
Abstract
Eosinophils are granule-containing leukocytes which develop in the bone marrow. For many years, eosinophils have been recognized as cytotoxic effector cells, but recent studies suggest that they perform additional immunomodulatory and homeostatic functions. Autophagy is a conserved intracellular process which preserves cellular homeostasis. Autophagy defects have been linked to the pathogenesis of many human disorders. Evidence for abnormal regulation of autophagy, including decreased or increased expression of autophagy-related (ATG) proteins, has been reported in several eosinophilic inflammatory disorders, such as Crohn’s disease, bronchial asthma, eosinophilic esophagitis, and chronic rhinosinusitis. Despite the increasing extent of research using preclinical models of immune cell-specific autophagy deficiency, the physiological relevance of autophagic pathway in eosinophils has remained unknown until recently. Owing to the increasing evidence that eosinophils play a role in keeping organismal homeostasis, the regulation of eosinophil functions is of considerable interest. Here, we discuss the most recent advances on the role of autophagy in eosinophils, placing particular emphasis on insights obtained in mouse models of infections and malignant diseases in which autophagy has genetically dismantled in the eosinophil lineage. These studies pointed to the possibility that autophagy-deficient eosinophils exaggerate inflammation. Therefore, the pharmacological modulation of the autophagic pathway in these cells could be used for therapeutic interventions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Eosinophils are granulocytes which are characterized by their avidity for the acidic dye eosin [1]. Relatively few mature eosinophils are found in the peripheral blood of healthy humans (less than 400 per mm3) [2]. Moreover, under homeostatic conditions, eosinophils reside primarily in all regions of the digestive system except the esophagus [3]. Eosinophils are also present in lymphoid organs such as thymus, lymph nodes, and spleen. A physiological infiltration of eosinophils is also seen in non-lymphoid organs such as adipose and mammary gland tissues as well as in the uterus [4]. Moreover, the so-called regulatory eosinophils have been observed in normal human and mouse lungs [5]. In response to inflammatory stimuli, eosinophil differentiation in the bone marrow is increased and eosinophils migrate towards inflammatory tissues where their lifespan is prolonged [6, 7]. When blood eosinophil numbers exceed 400 per mm3, the term eosinophilia applies. A threshold of 1500 eosinophils per mm3 is usually employed to define blood hypereosinophilia [8]. A cytokine-independent eosinophilia is caused by genetic changes within the eosinophil lineage, and the resulting diseases are classified as primary or intrinsic eosinophilic disorders [9].
The most typical eosinophil characteristic which discriminates them from other granulocytes (neutrophils and basophils) is the presence of large specific granules in the cytoplasm [10, 11]. A significant amount of mediators, including cytotoxic cationic proteins, cytokines, chemokines, and growth factors, are preformed and stored within eosinophil specific granules, where they are available for a rapid, stimulus-dependent release [12, 13]. Other characteristic organelles in eosinophils are primary granules, known to contain the Charcot-Leyden crystal protein (also known as galectin-10), and lipid bodies, which are production sites of inflammatory lipid mediators such as cysteinyl leukotrienes, thromboxanes, and prostaglandins [14, 15].
Contrary to neutrophils, which are essential for host defense, pharmacological or genetic depletion of eosinophils does not cause evident functional consequences [16]. Nevertheless, the fact that eosinophil lineage in all vertebrates survived the evolution pressure over several thousands of years demonstrates the importance of eosinophils in health and disease [17]. While eosinophils have been traditionally perceived as cytotoxic effector cells, recent studies have revealed their additional immunomodulatory and homeostatic activities. The role of eosinophils in immunity is still a matter of dispute and remains to be ambiguously defined. Preclinical mouse models mimicking human eosinopenia [18, 19] and hypereosinophilia [20, 21] as well as the ability to specifically knockout genes in the eosinophil lineage [22] allow to investigate the potential roles of eosinophils in health and disease. Moreover, targeted anti-eosinophil therapies allow to draw conclusions regarding the contribution of eosinophils for physiological and pathophysiological processes in humans [23].
Autophagy
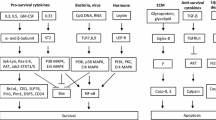
Macroautophagy (hereafter referred to as autophagy) is a highly dynamic intracellular degradation system by which cytoplasmic constituents are delivered to lysosomes for degradation [24, 25]. The main morphological feature of autophagy is the biogenesis of autophagosome, a unique double-membraned vesicle which encloses parts of the cytoplasm. The fusion between autophagosomes and lysosomes results in the formation of auto(phago)lysosomes, in which cargo is degraded by a number of hydrolytic enzymes. Breakdown products are then returned to the cytosol and reused [24, 25] (Fig. 1a).

Schematic representation of the mammalian autophagy pathway. a Upon initiation of autophagy, a small portion of the cytoplasm is enclosed by the phagophore (also known as isolation membrane) which may originate from an ER-based structure. Elongation of the phagophore is followed by the autophagosome completion and its fusion with the lysosome, where the engulfed contents are degraded within the autolysosome. b ULK and class III PI3K (VPS34) protein complexes are required for the phagophore initiation. In addition, ATG12 and LC3 ubiquitin-like conjugation systems are necessary for subsequent elongation of the phagophore and its closure. c Autophagy is largely regulated by the activity of mTORC1, which reflects cellular nutritional status. Sufficient amounts of amino acids and growth factors suppress autophagy due to the inactivation of ULK complex by mTORC1 activity. On the contrary, mTORC1 activity is inhibited by cellular stress such as energy deprivation, DNA damage, and hypoxia, leading to the release and activation of the ULK complex which initiates the formation of autophagosomes
The autophagic pathway is a central component of cellular stress response, and it assures a proper biological adaptation to the continually changing environment [26]. In general, autophagy has two principal functions: (1) provision of essential building blocks under the conditions of nutrient or energy deprivation and (2) maintenance of cellular homeostasis by elimination of damaged macromolecules and organelles [27]. Therefore, autophagy is a process which keeps individual cells in a healthy state. Any perturbation is expected to cause, or at least to contribute, to the pathogenesis of diseases. For instance, autophagy has been implicated in several pathologies, particularly neurodegeneration, cancer, inflammation, and infectious diseases [28,29,30]. Moreover, work in the last two decades has also demonstrated that autophagy is critical for multiple functions of the immune system such as removal of pathogens, differentiation of immune cells, antigen presentation, and regulation of inflammatory responses [31, 32].
The autophagic pathways have been characterized in excellent reviews, and general roles of autophagy in the regulation of immunity have been covered elsewhere [31,32,33,34,35,36,37]. Therefore, we only introduce the canonical autophagy pathway that requires autophagy-related (ATG) proteins. In the following, we discuss recently published work on the involvement of autophagy in eosinophil-associated diseases and review experimental work on the role of ATG proteins in eosinophil development and functions.
The molecular mechanism of autophagy
Understanding of the molecular mechanism of autophagy has progressively advanced since the identification of ATG genes. In mammals, the protein machinery is organized into functional units which are required for autophagosome biogenesis. Upon autophagy induction, the ULK complex (consisting of the protein kinase ULK1/2, ATG13, ATG101 and FIP200) is generated, arising from regions where ATG9 vesicles line up with the endoplasmic reticulum (ER) [38, 39]. ULK complex is required for the recruitment and activation of the class III phosphatidylinositol 3-kinase (PI3K) complex (consisting of the lipid kinase VPS34, VPS15, ATG14, and Beclin 1), locally producing phosphatidylinositol 3-phosphate (PI3P) [39, 40]. The function of PI3P could be the formation of a phagophore membrane by modifying the composition of the ER membrane and recruitment of PI3P effectors [41]. Two ubiquitin-like conjugation systems are essential for proper elongation and closure of the phagophore membrane. The ATG12-ATG5-ATG16L1 complex is located mainly on the outer side of the phagophore membrane, followed by its disassembly upon autophagosome completion [41]. In the next step, the ATG12-ATG5-ATG16L1 complex promotes microtubule-associated protein light chain 3 (LC3) conjugation with phosphatidylethanolamine (PE) on the expanding phagophore membrane. Lipidated LC3 (LC3-II) serves as a commonly used marker for detection of double-membrane organelles owing to its integration into both the inner and outer autophagosomal membrane [42, 43] (Fig. 1b).
Autophagic pathway is strongly regulated by the activity of mechanistic target of rapamycin complex 1 (mTORC1), which senses the levels of nutrients and growth factors [44]. A strong trigger for autophagy induction is the lack of amino acids, leading to inhibition of mTORC1 activity and activation of suppressed ULK complex [45, 46]. Another central autophagy regulator is an AMP-activated protein kinase (AMPK), a sensor of metabolic, oxidative, and genomic stress [47]. Under the conditions of inadequate glucose levels, hypoxia or DNA damage, the AMPK activity is induced and promotes autophagy either by mTORC1 inhibition or direct phosphorylation of ULK complex [48, 49]. In addition, class I PI3K responds to growth factor signaling and suppresses autophagy by positively regulating the mTORC1 activity [50] (Fig. 1c). In contrast, class III PI3K (VPS34) forms a protein complex and produces the phospholipid PI3P, contributing to the initiation and progression of autophagy [50].
The protein p62 (sequestosome 1/SQSTM1) is a specific substrate for autophagy, and reduced autophagy is often related to excessive accumulation of p62 [41]. Besides, p62 selectively delivers ubiquitinated proteins to the phagophore membrane following the interaction with LC3-II [51, 52]. Finally, fusion between autophagosomes and lysosomes results in degradation of the auto(phago)lysosomal contents by hydrolytic enzymes [53]. Despite the extensive involvement of ATG proteins in autophagy machinery, it has been shown that many ATGs also exhibit additional non-autophagic functions that are discussed elsewhere [54, 55].
