https://doi.org/10.4049/jimmunol.176.5.3115
Sim JA, Chaumont S, Jo J et al (2006) Altered hippocampal synaptic potentiation in P2X4 knock-out mice. J Neurosci 26:9006–9009. https://doi.org/10.1523/JNEUROSCI.2370-06.2006
Er-Lukowiak M, Duan Y, Rassendren F et al (2020) A P2rx7 passenger mutation affects the vitality and function of T cells in congenic mice. iScience 23:101870. https://doi.org/10.1016/j.isci.2020.101870
Solle M, Labasi J, Perregaux DG et al (2001) Altered cytokine production in mice lacking P2X7 receptors. J Biol Chem 276:125–132. https://doi.org/10.1074/jbc.M006781200
Xing S, Grol MW, Grutter PH et al (2016) Modeling interactions among individual P2 receptors to explain complex response patterns over a wide range of ATP concentrations. Front Physiol 7:294. https://doi.org/10.3389/fphys.2016.00294
Yamauchi K, Imai T, Shimazawa M et al (2017) Effects of ticagrelor in a mouse model of ischemic strokearticle. Sci Rep 7:12088. https://doi.org/10.1038/s41598-017-12205-w
Gao Y, Yu C, Pi S et al (2019) The role of P2Y 12 receptor in ischemic stroke of atherosclerotic origin. Cell Mol Life Sci 76:341–354. https://doi.org/10.1007/s00018-018-2937-2
Gelosa P, Lecca D, Fumagalli M et al (2014) Microglia is a key player in the reduction of stroke damage promoted by the new antithrombotic agent ticagrelor. J Cereb Blood Flow Metab 34:979–988. https://doi.org/10.1038/jcbfm.2014.45
Cserép C, Pósfai B, Lénárt N et al (2020) Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 367:528–537. https://doi.org/10.1126/science.aax6752
Chin Y, Kishi M, Sekino M et al (2013) Involvement of glial P2Y 1 receptors in cognitive deficit after focal cerebral stroke in a rodent model. J Neuroinflammation 10:95. https://doi.org/10.1186/1742-2094-10-95
Carmo MRS, Simões AP, Fonteles AA et al (2014) ATP P2Y1 receptors control cognitive deficits and neurotoxicity but not glial modifications induced by brain ischemia in mice. Eur J Neurosci 39:614–622. https://doi.org/10.1111/ejn.12435
Wen R-X, Shen H, Huang S-X et al (2020) P2Y6 receptor inhibition aggravates ischemic brain injury by reducing microglial phagocytosis. CNS Neurosci Ther 26:416–429. https://doi.org/10.1111/cns.13296
Milde S, Brown GC (2022) Knockout of the P2Y6 receptor prevents peri-infarct neuronal loss after transient, focal ischemia in mouse brain. Int J Mol Sci 23:2304. https://doi.org/10.3390/ijms23042304
Chen Y, Corriden R, Inoue Y et al (2006) ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 314:1792–1795. https://doi.org/10.1126/science.1132559
Alkayed F, Kashimata M, Koyama N et al (2012) P2Y11 purinoceptor mediates the ATP-enhanced chemotactic response of rat neutrophils. J Pharmacol Sci 120:288–295. https://doi.org/10.1254/jphs.12173FP
Kaufmann A, Musset B, Limberg SH et al (2005) “Host tissue damage” signal ATP promotes non-directional migration and negatively regulates toll-like receptor signaling in human monocytes. J Biol Chem 280:32459–32467. https://doi.org/10.1074/jbc.M505301200
Abbracchio M-P, Boeynaems J-M, Boyer JL, et al (2021) P2Y receptors in GtoPdb v.2021.3. IUPHAR/BPS Guide to Pharmacology CITE, 2021(3). https://doi.org/10.2218/gtopdb/F52/2021.3
Burnstock G (2016) An introduction to the roles of purinergic signalling in neurodegeneration, neuroprotection and neuroregeneration. Neuropharmacology 104:4–17. https://doi.org/10.1016/j.neuropharm.2015.05.031
Antonioli L, Pacher P, Vizi ES, Haskó G (2013) CD39 and CD73 in immunity and inflammation. Trends Mol Med 19:355–367. https://doi.org/10.1016/j.molmed.2013.03.005
Vieira C, Magalhães-cardoso MT, Ferreirinha F et al (2014) Feed-forward inhibition of CD73 and upregulation of adenosine deaminase contribute to the loss of adenosine neuromodulation in postinflammatory ileitis. Mediators Inflamm 2014:254640. https://doi.org/10.1155/2014/254640
Horenstein AL, Chillemi A, Zaccarello G et al (2013) A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology 2:e26246. https://doi.org/10.4161/onci.26246
