
Abstract
Pemphigoid diseases are a group of autoimmune blistering skin diseases defined by an immune response against certain components of the dermal-epidermal adhesion complex. They are prototypical, autoantibody-driven, organ-specific diseases with the emergence of inflammatory skin lesions dependent on the recruitment of immune cells, particularly granulocytes, into the skin. During an acute flare of disease, inflammatory skin lesions typically progressing from erythema through urticarial plaques to subepidermal blisters erosions erupt and, finally, completely resolve, thus illustrating that resolution of inflammation is continuously executed in pemphigoid disease patients and can be directly monitored on the skin. Despite these superb conditions for examining resolution in pemphigoid diseases as paradigm diseases for antibody-induced tissue inflammation, the mechanisms of resolution in pemphigoid are underinvestigated and still largely elusive. In the last decade, mouse models for pemphigoid diseases were developed, which have been instrumental to identify several key pathways for the initiation of inflammation in these diseases. More recently, also protective pathways, specifically IL-10 and C5aR2 signalling on the molecular level and Tregs on the cellular level, counteracting skin inflammation have been highlighted and may contribute to the continuous execution of resolution in pemphigoid diseases. The upstream orchestrators of this process are currently under investigation. Pemphigoid disease patients, particularly bullous pemphigoid patients, who are predominantly above 75 years of age, often succumb to the side effects of the immunosuppressive therapeutics nowadays still required to suppress the disease. Pemphigoid disease patients may therefore represent a group of patients benefiting most substantially from the introduction of non-immunosuppressive, proresolving therapeutics into the treatment regimens for their disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Definition of pemphigoid diseases
Organ-specific, IgG- and/or IgA-mediated immune responses are one of the most common pathomechanisms in the pathogenesis of autoimmune diseases. In humans, there are examples for this aberrant type of immune response leading to disease in almost any organ [1]. This also includes the skin where an IgG- and/or IgA-mediated immune response directed to specific components of the dermal-epidermal adhesion complex is the defining, common pathomechanistic principle of pemphigoid diseases, a group of autoimmune blistering skin diseases, which characteristically features the formation of subepidermal, dense blisters of the skin.
Seven different disease entities, namely bullous pemphigoid (BP), pemphigoid gestationis, mucous membrane pemphigoid, linear IgA disease, lichen planus pemphigoides, anti-p200 pemphigoid, and epidermolysis bullosa acquisita (EBA), belong to the group of pemphigoid disease entities. Although the diseases share many aspects of their histopathological and clinical presentation, they also partially differ in many aspects, including in their autoantigens (Table 1) and in the long-term course of disease (reviewed in [2,3,4]).
The by far most common and best examined pemphigoid disease is BP. We will therefore focus our brief overview on the clinical features and pathophysiology pemphigoid disease on BP. More detailed information on other pemphigoid diseases are available in numerous excellent reviews published on the clinical features and management of pemphigoid diseases in the last years [1,2,3,4].
Bullous pemphigoid
BP is the most frequent autoimmune blistering skin disease with a prevalence of approximately 21,000 patients, i.e. 260/million, in Germany [5] and an incidence varying between populations from 10 to 25 patients/million/year in Central Europe and the USA [6,7,8,9,10]. Registry data from the UK and Sweden even reveal incidences of about 70/million/year [11, 12]. BP is, notably, a disease of the elderly with most patients affected in the 8th and 9th decennium with a mean age at the time of diagnosis between 75 and 80 years [5, 10, 12]. Accordingly, incidences steeply rise with age to more than 200/million/year in individuals older than 80 years [6,7,8, 11].
BP is a chronic disease usually exhibiting an undulating, remitting-relapsing course. However, in the pre-corticosteroid era, about 70% of patients already succumbed to the first flare of disease [13]. Since the introduction of various immunosuppressants into the treatment of pemphigoid diseases, acute flares of disease can be usually therapeutically suppressed but disease relapses in 40% of patients within 6 months after the discontinuation of immunosuppressive therapy [14] and the 1-year mortality of BP patients after the first flare is approximately 20–40% and, thus, 2–3-fold that of age- and sex-matched peers [4, 14, 15]. The cause for the increased mortality of BP patients even under treatment is not entirely clear. Many patients, however, succumb to infections, which are certainly promoted by the immunosuppressive drugs employed for the treatment of BP. It appears evident that with the vast majority of BP patients being elderly above 80 years, the collective of BP patients is exceptionally susceptible to the unwanted side effects of immunosuppressive drugs.