Regulation of the function of immune cells by autophagy
The discovery of the ATG proteins was followed by in-depth investigation of the autophagy machinery and its critical involvement in the functioning of the immune system became evident [32]. The most direct approach of autophagy-dependent microbial removal is through xenophagy, a selective form of autophagy in which intracellular pathogens are targeted for autophagosomal sequestration [56,57,58,59,60]. Two possible mechanisms for the recognition of intracellular microbes by the autophagy machinery have been suggested. During xenophagy, the cytosolic bacteria are tagged by ubiquitin molecules and recognized by p62, which recruits the LC3-positive phagophores to capture the bacteria [61, 62]. Alternatively, ubiquitin is conjugated to host proteins on Salmonella-containing endosomes and binds with ATG16L1 independently of LC3-ubiquitin interaction through adaptor proteins [63]. In addition, p62 is able to deliver the ribosomal protein precursor Fau to autolysosomes where it is metabolized into bactericidal peptides. As a consequence, autophagic organelles are endowed with unique antimicrobial properties [64].
Autophagy is involved in the regulation of inflammatory responses. For example, it can suppress the immune response through the inhibition of inflammasomes, leading to reduced activation of caspase-1 and secretion of the pro-inflammatory cytokines IL-1β in IL-18 [65,66,67,68,69]. Moreover, autophagy has an influence on the homeostasis, survival, activation, proliferation, and differentiation of multiple cells of the immune system such as natural killer (NK) cells, macrophages, dendritic cells (DCs), as well as T and B cells. For instance, autophagy contributes to the maturation and antiviral activities of NK cells [70, 71]. Autophagy also participates in antigen presentation by DCs, forming a link between the innate and adaptive immune systems. The autophagic sequestration promotes the delivery and presentation of endogenous antigens on major histocompatibility complex (MHC) class II molecules, resulting in enhanced CD4+ T helper cell responses [72, 73]. On the contrary, autophagy enhances internalization and degradation of MHC class I molecules, leading to compromised MHC class I antigen presentation and attenuated response of cytotoxic CD8+ T cells [74]. Previous reports have also demonstrated the significance of autophagy for the development, survival and effector functions of T [75,76,77,78,79] and B cells [80,81,82,83,84]. Furthermore, it has been suggested that autophagy supports the release of β-hexosaminidase and histamine from mast cells [85]. The involvement of autophagy has also been demonstrated in neutrophil differentiation [86, 87] and effector functions [88].
In contrast to other cells of the immune system, the relevance of autophagy and ATG proteins for the biology of eosinophils has long remained elusive. However, insightful evidence was recently generated from experimental mouse models in which Atg5 was specifically depleted in the eosinophil lineage, resulting in eosinophil-specific autophagy deficiency [89]. As discussed below, these models allowed testing the function of eosinophils under in vitro conditions in the presence and absence of ATG5. Moreover, these mouse lines were used in preclinical models of bacterial infection and primary hypereosinophilic disease.
Role of autophagy in eosinophilic diseases
Interest in the role of autophagy in immunity was partially driven by the association of ATG genes with inflammatory disorders. Genetic alterations in autophagy may be hereditary, predisposing individuals to autoimmune, autoinflammatory, or infectious diseases. The involvement of autophagy has been observed in several eosinophilic inflammatory diseases, such as Crohn’s disease (CD), bronchial asthma, eosinophilic esophagitis (EoE), and chronic rhinosinusitis (CRS). Study models of eosinophilic inflammatory diseases and observed results are summarized in Table 1.
Crohn’s disease (CD)
Inflammatory bowel disease (IBD) encompasses two types of chronic inflammatory disorders of the gastrointestinal tract, CD, and ulcerative colitis (UC) [90]. Accumulating evidence suggests that inflammatory conditions in the intestine result from the abnormal immune response to enteric microbes in genetically predisposed individuals [91]. Genetic studies of IBD have made great progress since 242 risk loci have been identified through genome-wide association studies (GWAS) associated with the presence of IBD, highlighting some major disease-associated pathways [92]. Moreover, NOD2 has been identified as a major susceptibility gene [93, 94].
Two genes involved in autophagy, ATG16L1 and immunity-related GTPase M (IRGM), have been strongly associated with CD but not with UC, suggesting that autophagy is involved in the pathogenesis of CD [91]. A GWAS reported a single-nucleotide polymorphism (SNP) encoding a susceptibility variant of ATG16L1 gene (rs2241880, Thr300Ala) which is associated with a significant risk for CD [95, 96]. Since its discovery, SNP rs2241880 remained one of the most clinically important variants in CD and a large number of subsequent association studies have replicated the strong association of this genetic variation with CD [97,98,99,100]. The ATG16L1 Thr300Ala variant is associated with an excessive production of the pro-inflammatory cytokines IL-1β and IL-6 that drive the chronic inflammation observed in CD [101]. Moreover, this variant is more susceptible to cleavage by caspase-3, resulting in compromised clearance of Yersinia (Y.) enterocolitica and Salmonella (S.) typhimurium, as well as elevated cytokine production [102, 103].
IRGM has been demonstrated to induce autophagy and generate large autolysosomal organelles as a mechanism to inhibit the survival of intracellular Mycobacterium (M.) tuberculosis [104]. Recently, it has been reported that IRGM physically interacts with ULK1 and Beclin 1, promoting their assembly and thus controlling the arrangement of autophagy initiation complexes. In addition, IRGM forms a molecular complex with NOD2 and ATG16L1, modulating autophagic responses to pathogens [105]. A significant association has been reported between CD in various ethnic cohorts and sequence variants in the IRGM gene (SNPs rs13361189, rs4958847, and rs10065172) [100, 106,107,108,109]. The SNP rs13361189 was found to increase the risk of CD clinical sub-phenotypes such as ileal disease, perianal disease, and intestinal resection [110]. Interestingly, a gene-gene interaction analysis showed a significant two-way interaction between SNP rs2241880 (ATG16L1) and rs10065172 (IRGM), suggesting that ATG16L1 and IRGM work jointly toward CD pathogenesis [111].
Further insight into the role of ATG16L1 was obtained using genetically modified mice. Mice lacking Atg16L1 in hematopoietic cells revealed a strong susceptibility to acute colitis induced by dextran sulfate sodium, implying that Atg16L1 protects mice from intestinal inflammation [65]. Atg16L1 is also important in the biology of epithelial Paneth cells as Atg16L1-knockout Paneth cells demonstrated a defective granule exocytosis which might alter the intestinal microbiota [112]. However, despite the intense investigations of the IBD pathogenesis, the role of autophagy in eosinophils has not been addressed yet.
Bronchial asthma
Asthma is a heterogeneous disease characterized by chronic airway inflammation. Patients develop a variety of respiratory symptoms such as wheeze, breath shortness, chest tightness, cough, and expiratory airflow limitation [113]. Polymorphisms in ATG genes have suggested that a genetic predisposition may increase the chance to develop asthma. Since autophagy has been shown to regulate immune responses and inflammation, a possible association of genetic variants of ATG5 and ATG7 genes with childhood asthma was investigated. Two ATG5 SNPs, rs12201458 and rs510432, were significantly associated with asthma, the latter being functionally relevant by enhancing promotor activity [114].
Moreover, ATG5 gene expression was upregulated in nasal epithelial cells isolated from asthmatics with acute symptoms [114]. Similarly, the expression of ATG proteins (LC3-II, ATG4, ATG5-ATG12, ATG7) as well as the number of autophagic vacuoles was also increased in lung tissue from patients with chronic obstructive pulmonary disease (COPD) [115]. Another study investigated the potential association of SNPs in ATG genes (ULK1, SQSTM1, MAP1LC3B, BECN1, and ATG5) with asthma. SNP rs12212740 of ATG5 exhibited a positive association with asthma [116]. Examination of bronchial tissue from asthmatic patients demonstrated an increased number of autophagosomes in fibroblasts and epithelial cells compared with healthy individuals [116].
Similarly to structural cells, autophagy levels in sputum granulocytes, blood leukocytes, and blood eosinophils from patients with severe asthma were significantly increased as compared with subjects with non-severe asthma and healthy controls [117]. Interestingly, autophagy was induced in isolated blood eosinophils and human eosinophil-like (HL-60) cells in response to IL-5 treatment. To confirm that IL-5 induced autophagy rather than inhibited autophagosome degradation, inhibitors were used which blocked autolysosome degradation or fusion of autophagosome with lysosome [117]. These findings stimulated investigations in ovalbumin (OVA)-specific mouse model of allergic asthma [118]. OVA-challenged mice exhibited an increased expression of LC3-II in lung homogenates and a higher abundance of autophagosomes in cells of the bronchoalveolar lavage fluid (BALF), particularly in eosinophils. The eosinophil count in BALF also positively correlated with the LC3-II expression in lung homogenates, suggesting that autophagy is closely correlated with the severity of asthma as well as the eosinophilic inflammation. Inhibition of autophagy by intraperitoneal injection of 3-methyladenine (3-MA) and intranasal treatment with Atg5 shRNA led to a significantly improved airway hyperresponsiveness (AHR), reduced number of eosinophils and IL-5 levels in BALF, as well as improved histological inflammatory features [118]. However, it cannot be excluded that 3-MA also blocked cytokine signaling events in this model [119, 120]. Finally, intranasal administration of anti-IL-5 monoclonal antibody resulted in reduced LC3-II expression in lung homogenates, together with improved AHR and decreased eosinophil numbers in BALF [118]. Taken together, there is evidence that autophagy is induced in structural and inflammatory cells of the lungs in asthma, but it remains unclear how this phenomenon contributes to the pathogenesis of asthma. Therefore, it seems too early to propose novel therapeutic approaches for the treatment of asthma based on autophagy inhibition.