Sebastián-Serrano Á, De D-GL, Martínez-Frailes C et al (2015) Tissue-nonspecific alkaline phosphatase regulates purinergic transmission in the central nervous system during development and disease. CSBJ 13:95–100. https://doi.org/10.1016/j.csbj.2014.12.004
Langer D, Hammer K, Koszalka P et al (2008) Distribution of ectonucleotidases in the rodent brain revisited. Cell Tissue Res 334:199–217. https://doi.org/10.1007/s00441-008-0681-x
Niemelä J, Ifergan I, Yegutkin GG et al (2008) IFN-β regulates CD73 and adenosine expression at the blood-brain barrier. Eur J Immunol 38:2718–2726. https://doi.org/10.1002/eji.200838437
Mills JH, Thompson LF, Mueller C et al (2008) CD73 is required for efficient entry of lymphocytes into the central nervous system during experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 105:9325–9330. https://doi.org/10.1073/pnas.0711175105
Petrovic-Djergovic D, Hyman MC, Ray JJ et al (2012) Tissue-resident ecto-5’ nucleotidase (CD73) regulates leukocyte trafficking in the ischemic brain. J Immunol 188:2387–2398. https://doi.org/10.4049/jimmunol.1003671
Sauer AV, Brigida I, Carriglio N, Aiuti A (2012) Autoimmune dysregulation and purine metabolism in adenosine deaminase deficiency. Front Immunol 3:265. https://doi.org/10.3389/fimmu.2012.00265
Nagy JI, LaBella LA, Buss M, Daddona PE (1984) Immunohistochemistry of adenosine deaminase: implications for adenosine neurotransmission. Science 224:4645. https://doi.org/10.1126/science.6142530
Zhou Q, Yang D, Ombrello AK et al (2014) Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med 370:911–920. https://doi.org/10.1056/nejmoa1307361
Elbracht M, Mull M, Wagner N et al (2017) Stroke as initial manifestation of adenosine deaminase 2 deficiency. Neuropediatrics 48:111–114. https://doi.org/10.1055/s-0036-1597611
Gayle RB, Broekman MJ, Marcus AJ (1998) Inhibition of platelet function by recombinant soluble ecto-ADPase / CD39. J Clin Invest 101:1851–1859. https://doi.org/10.1172/jci1753
Kaczmarek E, Koziak K, Sévigny J et al (1996) Identification and characterization of CD39/vascular ATP diphosphohydrolase. J Biol Chem 271:33116–33122. https://doi.org/10.1074/jbc.271.51.33116
Marcus AJ, Broekman MJ, Drosopoulos JHF et al (1997) The endothelial cell ecto-ADPase responsible for inhibition of platelet function is CD39. J Clin Invest 99:1351–1360. https://doi.org/10.1172/JCI119294
Choudhri TF, Hoh BL, Zerwes HG et al (1998) Reduced microvascular thrombosis and improved outcome in acute murine stroke by inhibiting GP IIb/IIa receptor-mediated platelet aggregation. J Clin Invest 102:1301–1310. https://doi.org/10.1172/JCI3338
Pinsky DJ, Johan Broekman M, Peschon JJ et al (2002) Elucidation of the thromboregulatory role of CD39/ectoapyrase in the ischemic brain. J Clin Invest 109:1031–1040. https://doi.org/10.1172/JCI0210649
Baek AE, Sutton NR, Petrovic-Djergovic D et al (2017) Ischemic cerebroprotection conferred by myeloid lineage-restricted or global CD39 transgene expression. Circulation 135:2389–2402. https://doi.org/10.1161/CIRCULATIONAHA.116.023301
Schädlich IS, Schnapauff O, Pöls L et al (2022) Nt5e deficiency does not affect post-stroke inflammation and lesion size in a murine ischemia/reperfusion stroke model. iScience 25:104470. https://doi.org/10.1016/j.isci.2022.104470
Sommer CJ (2017) Ischemic stroke: experimental models and reality. Acta Neuropathol 133:245–261. https://doi.org/10.1007/s00401-017-1667-0
Munji RN, Soung AL, Weiner GA et al (2019) Profiling the mouse brain endothelial transcriptome in health and disease models reveals a core blood–brain barrier dysfunction module. Nat Neurosci 22:1892–1902. https://doi.org/10.1038/s41593-019-0497-x
Thompson LF, Eltzschig HK, Ibla JC et al (2004) Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med 200:1395–1405. https://doi.org/10.1084/jem.20040915
Deaglio S, Dwyer KM, Gao W et al (2007) Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 204:1257–1265. https://doi.org/10.1084/jem.20062512
Ohta A, Kini R, Ohta A et al (2012) The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol 3:190. https://doi.org/10.3389/fimmu.2012.00190
Zhang H, Xia Y, Ye Q et al (2018) In vivo expansion of regulatory T cells with IL-2/IL-2 antibody complex protects against transient ischemic stroke. J Neurosci 38:10168–10179. https://doi.org/10.1523/JNEUROSCI.3411-17.2018
Liang D, Zuo A, Zhao R et al (2016) CD73 expressed on γδ T cells shapes their regulatory effect in experimental autoimmune uveitis. PLoS One 11:e0150078. https://doi.org/10.1371/journal.pone.0150078
Gelderblom M, Weymar A, Bernreuther C et al (2012) Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood 120:3793–3802. https://doi.org/10.1182/blood-2012-02-412726
Guerreiro S, Privat AL, Bressac L, Toulorge D (2020) CD38 in neurodegeneration and neuroinflammation. Cells 9:471. https://doi.org/10.3390/cells9020471
Partida-Sánchez S, Goodrich S, Kusser K et al (2004) Regulation of dendritic cell trafficking by the ADP-ribosyl cyclase CD38: impact on the development of humoral immunity. Immunity 20:279–291. https://doi.org/10.1016/S1074-7613(04)00048-2
Partida-Sánchez S, Cockayne DA, Monard S et al (2001) Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat Med 7:1209–1216. https://doi.org/10.1038/nm1101-1209
Choe C, Lardong K, Gelderblom M et al (2011) CD38 exacerbates focal cytokine production, postischemic inflammation and brain injury after focal cerebral ischemia. PLoS One 6:e19046. https://doi.org/10.1371/journal.pone.0019046
Long A, Park JH, Klimova N et al (2017) CD38 knockout mice show significant protection against ischemic brain damage despite high level poly-ADP-ribosylation. Neurochem Res 42:283–293. https://doi.org/10.1007/s11064-016-2031-9
Young GS, Choleris E, Lund FE, Kirkland JB (2006) Decreased cADPR and increased NAD+ in the Cd38-/- mouse. Biochem Biophys Res Commun 346:188–192. https://doi.org/10.1016/j.bbrc.2006.05.100
Lautrup S, Sinclair DA, Mattson MP, Fang EF (2019) NAD+ in brain aging and neurodegenerative disorders. Cell Metab 30:630–655. https://doi.org/10.1016/j.cmet.2019.09.001
Verdin E (2015) NAD+ in aging, metabolism, and neurodegeneration. Science 350:1208–1213. https://doi.org/10.1126/science.aac4854
Hannawi Y, Ewees MG, Moore JT, Zweier JL (2022) Characterizing CD38 expression and enzymatic activity in the brain of spontaneously hypertensive stroke-prone rats. Front Pharmacol 13:881708. https://doi.org/10.3389/fphar.2022.881708
Zhu XH, Lu M, Lee BY et al (2015) In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci U S A 112:2876–2881. https://doi.org/10.1073/pnas.1417921112
Melani A, Pantoni L, Corsi C et al (1999) Striatal outflow of adenosine, excitatory amino acids, γ-aminobutyric acid, and taurine in awake freely moving rats after middle cerebral artery occlusion: Correlations with neurological deficit and histopathological damage. Stroke 30:2448–2455. https://doi.org/10.1161/01.STR.30.11.2448
Deussen A, Schrader J (1991) Cardiac adenosine production is linked to myocardial pO2. J Mol Cell Cardiol 23:495–504. https://doi.org/10.1016/0022-2828(91)90173-J
Schrader J (2022) Ectonucleotidases as bridge between the ATP and adenosine world: reflections on Geoffrey Burnstock. Purinergic Signal 18:193–198. https://doi.org/10.1007/s11302-022-09862-6
Naito Y, Lowenstein JM (1985) 5’-Nucleotidase from rat heart membranes. Inhibition by adenine nucleotides and related compounds. Biochem J 226:645–651. https://doi.org/10.1042/bj2260645
Decking UKM, Schlieper G, Kroll K, Schrader J (1997) Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ Res 81:154–164. https://doi.org/10.1161/01.res.81.2.154
Pignataro G, Maysami S, Studer FE et al (2008) Downregulation of hippocampal adenosine kinase after focal ischemia as potential endogenous neuroprotective mechanism. J Cereb Blood Flow Metab 28:17–23. https://doi.org/10.1038/sj.jcbfm.9600499