Chronic rhinosinusitis (CRS)
CRS is characterized by chronic inflammation of the sinonasal mucosa and clinically associated with sinus pressure, nasal congestion, and a decreased sense of smell persisting for more than 12 weeks [121]. CRS is often associated with pronounced eosinophil-dominant infiltration and inflammation and then classified as eosinophilic chronic rhinosinusitis (ECRS) [122].
The effect of autophagy on the development of ECRS was investigated in mice with a conditional knockout of Atg7 within myeloid cells (mainly neutrophils and macrophages), which was mediated using the LyzM-Cre (Lyz2-Cre) recombinase activity. An established mouse model of ECRS resulted in significantly increased eosinophil infiltration, epithelial hyperplasia, and mucosal thickening in Atg7flox/floxLyzM-Cre mice as compared with Atg7flox/flox mice, possibly owing to increased prostaglandin (PG) D2 production [123]. Interestingly, eosinophil infiltration and histological abnormalities were significantly improved following macrophage depletion in Atg7flox/floxLyzM-Cre mice with ECRS. The autophagy-deficient macrophages exacerbate the eosinophilic inflammation in ECRS, at least partially, through the release of elevated IL-1β levels. Therefore, results of this study suggest a protective role of macrophage autophagy on eosinophilic inflammation [123].
Eosinophilic esophagitis (EoE)
EoE is defined as a chronic, immune-mediated disorder resulting in esophageal dysfunction and eosinophil-predominant inflammation leading to tissue remodeling and fibrotic stricture [124]. A study performed on a pediatric patient cohort revealed upregulated ATG7 gene expression in esophageal biopsies from active EoE patients as compared with esophagus-healthy control individuals, EoE patients in remission and patients with gastroesophageal reflux disease (GERD) [125]. Therefore, ATG7 might be used as a valuable tissue biomarker of active EoE, and other ATG genes may be explored to potentially identify novel biomarkers for EoE diagnosis, monitoring, and prognosis. A recent study revealed a possible cytoprotective mechanism of autophagy which supports cellular redox balance and homeostasis following exposure to the inflammatory EoE environment, providing mechanistic insights into the role of autophagy in EoE pathogenesis [126]. Specifically, TNF-α and IL-13 have been identified as triggers of autophagy within the epithelium of the esophagus under in vitro conditions, including an esophageal organoid model. Inhibition of autophagic flux via chloroquine treatment augmented basal cell hyperplasia in these model systems. Moreover, this study has demonstrated increased autophagy in epithelial cells of the esophagus in EoE patients in vivo [126]. However, it should be noted that also this study did not investigated the role of autophagy in eosinophils.
Role of autophagy in eosinophil differentiation
Understanding the differentiation of eosinophils is crucial since many eosinophilic diseases are associated with increased production of eosinophils in the bone marrow. Eosinophils are continuously produced from eosinophil lineage-committed progenitors (EoPs) which derive from common myeloid progenitors (CMPs) in human [127] and granulocyte-monocyte progenitors (GMPs) in mice [128]. Terminally differentiated eosinophils are no longer mitotically active, and they are released into the circulation [129]. To keep cellular homeostasis, it is important that the differentiation process of eosinophils is tightly controlled [130].
Eosinophil differentiation is regulated by a complex network of transcription factors and extrinsic signals. During eosinopoiesis, a unique set of dynamic changes in the expression of transcription factors occurs, among which GATA-1, PU.1, and C/EBP members are the most critical ones [131, 132]. In addition, RhoH (small atypical GTPase) has been reported as a negative regulator of eosinophil differentiation, presumably due to dysregulated GATA-2 expression [133]. XBP1 has been identified as a highly selective and required transcription factor for eosinophil development [134]. It supports the survival of cells of the eosinophil lineage in a cell-intrinsic way while having no effect on basophils or neutrophils [134]. IL-5, IL-3, and granulocyte/macrophage colony-stimulating factor (GM-CSF) are recognized as the most relevant cytokines which stimulate eosinophil differentiation, activation, and survival [135]. A recent study implies IL-33 in the control of various stages of eosinophil differentiation mainly through the expansion of eosinophil precursors and upregulation of the IL-5 receptor α (IL-5Rα) on this population [136].
Until recently very little was known about the role of autophagy in eosinophils. Two studies investigated the involvement of mechanistic target of rapamycin (mTOR) on eosinophil hematopoiesis and asthma pathogenesis [137, 138]. The mTOR functions as a major nutrient-sensitive regulator of cell metabolism, balancing many anabolic and catabolic processes including protein synthesis and autophagy, respectively [44]. mTOR is a serine/threonine protein kinase that forms two distinct protein complexes, mTORC1 and mTORC2 [44]. Rapamycin selectively inhibits the activity of mTORC1, resulting in restricted eosinophil differentiation from mouse bone marrow cells and reduced cytokine production in vitro [137]. To examine the effect of rapamycin on allergic asthma, rapamycin was administered in a mouse model of OVA-induced allergic airway inflammation. Eosinophil numbers in BALF, peripheral blood, and bone marrow of rapamycin-treated mice were remarkably decreased, resulting in attenuated allergic airway inflammation and mucus production. However, rapamycin induced the accumulation of eosinophil progenitors in the bone marrow [137].
The same group subsequently investigated the function of mTOR in eosinophil differentiation and asthma pathogenesis using both genetic and pharmacological approaches. Treatment with torin-1, an inhibitor of mTORC1 and mTORC2, resulted in an enhanced in vitro eosinophil differentiation, as well as an increased size and number of eosinophil colony-forming units [138]. Similarly, Mtorflox/floxLyzM-Cre mice with a myeloid-specific knockout of mTOR exhibited an augmented production of eosinophil progenitors together with deteriorated allergic airway inflammation after OVA exposure [138]. Collectively, these data demonstrated the differential effects of mTOR in the regulation of eosinophil development, likely due to the distinct functions of mTORC1 and mTORC2 [138].
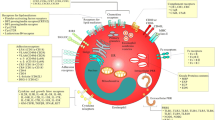
With the purpose of studying the developmental and functional consequences of autophagy deficiency in eosinophils, a novel mouse model with an eosinophil-specific knockout of Atg5 was generated (Atg5flox/floxeoCre mice) [120]. Cre recombinase is expressed only after commitment to the eosinophil lineage and is absolutely specific for eosinophils (eoCre mice) [22]. Results obtained from Atg5flox/floxeoCre mice showed elevated numbers of immature eosinophils in the bone marrow and a significant drop of mature eosinophils in the circulation [89] (Fig. 2). Knockout of Atg5 within eosinophils resulted in delayed and reduced eosinophil precursor proliferation and maturation under in vitro conditions. No abnormalities in cell death were observed in Atg5-knockout eosinophils. During in vitro eosinophil differentiation, a reduced phosphorylation of p38 and p44/42 mitogen-activated protein kinases (MAPKs) was detected in eosinophil precursors lacking Atg5, which might explain, at least partially, the observed phenotype. Eosinophil populations were further purified from the bone marrow of hypereosinophilic II5 (IL-5) transgenic mice (NJ.1638), showing downregulation of Gata-1, C/ebpε, Pu.1, and Trib1 transcription factors in the absence of Atg5, which might reflect the reduced and delayed eosinophil differentiation [89]. Moreover, the differentiation potential of Atg5-knockout eosinophil precursors was tested under pathologic conditions, employing an established mouse model of chronic eosinophilic leukemia (CEL). CEL has been initiated in mice by the combination of fusion protein FIP1L1-PDGFRα (F/P) expression and IL-5 overexpression [139], resulting in a less severe eosinophilia development in the absence of Atg5. Similar results were obtained in EoL-1 cells, a model of an established human CEL. Upon induced differentiation of EoL-1 cells, significantly lower levels of surface markers CD11b, Siglec-8 and CCR3 were observed in ATG5-knockout EoL-1 cells, indicating decreased maturation of eosinophil precursors [89] (Fig. 2). These observations suggest that targeting ATG5 within the eosinophil lineage might represent a possible future treatment of eosinophilic leukemia.

The role of ATG5 in eosinophil differentiation and effector functions. Atg5-knockout eosinophil precursors with suppressed autophagy exhibit a delayed and reduced proliferation, maturation, p38 and p44/42 MAPK activation, and a reduced expression of Gata-1, C/ebpε, Pu.1, and Trib1 transcription factors (TFs). A decrease in eosinophil differentiation results in reduced numbers of mature eosinophils in blood and peripheral tissues. The differentiation capacity of eosinophil precursors in the absence of Atg5/ATG5 is reduced in established mouse (FIP1L1-PDGFRα; F/P mice) and human eosinophil leukemia models (EoL-1 cells). Moreover, Atg5-knockout eosinophils exhibit enhanced degranulation, EET formation, bacterial killing, and signaling transduction following their activation in vitro. Mice with Atg5-knockout eosinophils have been shown to better clear a bacterial infection with C. rodentium. ATG5low-expressing human eosinophils demonstrate enhanced degranulation abilities in both tissues and blood
These findings were in contrast with the effect of Atg5-deficiency on neutrophil differentiation in Atg5flox/floxLyzM-Cre mice [86]. These mice exhibited an increased proliferation and differentiation of Atg5-knockout neutrophils, culminating in neutrophil accumulation in the circulation and lymphoid organs. Accelerated neutrophil differentiation was also observed upon shRNA-mediated Atg5 knockdown in Hoxb8 neutrophils [86]. Furthermore, autophagy was studied in early granulopoiesis in mice with a conditional deletion of Atg7 at the hematopoietic stem and progenitor level (Atg7flox/floxVav-Cre), which caused accumulation of immature neutrophils in the spleen, blood, and peritoneum [87]. An expanded population of immature myeloblasts and myelocyte precursors was also observed in Atg7flox/floxCebpa-Cre mice, expressing C/ebpα promotor predominantly at the GMP stage. Atg7-knockout neutrophil precursors were unable to shift from glycolytic activity toward mitochondrial respiration, demonstrating accumulation of lipid bodies and decreased ATP production. Inhibition of autophagy-mediated lipid degradation failed to provide free fatty acids to support mitochondrial respiration and ATP production, resulting in a defective neutrophil differentiation [87].