Morote-Garcia JC, Rosenberger P, Kuhlicke J, Eltzschig HK (2008) HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood 111:5571–5580. https://doi.org/10.1182/blood-2007-11-126763
Kitagawa H, Mori A, Shimada J et al (2002) Intracerebral adenosine infusion improves neurological outcome after transient focal ischemia in rats. Neurol Res 24:317–323. https://doi.org/10.1179/016164102101199819
Gaudin A, Yemisci M, Eroglu H et al (2014) Squalenoyl adenosine nanoparticles provide neuroprotection after stroke and spinal cord injury. Nat Nanotechnol 9:1054–1062. https://doi.org/10.1038/nnano.2014.274
Tatlisumak T, Takano K, Carano R et al (1998) Delayed treatment with an adenosine kinase inhibitor, GP683, attenuates infarct size in rats with temporary middle cerebral artery occlusion. Stroke 29:1952–1958. https://doi.org/10.1161/01.str.29.9.1952
Pignataro G, Simon RP, Boison D (2007) Transgenic overexpression of adenosine kinase aggravates cell death in ischemia. J Cereb Blood Flow Metab 27:1–5. https://doi.org/10.1038/sj.jcbfm.9600334
Pardridge M, Yoshikawa T, Kang Y, Miller L (1994) Blood-brain barrier transport and brain metabolism adenosine and adenosine analogs. J Pharmacol Exp Ther 268:14–18
Koeppen M, Eckle T, Eltzschig HK (2009) Selective deletion of the A1 adenosine receptor abolishes heart-rate slowing effects of intravascular adenosine in vivo. PLoS One 4:e6784. https://doi.org/10.1371/journal.pone.0006784
Jacobson KA, Gao ZG (2006) Adenosine receptors as therapeutic targets. Nat Rev Drug Discov 5:247–264. https://doi.org/10.1038/nrd1983
Latini S, Pedata F (2001) Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J Neurochem 79:463–484. https://doi.org/10.1046/j.1471-4159.2001.00607.x
Kull B, Svenningsson P, Fredholm BB (2000) Adenosine A(2A) receptors are colocalized with and activate G (olf) in rat striatum. Mol Pharmacol 58:771–777. https://doi.org/10.1124/mol.58.4.771
Wei CJ, Li W, Chen JF (2011) Normal and abnormal functions of adenosine receptors in the central nervous system revealed by genetic knockout studies. Biochim Biophys Acta 1808:1358–1379. https://doi.org/10.1016/j.bbamem.2010.12.018
Pedata F, Dettori I, Coppi E et al (2016) Purinergic signalling in brain ischemia. Neuropharmacology 104:105–130. https://doi.org/10.1016/j.neuropharm.2015.11.007
Fowler JC, Gervitz LM, Hamilton ME, Walker JA (2003) Systemic hypoxia and the depression of synaptic transmission in rat hippocampus after carotid artery occlusion. J Physiol 550:961–972. https://doi.org/10.1113/jphysiol.2003.039594
Gervitz LM, Lutherer LO, Davies DG et al (2001) Adenosine induces initial hypoxic-ischemic depression of synaptic transmission in the rat hippocampus in vivo. Am J Physiol - Regul Integr Comp Physiol 280:639–645. https://doi.org/10.1152/ajpregu.2001.280.3.r639
Gervitz LM, Davies DG, Omidvar K, Fowler JC (2003) The effect of acute hypoxemia and hypotension on adenosine-mediated depression of evoked hippocampal synaptic transmission. Exp Neurol 182:507–517. https://doi.org/10.1016/S0014-4886(03)00160-2
Arrigoni E, Crocker AJ, Saper CB et al (2005) Deletion of presynaptic adenosine A1 receptors impairs the recovery of synaptic transmission after hypoxia. Neuroscience 132:575–580. https://doi.org/10.1016/j.neuroscience.2004.12.009
Johansson B, Halldner L, Dunwiddie TV et al (2001) Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc Natl Acad Sci U S A 98:9407–9412. https://doi.org/10.1073/pnas.161292398
Sebastiao AM, De Mendonça A, Moreira T, Alexandre Ribeiro J (2001) Activation of synaptic NMDA receptors by action potential-dependent release of transmitter during hypoxia impairs recovery of synaptic transmission on reoxygenation. J Neurosci 21:8564–8571. https://doi.org/10.1523/jneurosci.21-21-08564.2001
von Lubitz DKJE, Marangos PJ (1990) Cerebral ischemia in gerbils: postischemic administration of cyclohexyl adenosine and 8-sulfophenyl-theophylline. J Mol Neurosci 2:53–59. https://doi.org/10.1007/BF02896926
Von Lubitz DKJE, Lin RCS, Melman N et al (1994) Chronic administration of selective adenosine A1 receptor agonist or antagonist in cerebral ischemia. Eur J Pharmacol 256:161–167. https://doi.org/10.1016/0014-2999(94)90241-0
Von Lubitz DKJE, Lin RCS, Paul IA et al (1996) Postischemic administration of adenosine amine congener (ADAC): analysis of recovery in gerbils. Eur J Pharmacol 316:171–179. https://doi.org/10.1016/S0014-2999(96)00667-X
Jacobson KA, Von Lubitz DKJE, Daly JW, Fredholm BB (1996) Adenosine receptor ligands: differences with acute versus chronic treatment. Trends Pharmacol Sci 17:108–113. https://doi.org/10.1016/0165-6147(96)10002-X
Coelho JE, Rebola N, Fragata I et al (2006) Hypoxia-induced desensitization and internalization of adenosine A 1 receptors in the rat hippocampus. Neuroscience 138:1195–1203. https://doi.org/10.1016/j.neuroscience.2005.12.012
Nakamura M, Nakakimura K, Matsumoto M, Sakabe T (2002) Rapid tolerance to focal cerebral ischemia in rats is attenuated by adenosine A1 receptor antagonist. J Cereb Blood Flow Metab 22:161–170. https://doi.org/10.1097/00004647-200202000-00004
Yoshida M, Nakakimura K, Cui YJ et al (2004) Adenosine A1 receptor antagonist and mitochondrial ATP-sensitive potassium channel blocker attenuate the tolerance to focal cerebral ischemia in rats. J Cereb Blood Flow Metab 24:771–779. https://doi.org/10.1097/01.WCB.0000122742.72175.1B
Martire A, Lambertucci C, Pepponi R et al (2019) Neuroprotective potential of adenosine A 1 receptor partial agonists in experimental models of cerebral ischemia. J Neurochem 149:211–230. https://doi.org/10.1111/jnc.14660
Gao Y, Phillis JW (1994) CGS 15943, An adenosine A2 receptor antagonist, reduces cerebral ischemic injury in the mongolian gerbil. Life Sci 55:61–65. https://doi.org/10.1016/0024-3205(94)00889-2
Phillis JW (1995) The effects of selective A1 and A2a adenosine receptor antagonists on cerebral ischemic injury in the gerbil. Brain Res 705:79–84. https://doi.org/10.1016/0006-8993(95)01153-6
Higashi H, Meno JR, Marwaha AS, Winn HR (2002) Hippocampal injury and neurobehavioral deficits following hyperglycemic cerebral ischemia: effect of theophylline and ZM 241385. J Neurosurg 96:117–126. https://doi.org/10.3171/jns.2002.96.1.0117
Melani A, Pantoni L, Bordoni F et al (2003) The selective A2A receptor antagonist SCH 58261 reduces striatal transmitter outflow, turning behavior and ischemic brain damage induced by permanent focal ischemia in the rat. Brain Res 959:243–250. https://doi.org/10.1016/S0006-8993(02)03753-8
Melani A, Gianfriddo M, Vannucchi MG et al (2006) The selective A2A receptor antagonist SCH 58261 protects from neurological deficit, brain damage and activation of p38 MAPK in rat focal cerebral ischemia. Brain Res 1073–1074:470–480. https://doi.org/10.1016/j.brainres.2005.12.010
Monopoli A, Lozza G, Forlani A et al (1998) Blockade of adenosine A2A receptors by SCH 58261 results in neuroprotective effects in cerebral ischaemia in rats. Neuroreport 9:3955–3959. https://doi.org/10.1097/00001756-199812010-00034
Melani A, Dettori I, Corti F et al (2015) Time-course of protection by the selective A2A receptor antagonist SCH58261 after transient focal cerebral ischemia. Neurol Sci 36:1441–1448. https://doi.org/10.1007/s10072-015-2160-y
Gui L, Duan W, Tian H et al (2009) Adenosine A2A receptor deficiency reduces striatal glutamate outflow and attenuates brain injury induced by transient focal cerebral ischemia in mice. Brain Res 1297:185–193. https://doi.org/10.1016/j.brainres.2009.08.050
Chen JF, Huang Z, Ma J et al (1999) A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J Neurosci 19:9192–9200. https://doi.org/10.1523/jneurosci.19-21-09192.1999
Zhou Y, Zeng X, Li G et al (2019) Inactivation of endothelial adenosine A2A receptors protects mice from cerebral ischaemia-induced brain injury. Br J Pharmacol 176:2250–2263. https://doi.org/10.1111/bph.14673