Interestingly, it has been reported that the p38 MAPK activity differentially regulates eosinophil and neutrophil differentiation, potentially through the modulation of C/EBPα transcriptional activity [140]. A recent study showed that Trib1 expression favors eosinophil development by restraining neutrophil lineage commitment by modulating C/EBPα, partly clarifying the regulation of granulocytic lineage selection and identity [141]. In addition, single-cell transcriptome analysis has determined two different myeloid progenitor subsets which separate early in the hematopoietic development. Subsets can be differentiated according to the presence (eosinophils, mast cells, megakaryocytes, erythrocytes) or absence (neutrophils, lymphocytes, monocytes) of Gata1 expression [142]. The potential involvement of autophagy in the segregation and regulation of these two distinct myeloid progenitor-differentiation pathways has not been established yet and could be the subject for future studies.
Role of autophagy in eosinophil effector functions
Activated eosinophils exert their effector functions mainly through degranulation and formation of eosinophil extracellular traps (EETs). While both degranulation and EETs are important innate immune effector functions against pathogens, they can also cause significant immunopathologies [14].
Degranulation
Eosinophil specific granules are rich in four major cationic proteins: major basic protein (MBP), eosinophil peroxidase (EPX), and the ribonucleases eosinophil cationic protein (ECP) and eosinophil-derived neurotoxin (EDN) [143]. Eosinophils release granule contents into the extracellular space through three commonly observed pathways, namely, piecemeal degranulation (PMD), exocytosis, or cytolysis [17]. During PMD, the specific granules are progressively emptied and eosinophil sombrero vesicles (EoSVs) transfer the selected secretory cargo to plasma membrane [144, 145]. Granules can release their entire contents following granule fusion with the plasma membrane in a process called exocytosis, which can be preceded by intracellular granule-granule fusion [146]. Eosinophil cytolysis is characterized by the disintegration of cytoplasmic membrane and release of nuclear DNA cloud together with intact granules, which are deposited in tissues upon cell lysis [147]. It has been demonstrated that eosinophil cytolysis is dependent on receptor-interacting protein kinase 3 (RIPK3)-mixed lineage kinase-like (MLKL) signaling pathway and can be counterregulated by autophagy induction, perhaps opening up new ways for therapeutic interventions [148]. PMD and cytolysis have been frequently reported to be associated with eosinophilic diseases such as asthma, nasal polyps, IBD, allergic rhinitis, and EoE [149,150,151]. On the contrary, exocytosis is rarely documented during inflammatory responses, but has been observed during the interaction of eosinophils with helminths [152] and certain environmental fungi [153].
The effect of autophagy on eosinophil degranulation has been studied in Atg5-knockout eosinophils, which intriguingly exhibited an enhanced capacity to degranulate in vitro as measured by increased CD63 surface expression following GM-CSF priming and C5a stimulation [89] (Fig. 2). Moreover, an experimental in vivo mouse model of bacterial infection with Citrobacter (C.) rodentium was used to test the infiltrating colonic eosinophils for their activation and degranulation status. Eosinophils lacking Atg5 exhibited higher Siglec-F and CD63 surface expression levels, supporting the in vitro observations [89]. In addition, the expression of ATG5 in human eosinophils was analyzed together with their degranulation status in human eosinophilic tissues. Patients with angiolymphoid hyperplasia, EoE, and sebaceous gland carcinoma demonstrated a positive correlation between ATG5 expression and intracellular EPX levels in tissue eosinophils, suggesting increased degranulation in ATG5low-expressing eosinophils. These data were supported by observations in hypereosinophilic syndrome (HES) patients, in which a significant negative correlation between ATG5 (and ATG7) mRNA expression in blood eosinophils and secreted EDN levels in plasma was found [89] (Fig. 2). These data supported the concept that Atg5-knockout eosinophils in mice and ATG5low-expressing human eosinophils are more susceptible to degranulation.
In contrast to eosinophils, autophagy was reported to be crucial for the degranulation of mouse neutrophils [88] and mast cells [85]. Mice with autophagy deficiency in myeloid cell lineage (Atg7flox/floxLyzM-Cre) showed reduced severity of several neutrophil-mediated inflammatory and autoimmune disease models, including PMA-induced ear inflammation, LPS-induced breakdown of blood-brain barrier, and experimental autoimmune encephalomyelitis. The most likely mechanism was suggested to be a reduced NADPH oxidase-mediated production of reactive oxygen species (ROS) in Atg7-knockout neutrophils [88]. These findings suggest that autophagy has differential effects on eosinophil and neutrophil degranulation. It has also been reported that in contrast to neutrophils, which are absolutely required for antibacterial defense, pharmacological or genetic ablation of eosinophils does not result in obvious functional consequences [16].
Extracellular trap (ET) formation
Eosinophils are able to form ETs which consist of mitochondrial DNA and cationic granule proteins released from activated cells [154]. EETs perform antibacterial functions, and they enable the accumulation of toxic granule proteins directly onto pathogens captured in the DNA scaffold, limiting the damage of surrounding host tissues [155]. The formation of EETs has been demonstrated in various infectious, allergic, and autoimmune eosinophilic disorders [154, 156,157,158,159].
In addition to eosinophils, activated neutrophils are also able to form similar extracellular DNA structures, known as neutrophil extracellular traps (NETs) [160, 161]. Interestingly, the release of mitochondrial DNA does not require cell death neither does it limit the viability of the granulocytes [161]. On the other hand, neutrophil death-dependent mechanisms have also been described, and the scientific dispute regarding the requirement of cell death for NET formation is ongoing [155]. It has been demonstrated that NET formation by viable neutrophils depends on the activity of NADPH oxidase, cytoskeleton rearrangements, and glycolytic ATP production [162, 163].
A recent study reported the requirement for autophagy in the formation of EETs in the airway of asthmatic mice. Treatment with the autophagy inhibitor 3-MA attenuated EET formation and improved the lung inflammation, mitochondrial metabolism, and oxidative stress in OVA-challenged mice [164]. Moreover, treatment of neutrophils with autophagy inhibitor wortmannin reduced NET formation by activated neutrophils [165, 166]. 3-MA and wortmannin have been widely used as autophagy inhibitors based on their inhibitory effect on class III PI3K activity, which is known to be essential for autophagy induction [50]. The same inhibitors, however, are also reported to block class I PI3Ks which contribute to the activation of NADPH oxidase [167,168,169]. Therefore, decreased formation of EETs following 3-MA and wortmannin treatment can also be explained by inhibition of ROS production. A recent study has investigated this issue in details, and the reported results suggest that PI3K inhibitors, such as 3-MA and wortmannin, block both EET and NET formation in an autophagy-independent manner [120]. In addition, both Atg5-knockout eosinophils and Atg5-knockout neutrophils were fully capable to release extracellular DNA after stimulation, demonstrating that autophagy is not required for both EET and NET formation [120]. A careful quantitative analysis revealed that Atg5-knockout eosinophils exhibit an even increased ability of EET formation compared with control eosinophils in vitro, supporting the surprising findings of augmented degranulation in eosinophils lacking Atg5 [89]. Eosinophils lacking Atg5 also demonstrated elevated in vitro bacterial killing of Escherichia (E.) coli, suggesting increased effector function of eosinophils in the absence of Atg5. Moreover, the antibacterial defense of Atg5-knockout eosinophils was tested under in vivo conditions in the C. rodentium model. An improved local and systemic clearance of C. rodentium was observed in Atg5flox/floxeoCre mice, together with an enhanced ability to form EETs [89]. Atg5-knockout eosinophils demonstrated an increased activity of Stat3, p38, and p44/42 signaling pathways following cytokine stimulation, providing a possible explanation for enhanced eosinophil effector functions in the absence of Atg5 [89].
Tumor-associated tissue eosinophilia is often observed in cancer patients and studies suggest their involvement in tumoricidal activities. Upon interaction with a colorectal carcinoma cell line Colo-205, eosinophils released their granule contents such as ECP, EDN, TNF-α, and granzyme A, which exerted cytotoxic responses against tumor cells [170]. Moreover, ablation of eosinophils severely compromised antitumor immunity in a colorectal cancer (CRC) mouse model, most likely owing to impaired Th1 and CD8+ T cell responses. On the other hand, CRC patients with enhanced eosinophil tumor infiltration demonstrated robust CD8 T cell infiltrates, resulting in a better prognosis compared with patients with low-eosinophil infiltrating tumors [171]. Interestingly, the blockade of autophagy enhanced T helper 9 (Th9) cell anticancer functions in vivo, and mice with T cell-specific deletion of Atg5 exhibited reduced tumor growth in an IL-9-dependent manner [172]. Finally, it would be interesting to investigate the functional consequences of Atg5-knockout eosinophils in different disease models and explore their potential for cancer immunotherapy.