Ito M, Aswendt M, Lee AG et al (2018) RNA-sequencing analysis revealed a distinct motor cortex transcriptome in spontaneously recovered mice after stroke. Stroke 49:2191–2199. https://doi.org/10.1161/STROKEAHA.118.021508
Marcoli M, Raiteri L, Bonfanti A et al (2003) Sensitivity to selective adenosine A1 and A2A receptor antagonists of the release of glutamate induced by ischemia in rat cerebrocortical slices. Neuropharmacology 45:201–210. https://doi.org/10.1016/S0028-3908(03)00156-4
Marcoli M, Bonfanti A, Roccatagliata P et al (2004) Glutamate efflux from human cerebrocortical slices during ischemia: Vesicular-like mode of glutamate release and sensitivity to A2A adenosine receptor blockade. Neuropharmacology 47:884–891. https://doi.org/10.1016/j.neuropharm.2004.06.022
Nishizaki T, Nagai K, Nomura T et al (2002) A new neuromodulatory pathway with a glial contribution mediated via A2a adenosine receptors. Glia 39:133–147. https://doi.org/10.1002/glia.10100
Pinto-Duarte A, Coelho JE, Cunha RA et al (2005) Adenosine A2A receptors control the extracellular levels of adenosine through modulation of nucleoside transporters activity in the rat hippocampus. J Neurochem 93:595–604. https://doi.org/10.1111/j.1471-4159.2005.03071.x
Pintor A, Galluzzo M, Grieco R et al (2004) Adenosine A2A receptor antagonists prevent the increase in striatal glutamate levels induced by glutamate uptake inhibitors. J Neurochem 89:152–156. https://doi.org/10.1111/j.1471-4159.2003.02306.x
Matos M, Augusto E, Dos S-RA et al (2012) Adenosine A 2A receptors modulate glutamate uptake in cultured astrocytes and gliosomes. Glia 60:702–716. https://doi.org/10.1002/glia.22290
Ciruela F, Casadó V, Rodrigues RJ et al (2006) Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J Neurosci 26:2080–2087. https://doi.org/10.1523/JNEUROSCI.3574-05.2006
Melani A, Cipriani S, Vannucchi MG et al (2009) Selective adenosine A2a receptor antagonism reduces JNK activation in oligodendrocytes after cerebral ischaemia. Brain 132:1480–1495. https://doi.org/10.1093/brain/awp076
Orr AG, Orr AL, Li XJ et al (2009) Adenosine A2A receptor mediates microglial process retraction. Nat Neurosci 12:872–878. https://doi.org/10.1038/nn.2341
Von Lubitz DKJE, Lin RCS, Jacobson KA (1995) Cerebral ischemia in gerbils: effects of acute and chronic treatment with adenosine A2A receptor agonist and antagonist. Eur J Pharmacol 287:295–302. https://doi.org/10.1016/0014-2999(95)00498-X
Melani A, Corti F, Cellai L et al (2014) Low doses of the selective adenosine A2A receptor agonist CGS21680 are protective in a rat model of transient cerebral ischemia. Brain Res 1551:59–72. https://doi.org/10.1016/j.brainres.2014.01.014
Sebastião AM, Ribeiro JA (2009) Triggering neurotrophic factor actions through adenosine A2A receptor activation: implications for neuroprotection. Br J Pharmacol 158:15–22. https://doi.org/10.1111/j.1476-5381.2009.00157.x
Planas AM (2018) Role of immune cells migrating to the ischemic brain. Stroke 49:2261–2267. https://doi.org/10.1161/STROKEAHA.118.021474
Iadecola C, Buckwalter MS, Anrather J (2020) Immune responses to stroke: mechanisms, modulation, and therapeutic potential. J Clin Invest 130:2777–2788. https://doi.org/10.1172/JCI135530
Ohta A, Sitkovsky M (2001) Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 414:916–920. https://doi.org/10.1038/414916a
Haskó G, Linden J, Cronstein B, Pacher P (2008) Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov 7:759–770. https://doi.org/10.1038/nrd2638
Pasquini S, Vincenzi F, Casetta I et al (2020) Adenosinergic System Involvement in Ischemic Stroke Patients’ Lymphocytes. Cells 9:1072. https://doi.org/10.3390/cells9051072
Pedata F, Pugliese AM, Coppi E et al (2014) Adenosine A2A receptors modulate acute injury and neuroinflammation in brain ischemia. Mediators Inflamm 2014:805198. https://doi.org/10.1155/2014/805198
Gelderblom M, Leypoldt F, Steinbach K et al (2009) Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 40:1849–1857. https://doi.org/10.1161/STROKEAHA.108.534503
Yang D, Zhang Y, Nguyen HG et al (2006) The A2B adenosine receptor protects against inflammation and excessive vascular adhesion. J Clin Invest 116:1913–1923. https://doi.org/10.1172/JCI27933
Haskó G, Csóka B, Németh ZH et al (2009) A2B adenosine receptors in immunity and inflammation. Trends Immunol 30:263–270. https://doi.org/10.1016/j.it.2009.04.001
Eltzschig HK, Ibla JC, Furuta GT et al (2003) Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A 2B receptors. J Exp Med 198:783–796. https://doi.org/10.1084/jem.20030891
Kong T, Westerman KA, Faigle M et al (2006) HIF-dependent induction of adenosine A2B receptor in hypoxia. FASEB J 20:2242–2250. https://doi.org/10.1096/fj.06-6419com
Eckle T, Faigle M, Grenz A et al (2008) A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood 111:2024–2035. https://doi.org/10.1182/blood-2007-10-117044
Dettori I, Gaviano L, Ugolini F et al (2021) Protective effect of adenosine A2B receptor agonist, BAY60-6583, against transient focal brain ischemia in rat. Front Pharmacol 11:588757. https://doi.org/10.3389/fphar.2020.588757
Gu LZ, Huang BS, Shen W et al (2013) Early activation of nSMase2/ceramide pathway in astrocytes is involved in ischemia-associated neuronal damage via inflammation in rat hippocampi. J Neuroinflammation 10:109. https://doi.org/10.1186/1742-2094-10-109
Chen G, Harvey BK, Shen H et al (2006) Activation of adenosine A3 receptors reduces ischemic brain injury in rodents. J Neurosci Res 84:1848–1855. https://doi.org/10.1002/jnr.21071
Choi IY, Lee JC, Ju C et al (2011) A3 adenosine receptor agonist reduces brain ischemic injury and inhibits inflammatory cell migration in rats. Am J Pathol 179:2042–2052. https://doi.org/10.1016/j.ajpath.2011.07.006
Liston TE, Hama A, Boltze J et al (2022) Adenosine A1R/A3R (adenosine A1 and A3 receptor) agonist AST-004 reduces brain infarction in a nonhuman primate model of stroke. Stroke 53:238–248. https://doi.org/10.1161/STROKEAHA.121.036396
Pugliese AM, Coppi E, Volpini R et al (2007) Role of adenosine A3 receptors on CA1 hippocampal neurotransmission during oxygen-glucose deprivation episodes of different duration. Biochem Pharmacol 74:768–779. https://doi.org/10.1016/j.bcp.2007.06.003
Palmer TM, Benovic JL, Stiles GL (1995) Agonist-dependent phosphorylation and desensitization of the rat A3 adenosine receptor: evidence for a G-protein-coupled receptor kinase-mediated mechanism. J Biol Chem 270:29607–29613. https://doi.org/10.1074/jbc.270.49.29607
Trincavelli ML, Tuscano D, Cecchetti P et al (2000) Agonist-induced internalization and recycling of the human A3 adenosine receptors: role in receptor desensitization and resensitization. J Neurochem 75:1493–1501. https://doi.org/10.1046/j.1471-4159.2000.0751493.x
Von Lubitz DK, Lin RC, PP J et al (1994) Adenosine A3 receptor stimulation and cerebral ischemia. Eur J Pharmacol 263:59–67. https://doi.org/10.1016/0014-2999(94)90523-1
Ochaion A, Bar-Yehuda S, Cohen S et al (2009) The anti-inflammatory target A3 adenosine receptor is over-expressed in rheumatoid arthritis, psoriasis and Crohn’s disease. Cell Immunol 258:115–122. https://doi.org/10.1016/j.cellimm.2009.03.020
Fishman P, Bar-Yehuda S, Madi L et al (2006) The PI3K-NF-κB signal transduction pathway is involved in mediating the anti-inflammatory effect of IB-MECA in adjuvant-induced arthritis. Arthritis Res Ther 8:1–9. https://doi.org/10.1186/ar1887
Fishman P, Bar-Yehuda S, Liang BT, Jacobson KA (2012) Pharmacological and therapeutic effects of A 3 adenosine receptor agonists. Drug Discov Today 17:359–366. https://doi.org/10.1016/j.drudis.2011.10.007
Silverman MH, Strand V, Markovits D et al (2008) Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: Data from a phase II clinical trial. J Rheumatol 35:41–48