Concluding remarks
The findings summarized in this review article highlight the autophagic pathway as a protective mechanism of cells which contributes to the limitation of disease severity in eosinophilic diseases. Autophagy seems to secure the function of parenchymal cells under inflammatory conditions. This process appears to be particularly important in epithelial cells to maintain their barrier function. The generation of conditional/promoter-specific knockout mice has enabled researcher to investigate the role of autophagy in a cell-type specific manner. Recently, such experimental models were also developed to study the cell-type inherent function of autophagy in eosinophils. Surprisingly, and in contrast to other immune cells, eosinophils enhance effector functions when autophagy is impaired. Therefore, drug-induced inhibition of autophagy in chronic inflammatory eosinophilic diseases does not seem to be indicated because of two possible unwanted effects: (1) reduced epithelial barrier function and (2) increased eosinophil-mediated immunopathology. In contrast, however, blocking the autophagic pathway in eosinophils might be beneficial in eosinophilic tumors. Therefore, pharmacological impairment of autophagy might also be an option for the therapy of eosinophilic leukemias. Clearly, additional experimental work, including the analyses of human cells and tissues, is required to identify drug targets suitable for the modulation of the autophagic pathway as preventive or therapeutic intervention in eosinophilic diseases and other human pathologies.
References
Radonjic-Hosli S, Simon HU (2014) Eosinophils. Chem Immunol Allergy 100:193–204
Rosenberg HF, Dyer KD, Foster PS (2013) Eosinophils: changing perspectives in health and disease. Nat Rev Immunol 13:9–22
Bochner BS (2018) The eosinophil: For better or worse, in sickness and in health. Ann Allergy Asthma Immunol 121:150–155
Shah K, Ignacio A, McCoy KD, Harris NL (2020) The emerging roles of eosinophils in mucosal homeostasis. Mucosal Immunol 13:574–583
Mesnil C, Raulier S, Paulissen G, Xiao X, Birrell MA, Pirottin D, Janss T, Starkl P, Ramery E, Henket M, Schleich FN, Radermecker M, Thielemans K, Gillet L, Thiry M, Belvisi MG, Louis R, Desmet C, Marichal T, Bureau F (2016) Lung-resident eosinophils represent a distinct regulatory eosinophil subset. J Clin Invest 126:3279–3295
Simon HU, Yousefi S, Schranz C, Schapowal A, Bachert C, Blaser K (1997) Direct demonstration of delayed eosinophil apoptosis as a mechanism causing tissue eosinophilia. J Immunol 158:3902–3908
Weller PF, Spencer LA (2017) Functions of tissue-resident eosinophils. Nat Rev Immunol 17:746–760
Simon HU, Rothenberg ME, Bochner BS, Weller PF, Wardlaw AJ, Wechsler ME, Rosenwasser LJ, Roufosse F, Gleich GJ, Klion AD (2010) Refining the definition of hypereosinophilic syndrome. J Allergy Clin Immunol 126:45–49
Simon D, Simon HU (2007) Eosinophilic disorders. J Allergy Clin Immunol 119:1291–1300
Shamri R, Xenakis JJ, Spencer LA (2011) Eosinophils in innate immunity: an evolving story. Cell Tissue Res 343:57–83
Melo RCN, Weller PF (2018) Contemporary understanding of the secretory granules in human eosinophils. J Leukoc Biol 104:85–93
Davoine F, Lacy P (2014) Eosinophil cytokines, chemokines, and growth factors: emerging roles in immunity. Front Immunol 5:570
Spencer LA, Bonjour K, Melo RC, Weller PF (2014) Eosinophil secretion of granule-derived cytokines. Front Immunol 5:496
Acharya KR, Ackerman SJ (2014) Eosinophil granule proteins: form and function. J Biol Chem 289:17406–17415
Melo RC, Weller PF (2014) Unraveling the complexity of lipid body organelles in human eosinophils. J Leukoc Biol 96:703–712
Gleich GJ, Klion AD, Lee JJ, Weller PF (2013) The consequences of not having eosinophils. Allergy 68:829–835
Simon HU, Yousefi S, Germic N, Arnold IC, Haczku A, Karaulov AV, Simon D, Rosenberg HF (2020) The cellular functions of eosinophils: Collegium Internationale Allergologicum (CIA) update 2020. Int Arch Allergy Immunol 181:11–23
Yu C, Cantor AB, Yang H, Browne C, Wells RA, Fujiwara Y, Orkin SH (2002) Targeted deletion of a high-affinity GATA-binding site in the GATA-1 promoter leads to selective loss of the eosinophil lineage in vivo. J Exp Med 195:1387–1395
Lee JJ, Dimina D, Macias MP, Ochkur SI, McGarry MP, O'Neill KR, Protheroe C, Pero R, Nguyen T, Cormier SA, Lenkiewicz E, Colbert D, Rinaldi L, Ackerman SJ, Irvin CG, Lee NA (2004) Defining a link with asthma in mice congenitally deficient in eosinophils. Science 305:1773–1776
Dent LA, Strath M, Mellor AL, Sanderson CJ (1990) Eosinophilia in transgenic mice expressing interleukin 5. J Exp Med 172:1425–1431
Lee NA, McGarry MP, Larson KA, Horton MA, Kristensen AB, Lee JJ (1997) Expression of IL-5 in thymocytes/T cells leads to the development of a massive eosinophilia, extramedullary eosinophilopoiesis, and unique histopathologies. J Immunol 158:1332–1344
Doyle AD, Jacobsen EA, Ochkur SI, Willetts L, Shim K, Neely J, Kloeber J, Lesuer WE, Pero RS, Lacy P, Moqbel R, Lee NA, Lee JJ (2013) Homologous recombination into the eosinophil peroxidase locus generates a strain of mice expressing Cre recombinase exclusively in eosinophils. J Leukoc Biol 94:17–24
Radonjic-Hoesli S, Valent P, Klion AD, Wechsler ME, Simon HU (2015) Novel targeted therapies for eosinophil-associated diseases and allergy. Annu Rev Pharmacol Toxicol 55:633–656
Feng Y, He D, Yao Z, Klionsky DJ (2014) The machinery of macroautophagy. Cell Res 24:24–41
Morishita H, Mizushima N (2019) Diverse cellular roles of autophagy. Annu Rev Cell Dev Biol 35:453–475
Kroemer G, Marino G, Levine B (2010) Autophagy and the integrated stress response. Mol Cell 40:280–293
Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19:349–364
Arroyo DS, Gaviglio EA, Peralta Ramos JM, Bussi C, Rodriguez-Galan MC, Iribarren P (2014) Autophagy in inflammation, infection, neurodegeneration and cancer. Int Immunopharmacol 18:55–65
Matsuzawa-Ishimoto Y, Hwang S, Cadwell K (2018) Autophagy and inflammation. Annu Rev Immunol 36:73–101
Yang Y, Klionsky DJ (2020) Autophagy and disease: unanswered questions. Cell Death Differ 27:858–871
Deretic V, Kimura T, Timmins G, Moseley P, Chauhan S, Mandell M (2015) Immunologic manifestations of autophagy. J Clin Invest 125:75–84
Shibutani ST, Saitoh T, Nowag H, Munz C, Yoshimori T (2015) Autophagy and autophagy-related proteins in the immune system. Nat Immunol 16:1014–1024
Levine B, Mizushima N, Virgin HW (2011) Autophagy in immunity and inflammation. Nature 469:323–335
Deretic V, Saitoh T, Akira S (2013) Autophagy in infection, inflammation and immunity. Nat Rev Immunol 13:722–737
Germic N, Frangez Z, Yousefi S, Simon HU (2019) Regulation of the innate immune system by autophagy: neutrophils, eosinophils, mast cells, NK cells. Cell Death Differ 26:703–714
Kuballa P, Nolte WM, Castoreno AB, Xavier RJ (2012) Autophagy and the immune system. Annu Rev Immunol 30:611–646
Clarke AJ, Simon AK (2019) Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat Rev Immunol 19:170–183
Karanasios E, Walker SA, Okkenhaug H, Manifava M, Hummel E, Zimmermann H, Ahmed Q, Domart MC, Collinson L, Ktistakis NT (2016) Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat Commun 7:12420
Simon HU, Friis R, Tait SW, Ryan KM (2017) Retrograde signaling from autophagy modulates stress responses. Sci Signal 10:2791
Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL (2013) ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 15:741–750
Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27:107–132
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19:5720–5728
Choi AM, Ryter SW, Levine B (2013) Autophagy in human health and disease. N Engl J Med 368:1845–1846
Saxton RA, Sabatini DM (2017) mTOR signaling in growth, metabolism, and disease. Cell 168:960–976
Gonzalez A, Hall MN (2017) Nutrient sensing and TOR signaling in yeast and mammals. EMBO J 36:397–408
Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320:1496–1501
Sanli T, Steinberg GR, Singh G, Tsakiridis T (2014) AMP-activated protein kinase (AMPK) beyond metabolism: a novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol Ther 15:156–169
Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30:214–226
Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132–141
Yu X, Long YC, Shen HM (2015) Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy 11:1711–1728
Anding AL, Baehrecke EH (2017) Cleaning house: selective autophagy of organelles. Dev Cell 41:10–22
Stolz A, Ernst A, Dikic I (2014) Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16:495–501