Hubert S, Rissiek B, Klages K et al (2010) Extracellular NAD+ shapes the Foxp3+ regulatory T cell compartment through the ART2–P2X7 pathway. J Exp Med 207:2561–2568. https://doi.org/10.1084/jem.20091154
Winzer R, Serracant-Prat A, Brock VJ et al (2022) P2X7 is expressed on human innate-like T lymphocytes and mediates susceptibility to ATP-induced cell death. Eur J Immunol 52:1805–1818. https://doi.org/10.1002/eji.202249932
Jacobson KA, Jarvis MF, Williams M (2002) Purine and pyrimidine (P2) receptors as drug targets. J Med Chem 45:4057–4093. https://doi.org/10.1021/jm020046y
Adriouch S, Bannas P, Schwarz N et al (2008) ADP-ribosylation at R125 gates the P2X7 ion channel by presenting a covalent ligand to its nucleotide binding site. FASEB J 22:861–869. https://doi.org/10.1096/fj.07-9294com
Haag F, Koch-Nolte F, Kühl M et al (1994) Premature stop codons inactivate the RT6 genes of the human and chimpanzee species. J Mol Biol 243:537–546. https://doi.org/10.1006/jmbi.1994.1680
Dreisig K, Sund L, Dommer MW et al (2018) Human P2Y11 expression level affects human P2X7 receptor-mediated cell death. Front Cell Neurosci 9:1159. https://doi.org/10.3389/fimmu.2018.01159
Schneider E, Winzer R, Rissiek A et al (2021) CD73-mediated adenosine production by CD8 T cell-derived extracellular vesicles constitutes an intrinsic mechanism of immune suppression. Nat Commun 12:5911. https://doi.org/10.1038/s41467-021-26134-w
Raczkowski F, Rissiek A, Ricklefs I et al (2018) CD39 is upregulated during activation of mouse and human T cells and attenuates the immune response to Listeria monocytogenes. PLoS One 13:e0197151. https://doi.org/10.1371/journal.pone.0197151
Baroja-Mazo A, Revilla-Nuin B, De Bejar Á et al (2019) Extracellular adenosine reversibly inhibits the activation of human regulatory T cells and negatively influences the achievement of the operational tolerance in liver transplantation. Am J Transplant 19:48–61. https://doi.org/10.1111/ajt.15023
Ohta A, Sitkovsky M (2014) Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol 5:304. https://doi.org/10.3389/fimmu.2014.00304
Zarek PE, Huang C-T, Lutz ER et al (2008) A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood 111:251–259. https://doi.org/10.1182/blood-2007-03-081646
Gomes JR, Cabrito I, Soares HR et al (2018) Delivery of an anti-transthyretin nanobody to the brain through intranasal administration reveals transthyretin expression and secretion by motor neurons. J Neurochem 145:393–408. https://doi.org/10.1111/jnc.14332
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Council) through SFB 1328 (Project-ID 335447717 to TM (A13), ET (A14), and BR (Z02)) and FOR 2879 Immunostroke (Project-ID 405358801 to TM (A1) and ET (C1)) and a grant from “Hermann und Lilly Schilling-Stiftung für Medizinische Forschung” to TM.
Author information
Authors and Affiliations
Contributions
ISS and BR assembled the figures; all authors participated in the writing process and approved the final version of the manuscript. JS provided the results of the human/mouse purinergic key player comparative expression analyses shown in Fig. 1.
Corresponding author
Ethics declarations
Conflict of interest
TM and BR are co-inventors on patent applications for CD73- and CD39-specific nanobodies.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is a contribution to the special issue on: Immunopathology of Stroke - Guest Editors: Arthur Liesz and Tim Magnus
Supplementary information
Supplementary materials 1:
Supplementary Table 1 Knockout mice for purinergic signaling investigated in brain ischemia. Supplementary Table 2 Pharmacological modification of AR signaling in brain ischemia. (DOCX 80 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schädlich, I.S., Winzer, R., Stabernack, J. et al. The role of the ATP-adenosine axis in ischemic stroke. Semin Immunopathol 45, 347–365 (2023). https://doi.org/10.1007/s00281-023-00987-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-023-00987-3