Zhao YG, Zhang H (2019) Autophagosome maturation: an epic journey from the ER to lysosomes. J Cell Biol 218:757–770
Levine B, Kroemer G (2019) Biological functions of autophagy genes: a disease perspective. Cell 176:11–42
Galluzzi L, Green DR (2019) Autophagy-independent functions of the autophagy machinery. Cell 177:1682–1699
Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, Hamada S, Yoshimori T (2004) Autophagy defends cells against invading group A Streptococcus. Science 306:1037–1040
Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V (2004) Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119:753–766
Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C (2005) Escape of intracellular Shigella from autophagy. Science 307:727–731
Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH (2006) Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem 281:11374–11383
Py BF, Lipinski MM, Yuan J (2007) Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy 3:117–125
Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, Mimuro H, Nakagawa I, Yanagawa T, Ishii T, Kakizuka A, Sztul E, Chakraborty T, Sasakawa C (2009) Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol 11:1233–1240
Kimmey JM, Stallings CL (2016) Bacterial pathogens versus autophagy: implications for therapeutic interventions. Trends Mol Med 22:1060–1076
Fujita N, Morita E, Itoh T, Tanaka A, Nakaoka M, Osada Y, Umemoto T, Saitoh T, Nakatogawa H, Kobayashi S, Haraguchi T, Guan JL, Iwai K, Tokunaga F, Saito K, Ishibashi K, Akira S, Fukuda M, Noda T, Yoshimori T (2013) Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J Cell Biol 203:115–128
Ponpuak M, Davis AS, Roberts EA, Delgado MA, Dinkins C, Zhao Z, Virgin HW, Kyei GB, Johansen T, Vergne I, Deretic V (2010) Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity 32:329–341
Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456:264–268
Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, Kornfeld H, Fitzgerald KA, Lavelle EC (2011) Autophagy controls IL-1β secretion by targeting pro-IL-1β for degradation. J Biol Chem 286:9587–9597
Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12:222–230
Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH (2012) Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 13:255–263
Pu Q, Gan C, Li R, Li Y, Tan S, Li X, Wei Y, Lan L, Deng X, Liang H, Ma F, Wu M (2017) Atg7 deficiency intensifies inflammasome activation and pyroptosis in pseudomonas sepsis. J Immunol 198:3205–3213
O'Sullivan TE, Geary CD, Weizman OE, Geiger TL, Rapp M, Dorn GW 2nd, Overholtzer M, Sun JC (2016) Atg5 is essential for the development and survival of innate lymphocytes. Cell Rep 15:1910–1919
Wang S, Xia P, Huang G, Zhu P, Liu J, Ye B, Du Y, Fan Z (2016) FoxO1-mediated autophagy is required for NK cell development and innate immunity. Nat Commun 7:11023
Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, Mizushima N, Grinstein S, Iwasaki A (2010) In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 32:227–239
Schmid D, Pypaert M, Munz C (2007) Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 26:79–92
Loi M, Muller A, Steinbach K, Niven J, Barreira da Silva R, Paul P, Ligeon LA, Caruso A, Albrecht RA, Becker AC, Annaheim N, Nowag H, Dengjel J, Garcia-Sastre A, Merkler D, Munz C, Gannage M (2016) Macroautophagy proteins control MHC class I levels on dendritic cells and shape anti-viral CD8+ T cell responses. Cell Rep 15:1076–1087
Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW (2007) A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med 204:25–31
Kovacs JR, Li C, Yang Q, Li G, Garcia IG, Ju S, Roodman DG, Windle JJ, Zhang X, Lu B (2012) Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ 19:144–152
Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, Konieczny BT, Bruinsma MW, Martinez J, Pearce EL, Green DR, Jones DP, Virgin HW, Ahmed R (2014) Autophagy is essential for effector CD8+ T cell survival and memory formation. Nat Immunol 15:1152–1161
Jia W, He MX, McLeod IX, Guo J, Ji D, He YW (2015) Autophagy regulates T lymphocyte proliferation through selective degradation of the cell-cycle inhibitor CDKN1B/p27Kip1. Autophagy 11:2335–2345
Wei J, Long L, Yang K, Guy C, Shrestha S, Chen Z, Wu C, Vogel P, Neale G, Green DR, Chi H (2016) Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol 17:277–285
Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK, Mizushima NN, Iwasaki A, He YW, Swat W, Virgin HW (2008) The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy 4:309–314
Conway KL, Kuballa P, Khor B, Zhang M, Shi HN, Virgin HW, Xavier RJ (2013) ATG5 regulates plasma cell differentiation. Autophagy 9:528–537
Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, Fagioli C, Merlini A, Mariani E, Pasqualetto E, Orfanelli U, Ponzoni M, Sitia R, Casola S, Cenci S (2013) Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol 14:298–305
Martinez-Martin N, Maldonado P, Gasparrini F, Frederico B, Aggarwal S, Gaya M, Tsui C, Burbage M, Keppler SJ, Montaner B, Jefferies HB, Nair U, Zhao YG, Domart MC, Collinson L, Bruckbauer A, Tooze SA, Batista FD (2017) A switch from canonical to noncanonical autophagy shapes B cell responses. Science 355:641–647
Clarke AJ, Riffelmacher T, Braas D, Cornall RJ, Simon AK (2018) B1a B cells require autophagy for metabolic homeostasis and self-renewal. J Exp Med 215:399–413
Ushio H, Ueno T, Kojima Y, Komatsu M, Tanaka S, Yamamoto A, Ichimura Y, Ezaki J, Nishida K, Komazawa-Sakon S, Niyonsaba F, Ishii T, Yanagawa T, Kominami E, Ogawa H, Okumura K, Nakano H (2011) Crucial role for autophagy in degranulation of mast cells. J Allergy Clin Immunol 127:1267–1276
Rozman S, Yousefi S, Oberson K, Kaufmann T, Benarafa C, Simon HU (2015) The generation of neutrophils in the bone marrow is controlled by autophagy. Cell Death Differ 22:445–456
Riffelmacher T, Clarke A, Richter FC, Stranks A, Pandey S, Danielli S, Hublitz P, Yu Z, Johnson E, Schwerd T, McCullagh J, Uhlig H, Jacobsen SEW, Simon AK (2017) Autophagy-dependent generation of free fatty acids is critical for normal neutrophil differentiation. Immunity 47:466–480
Bhattacharya A, Wei Q, Shin JN, Abdel Fattah E, Bonilla DL, Xiang Q, Eissa NT (2015) Autophagy is required for neutrophil-mediated inflammation. Cell Rep 12:1731–1739
Germic N, Hosseini A, Stojkov D, Oberson K, Claus M, Benarafa C, Calzavarini S, Angelillo-Scherrer A, Arnold IC, Muller A, Riether C, Yousefi S, Simon HU (2021) ATG5 promotes eosinopoiesis, but inhibits eosinophil effector functions. Blood. https://doi.org/10.1182/blood.2020010208
Abraham C, Cho JH (2009) Inflammatory bowel disease. N Engl J Med 361:2066–2078
de Souza HS, Fiocchi C (2016) Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol 13:13–27
Mirkov MU, Verstockt B, Cleynen I (2017) Genetics of inflammatory bowel disease: beyond NOD2. Lancet Gastroenterol Hepatol 2:224–234
Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O'Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel JF, Sahbatou M, Thomas G (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 411:599–603
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nunez G, Cho JH (2001) A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 411:603–606
Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, Gunther S, Prescott NJ, Onnie CM, Hasler R, Sipos B, Folsch UR, Lengauer T, Platzer M, Mathew CG, Krawczak M, Schreiber S (2007) A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet 39:207–211
Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, Shugart YY, Griffiths AM, Targan SR, Ippoliti AF, Bernard EJ, Mei L, Nicolae DL, Regueiro M, Schumm LP, Steinhart AH, Rotter JI, Duerr RH, Cho JH, Daly MJ, Brant SR (2007) Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 39:596–604
Baldassano RN, Bradfield JP, Monos DS, Kim CE, Glessner JT, Casalunovo T, Frackelton EC, Otieno FG, Kanterakis S, Shaner JL, Smith RM, Eckert AW, Robinson LJ, Onyiah CC, Abrams DJ, Chiavacci RM, Skraban R, Devoto M, Grant SF, Hakonarson H (2007) Association of the T300A non-synonymous variant of the ATG16L1 gene with susceptibility to paediatric Crohn's disease. Gut 56:1171–1173
Cummings JR, Cooney R, Pathan S, Anderson CA, Barrett JC, Beckly J, Geremia A, Hancock L, Guo C, Ahmad T, Cardon LR, Jewell DP (2007) Confirmation of the role of ATG16L1 as a Crohn's disease susceptibility gene. Inflamm Bowel Dis 13:941–946
Glas J, Konrad A, Schmechel S, Dambacher J, Seiderer J, Schroff F, Wetzke M, Roeske D, Torok HP, Tonenchi L, Pfennig S, Haller D, Griga T, Klein W, Epplen JT, Folwaczny C, Lohse P, Goke B, Ochsenkuhn T, Mussack T, Folwaczny M, Muller-Myhsok B, Brand S (2008) The ATG16L1 gene variants rs2241879 and rs2241880 (T300A) are strongly associated with susceptibility to Crohn's disease in the German population. Am J Gastroenterol 103:682–691
Palomino-Morales RJ, Oliver J, Gomez-Garcia M, Lopez-Nevot MA, Rodrigo L, Nieto A, Alizadeh BZ, Martin J (2009) Association of ATG16L1 and IRGM genes polymorphisms with inflammatory bowel disease: a meta-analysis approach. Genes Immun 10:356–364
Plantinga TS, Crisan TO, Oosting M, van de Veerdonk FL, de Jong DJ, Philpott DJ, van der Meer JW, Girardin SE, Joosten LA, Netea MG (2011) Crohn's disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut 60:1229–1235
Murthy A, Li Y, Peng I, Reichelt M, Katakam AK, Noubade R, Roose-Girma M, DeVoss J, Diehl L, Graham RR, van Lookeren CM (2014) A Crohn's disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 506:456–462
Lassen KG, Kuballa P, Conway KL, Patel KK, Becker CE, Peloquin JM, Villablanca EJ, Norman JM, Liu TC, Heath RJ, Becker ML, Fagbami L, Horn H, Mercer J, Yilmaz OH, Jaffe JD, Shamji AF, Bhan AK, Carr SA, Daly MJ, Virgin HW, Schreiber SL, Stappenbeck TS, Xavier RJ (2014) Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U S A 111:7741–7746
Singh SB, Davis AS, Taylor GA, Deretic V (2006) Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313:1438–1441
Chauhan S, Mandell MA, Deretic V (2015) IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol Cell 58:507–521
Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, Roberts RG, Nimmo ER, Cummings FR, Soars D, Drummond H, Lees CW, Khawaja SA, Bagnall R, Burke DA, Todhunter CE, Ahmad T, Onnie CM, McArdle W, Strachan D, Bethel G, Bryan C, Lewis CM, Deloukas P, Forbes A, Sanderson J, Jewell DP, Satsangi J, Mansfield JC, Wellcome Trust Case Control C, Cardon L, Mathew CG (2007) Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet 39:830–832
McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, Zody MC, Hall JL, Brant SR, Cho JH, Duerr RH, Silverberg MS, Taylor KD, Rioux JD, Altshuler D, Daly MJ, Xavier RJ (2008) Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nat Genet 40:1107–1112
Roberts RL, Hollis-Moffatt JE, Gearry RB, Kennedy MA, Barclay ML, Merriman TR (2008) Confirmation of association of IRGM and NCF4 with ileal Crohn's disease in a population-based cohort. Genes Immun 9:561–565
Weersma RK, Stokkers PC, Cleynen I, Wolfkamp SC, Henckaerts L, Schreiber S, Dijkstra G, Franke A, Nolte IM, Rutgeerts P, Wijmenga C, Vermeire S (2009) Confirmation of multiple Crohn's disease susceptibility loci in a large Dutch-Belgian cohort. Am J Gastroenterol 104:630–638
Rufini S, Ciccacci C, Di Fusco D, Ruffa A, Pallone F, Novelli G, Biancone L, Borgiani P (2015) Autophagy and inflammatory bowel disease: Association between variants of the autophagy-related IRGM gene and susceptibility to Crohn's disease. Dig Liver Dis 47:744–750
Hoefkens E, Nys K, John JM, Van Steen K, Arijs I, Van der Goten J, Van Assche G, Agostinis P, Rutgeerts P, Vermeire S, Cleynen I (2013) Genetic association and functional role of Crohn disease risk alleles involved in microbial sensing, autophagy, and endoplasmic reticulum (ER) stress. Autophagy 9:2046–2055
Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, Stone CD, Brunt EM, Xavier RJ, Sleckman BP, Li E, Mizushima N, Stappenbeck TS, Virgin HW (2008) A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456:259–263
Pavord ID, Beasley R, Agusti A, Anderson GP, Bel E, Brusselle G, Cullinan P, Custovic A, Ducharme FM, Fahy JV, Frey U, Gibson P, Heaney LG, Holt PG, Humbert M, Lloyd CM, Marks G, Martinez FD, Sly PD, von Mutius E, Wenzel S, Zar HJ, Bush A (2018) After asthma: redefining airways diseases. Lancet 391:350–400
Martin LJ, Gupta J, Jyothula SS, Butsch Kovacic M, Biagini Myers JM, Patterson TL, Ericksen MB, He H, Gibson AM, Baye TM, Amirisetty S, Tsoras AM, Sha Y, Eissa NT, Hershey GK (2012) Functional variant in the autophagy-related 5 gene promotor is associated with childhood asthma. PLoS One 7:33454
Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ, Yousem SA, Nakahira K, Pilewski JM, Lee JS, Zhang Y, Ryter SW, Choi AM (2008) Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One 3:3316
Poon AH, Chouiali F, Tse SM, Litonjua AA, Hussain SN, Baglole CJ, Eidelman DH, Olivenstein R, Martin JG, Weiss ST, Hamid Q, Laprise C (2012) Genetic and histologic evidence for autophagy in asthma pathogenesis. J Allergy Clin Immunol 129:569–571
Ban GY, Pham DL, Trinh TH, Lee SI, Suh DH, Yang EM, Ye YM, Shin YS, Chwae YJ, Park HS (2016) Autophagy mechanisms in sputum and peripheral blood cells of patients with severe asthma: a new therapeutic target. Clin Exp Allergy 46:48–59
Liu JN, Suh DH, Trinh HK, Chwae YJ, Park HS, Shin YS (2016) The role of autophagy in allergic inflammation: a new target for severe asthma. Exp Mol Med 48:243
Lin YC, Kuo HC, Wang JS, Lin WW (2012) Regulation of inflammatory response by 3-methyladenine involves the coordinative actions on Akt and glycogen synthase kinase 3β rather than autophagy. J Immunol 189:4154–4164
Germic N, Stojkov D, Oberson K, Yousefi S, Simon HU (2017) Neither eosinophils nor neutrophils require ATG5-dependent autophagy for extracellular DNA trap formation. Immunology 152:517–525
Stevens WW, Lee RJ, Schleimer RP, Cohen NA (2015) Chronic rhinosinusitis pathogenesis. J Allergy Clin Immunol 136:1442–1453
Fujieda S, Imoto Y, Kato Y, Ninomiya T, Tokunaga T, Tsutsumiuchi T, Yoshida K, Kidoguchi M, Takabayashi T (2019) Eosinophilic chronic rhinosinusitis. Allergol Int 68:403–412
Choi GE, Yoon SY, Kim JY, Kang DY, Jang YJ, Kim HS (2018) Autophagy deficiency in myeloid cells exacerbates eosinophilic inflammation in chronic rhinosinusitis. J Allergy Clin Immunol 141:938–950
Liacouras CA, Furuta GT, Hirano I, Atkins D, Attwood SE, Bonis PA, Burks AW, Chehade M, Collins MH, Dellon ES, Dohil R, Falk GW, Gonsalves N, Gupta SK, Katzka DA, Lucendo AJ, Markowitz JE, Noel RJ, Odze RD, Putnam PE, Richter JE, Romero Y, Ruchelli E, Sampson HA, Schoepfer A, Shaheen NJ, Sicherer SH, Spechler S, Spergel JM, Straumann A, Wershil BK, Rothenberg ME, Aceves SS (2011) Eosinophilic esophagitis: updated consensus recommendations for children and adults. J Allergy Clin Immunol 128:3–20
Merves JF, Whelan KA, Benitez AJ, Muir AB, Furuta GT, Wang ML, Falk GW, Spergel JM, Nakagawa H (2016) ATG7 gene expression as a novel tissue biomarker in eosinophilic esophagitis. Am J Gastroenterol 111:151–153
Whelan KA, Merves JF, Giroux V, Tanaka K, Guo A, Chandramouleeswaran PM, Benitez AJ, Dods K, Que J, Masterson JC, Fernando SD, Godwin BC, Klein-Szanto AJ, Chikwava K, Ruchelli ED, Hamilton KE, Muir AB, Wang ML, Furuta GT, Falk GW, Spergel JM, Nakagawa H (2017) Autophagy mediates epithelial cytoprotection in eosinophilic oesophagitis. Gut 66:1197–1207
Mori Y, Iwasaki H, Kohno K, Yoshimoto G, Kikushige Y, Okeda A, Uike N, Niiro H, Takenaka K, Nagafuji K, Miyamoto T, Harada M, Takatsu K, Akashi K (2009) Identification of the human eosinophil lineage-committed progenitor: revision of phenotypic definition of the human common myeloid progenitor. J Exp Med 206:183–193
Iwasaki H, Mizuno S, Mayfield R, Shigematsu H, Arinobu Y, Seed B, Gurish MF, Takatsu K, Akashi K (2005) Identification of eosinophil lineage-committed progenitors in the murine bone marrow. J Exp Med 201:1891–1897
Hassani M, van Staveren S, van Grinsven E, Bartels M, Tesselaar K, Leijte G, Kox M, Pickkers P, Vrisekoop N, Koenderman L (2020) Characterization of the phenotype of human eosinophils and their progenitors in the bone marrow of healthy individuals. Haematologica 105:52–56
Geering B, Stoeckle C, Conus S, Simon HU (2013) Living and dying for inflammation: neutrophils, eosinophils, basophils. Trends Immunol 34:398–409
Rothenberg ME, Hogan SP (2006) The eosinophil. Annu Rev Immunol 24:147–174
Klion AD, Ackerman SJ, Bochner BS (2020) Contributions of eosinophils to human health and disease. Annu Rev Pathol 15:179–209
Stoeckle C, Geering B, Yousefi S, Rozman S, Andina N, Benarafa C, Simon HU (2016) RhoH is a negative regulator of eosinophilopoiesis. Cell Death Differ 23:1961–1972
Bettigole SE, Lis R, Adoro S, Lee AH, Spencer LA, Weller PF, Glimcher LH (2015) The transcription factor XBP1 is selectively required for eosinophil differentiation. Nat Immunol 16:829–837
Fulkerson PC, Rothenberg ME (2018) Eosinophil development, disease involvement, and therapeutic suppression. Adv Immunol 138:1–34
Johnston LK, Hsu CL, Krier-Burris RA, Chhiba KD, Chien KB, McKenzie A, Berdnikovs S, Bryce PJ (2016) IL-33 precedes IL-5 in regulating eosinophil commitment and is required for eosinophil homeostasis. J Immunol 197:3445–3453
Hua W, Liu H, Xia LX, Tian BP, Huang HQ, Chen ZY, Ju ZY, Li W, Chen ZH, Shen HH (2015) Rapamycin inhibition of eosinophil differentiation attenuates allergic airway inflammation in mice. Respirology 20:1055–1065
Zhu C, Xia L, Li F, Zhou L, Weng Q, Li Z, Wu Y, Mao Y, Zhang C, Wu Y, Li M, Ying S, Chen Z, Shen H, Li W (2018) mTOR complexes differentially orchestrates eosinophil development in allergy. Sci Rep 8:6883
Yamada Y, Rothenberg ME, Lee AW, Akei HS, Brandt EB, Williams DA, Cancelas JA (2006) The FIP1L1-PDGFRA fusion gene cooperates with IL-5 to induce murine hypereosinophilic syndrome (HES)/chronic eosinophilic leukemia (CEL)-like disease. Blood 107:4071–4079
Geest CR, Buitenhuis M, Laarhoven AG, Bierings MB, Bruin MC, Vellenga E, Coffer PJ (2009) p38 MAP kinase inhibits neutrophil development through phosphorylation of C/EBPα on serine 21. Stem Cells 27:2271–2282
Mack EA, Stein SJ, Rome KS, Xu L, Wertheim GB, Melo RCN, Pear WS (2019) Trib1 regulates eosinophil lineage commitment and identity by restraining the neutrophil program. Blood 133:2413–2426
Drissen R, Buza-Vidas N, Woll P, Thongjuea S, Gambardella A, Giustacchini A, Mancini E, Zriwil A, Lutteropp M, Grover A, Mead A, Sitnicka E, Jacobsen SEW, Nerlov C (2016) Distinct myeloid progenitor-differentiation pathways identified through single-cell RNA sequencing. Nat Immunol 17:666–676
Gigon L, Yousefi S, Karaulov A, Simon HU (2021) Mechanisms of toxicity mediated by neutrophil and eosinophil granule proteins. Allergol Int 70:30–38
Dvorak AM, Furitsu T, Letourneau L, Ishizaka T, Ackerman SJ (1991) Mature eosinophils stimulated to develop in human cord blood mononuclear cell cultures supplemented with recombinant human interleukin-5. Part I. Piecemeal degranulation of specific granules and distribution of Charcot-Leyden crystal protein. Am J Pathol 138:69–82
Melo RC, Perez SA, Spencer LA, Dvorak AM, Weller PF (2005) Intragranular vesiculotubular compartments are involved in piecemeal degranulation by activated human eosinophils. Traffic 6:866–879
Hafez I, Stolpe A, Lindau M (2003) Compound exocytosis and cumulative fusion in eosinophils. J Biol Chem 278:44921–44928
Erjefalt JS, Andersson M, Greiff L, Korsgren M, Gizycki M, Jeffery PK, Persson GA (1998) Cytolysis and piecemeal degranulation as distinct modes of activation of airway mucosal eosinophils. J Allergy Clin Immunol 102:286–294
Radonjic-Hoesli S, Wang X, de Graauw E, Stoeckle C, Styp-Rekowska B, Hlushchuk R, Simon D, Spaeth PJ, Yousefi S, Simon HU (2017) Adhesion-induced eosinophil cytolysis requires the receptor-interacting protein kinase 3 (RIPK3)-mixed lineage kinase-like (MLKL) signaling pathway, which is counterregulated by autophagy. J Allergy Clin Immunol 140:1632–1642
Ahlstrom-Emanuelsson CA, Greiff L, Andersson M, Persson CG, Erjefalt JS (2004) Eosinophil degranulation status in allergic rhinitis: observations before and during seasonal allergen exposure. Eur Respir J 24:750–757
Erjefalt JS, Greiff L, Andersson M, Adelroth E, Jeffery PK, Persson CG (2001) Degranulation patterns of eosinophil granulocytes as determinants of eosinophil driven disease. Thorax 56:341–344
Saffari H, Hoffman LH, Peterson KA, Fang JC, Leiferman KM, Pease LF 3rd, Gleich GJ (2014) Electron microscopy elucidates eosinophil degranulation patterns in patients with eosinophilic esophagitis. J Allergy Clin Immunol 133:1728–1734
Scepek S, Lindau M (1993) Focal exocytosis by eosinophils--compound exocytosis and cumulative fusion. EMBO J 12:1811–1817
Inoue Y, Matsuwaki Y, Shin SH, Ponikau JU, Kita H (2005) Nonpathogenic, environmental fungi induce activation and degranulation of human eosinophils. J Immunol 175:5439–5447
Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, Schmid I, Straumann A, Reichenbach J, Gleich GJ, Simon HU (2008) Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med 14:949–953
Yousefi S, Stojkov D, Germic N, Simon D, Wang X, Benarafa C, Simon HU (2019) Untangling "NETosis" from NETs. Eur J Immunol 49:221–227
Simon D, Radonjic-Hosli S, Straumann A, Yousefi S, Simon HU (2015) Active eosinophilic esophagitis is characterized by epithelial barrier defects and eosinophil extracellular trap formation. Allergy 70:443–452
Gevaert E, Zhang N, Krysko O, Lan F, Holtappels G, De Ruyck N, Nauwynck H, Yousefi S, Simon HU, Bachert C (2017) Extracellular eosinophilic traps in association with Staphylococcus aureus at the site of epithelial barrier defects in patients with severe airway inflammation. J Allergy Clin Immunol 139:1849–1860
Hwang CS, Park SC, Cho HJ, Park DJ, Yoon JH, Kim CH (2019) Eosinophil extracellular trap formation is closely associated with disease severity in chronic rhinosinusitis regardless of nasal polyp status. Sci Rep 9:8061
Yousefi S, Simon D, Stojkov D, Karsonova A, Karaulov A, Simon HU (2020) In vivo evidence for extracellular DNA trap formation. Cell Death Dis 11:300
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303:1532–1535
Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU (2009) Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ 16:1438–1444
Stojkov D, Amini P, Oberson K, Sokollik C, Duppenthaler A, Simon HU, Yousefi S (2017) ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J Cell Biol 216:4073–4090
Amini P, Stojkov D, Felser A, Jackson CB, Courage C, Schaller A, Gelman L, Soriano ME, Nuoffer JM, Scorrano L, Benarafa C, Yousefi S, Simon HU (2018) Neutrophil extracellular trap formation requires OPA1-dependent glycolytic ATP production. Nat Commun 9:2958
Silveira JS, Antunes GL, Kaiber DB, da Costa MS, Ferreira FS, Marques EP, Schmitz F, Gassen RB, Breda RV, Wyse ATS, Stein RT, Pitrez PM, da Cunha AA (2020) Autophagy induces eosinophil extracellular traps formation and allergic airway inflammation in a murine asthma model. J Cell Physiol 235:267–280
Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, Noppen S, Delforge M, Willems J, Vandenabeele P (2011) Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res 21:290–304
Kenno S, Perito S, Mosci P, Vecchiarelli A, Monari C (2016) Autophagy and reactive oxygen species are involved in neutrophil extracellular traps release induced by C. albicans morphotypes. Front Microbiol 7:879
Gao XP, Zhu X, Fu J, Liu Q, Frey RS, Malik AB (2007) Blockade of class IA phosphoinositide 3-kinase in neutrophils prevents NADPH oxidase activation- and adhesion-dependent inflammation. J Biol Chem 282:6116–6125
Geering B, Gurzeler U, Federzoni E, Kaufmann T, Simon HU (2011) A novel TNFR1-triggered apoptosis pathway mediated by class IA PI3Ks in neutrophils. Blood 117:5953–5962
Song Z, Hudik E, Le Bars R, Roux B, Dang PM, El Benna J, Nusse O, Dupre-Crochet S (2020) Class I phosphoinositide 3-kinases control sustained NADPH oxidase activation in adherent neutrophils. Biochem Pharmacol 178:114088
Legrand F, Driss V, Delbeke M, Loiseau S, Hermann E, Dombrowicz D, Capron M (2010) Human eosinophils exert TNF-α and granzyme A-mediated tumoricidal activity toward colon carcinoma cells. J Immunol 185:7443–7451
Arnold IC, Artola-Boran M, Gurtner A, Bertram K, Bauer M, Frangez Z, Becher B, Kopf M, Yousefi S, Simon HU, Tzankov A, Muller A (2020) The GM-CSF-IRF5 signaling axis in eosinophils promotes antitumor immunity through activation of type 1 T cell responses. J Exp Med 217:20190706
Rivera Vargas T, Cai Z, Shen Y, Dosset M, Benoit-Lizon I, Martin T, Roussey A, Flavell RA, Ghiringhelli F, Apetoh L (2017) Selective degradation of PU.1 during autophagy represses the differentiation and antitumour activity of TH9 cells. Nat Commun 8:559
Funding
Open Access funding provided by Universität Bern. The experimental work in the laboratories of the authors is supported by the Swiss National Science Foundation to S.Y. (grant number 31003A_173215) and H.U.S. (grant number 310030_184816). The authors acknowledge financial support by the Russian Government Program “Recruitment of the Leading Scientists into the Russian Institutions of Higher Education” (grant to H.U.S.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
H.U.S. is a consultant for GlaxoSmithKline. The other authors declare that they have no conflict of interest.
Additional information
This article is a contribution to the Special issue on: Eosinophils - Guest Editor: Hans-Uwe Simon
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Germic, N., Hosseini, A., Yousefi, S. et al. Regulation of eosinophil functions by autophagy. Semin Immunopathol 43, 347–362 (2021). https://doi.org/10.1007/s00281-021-00860-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-021-00860-1