BP is defined by the formation of autoantibodies against the hemidesmosomal protein BP180, also termed type XVII collagen (Col17) [2,3,4, 16]. About half of BP patients, additionally, form autoantibodies against BP230, another hemidesmosomal protein, probably due to epitope spreading, but the pathogenic significance of anti-BP230 autoantibodies is still not fully elucidated [17,18,19]. Like in all pemphigoid diseases, the deposition of autoantibodies at the dermal-epidermal junction (DEJ) alone does not precipitate inflammatory skin lesions. The latter require the marked recruitment of immune cells, particularly of granulocytes, into the dermis. Apart from old age, the greatest risk factors appear to be debilitating neurological disorders that affect a third to half of the BP patients and usually precede the autoimmune skin disease [20,21,22,23], and the use of diuretics, psycholeptics, and dipeptidyl-peptidase IV inhibitors (gliptins) [20, 24,25,26,27,28].
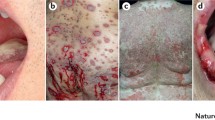
The clinical presentation of BP is variable but in its typical and most common clinical presentation, it exhibits the eruption of inflammatory skin lesions with the individual skin lesion progressing from erythema through urticarial plaques, blisters, and erosions to uninflamed and scarlessly re-epithelizing skin [2, 29] (Fig. 1). Thus, herein, the latter stage represents a state of resolving or resolved skin inflammation on track to reach the reconstitutio ad integrum. The different stages of lesion progression often co-exist in the individual patient during an acute flare of disease. This description of the inflammatory skin lesions in BP already implicates that, although autoantibodies usually deposit at the dermal-epidermal junction throughout the entire skin, in most cases, skin lesions do not involve the entire skin at the same time but only erupt in certain areas scattered all over the body but often sparing the face. The reasons for this selective eruption of skin lesions are still elusive. Of interest, data from the pre-corticosteroid area indicate that one third of BP patients will recover from the disease within 5 years without specific treatment [13]. Collectively, these clinical observations, however, strongly advocate the existence of endogenous, proresolving, and anti-inflammatory mechanisms first counteracting the emergence of skin inflammation and later, terminating (“resolving”) it, where it finally could erupt.

Typical co-existence of inflammatory skin lesions in different stages of development, including an emerging skin lesions in the shape of an urticarial plaque (blue arrow), a skin blister (red arrow), an erosion (yellow arrow), a resolving/healing erosion (grey arrow), and a resolved/healed skin lesions (black arrow) on the medial aspect of the upper arm of a BP patient during an acute flare
Diagnosis is based on (i) a compatible clinical picture, (ii) linear deposits of IgG and/or complement C3 at the dermal-epidermal junction by direct immunofluorescence (IF) microscopy of a perilesional skin biopsy, and (iii) the detection of serum autoantibodies against BP180 and/or BP230 [15, 30,31,32,33,34,35,36].
Treatment of moderate and severe BP relies on the long-term use of systemic corticosteroids usually combined with potentially corticosteroid-sparing immunomodulants such as doxycycline and dapsone or immunosuppressants such as methotrexate, azathioprine, and mycophenolate [15, 36,37,38,39]. In severe or recalcitrant patients, adjuvant rituximab, immunoadsorption, or high-dose intravenous immunoglobulins can be employed [15, 37, 40,41,42,43].
While several mouse models of pemphigoid diseases have been instrumental in uncovering some of the key proinflammatory pathways driving the effector phase of these diseases, research into these protective mechanisms is still in its infancy. The few insights gained to this point into these protective mechanisms will be summarized following a brief update on the pathophysiology of BP.
Pathophysiology
Pathology in BP is driven by autoantibodies against the two hemidesmosomal proteins BP180 (also termed type XVII collagen, Col17) and BP230 (reviewed in [2,3,4, 16, 44]). In addition, to anti-Col17 IgG about 40 and 60% of BP patients develop IgE and IgA anti-Col17 reactivity, respectively [18, 45,46,47,48,49,50,51]. BP230 is recognized by 50–70% of BP sera [32, 34, 52,53,54,55,56].
While in BP, a large body of evidence has been assembled to describe the pathogenic importance of autoantibodies and various mechanisms that mediate tissue destruction of anti-Col17 IgG (detailed below), data about the cellular immune response, undoubtedly essential in every autoimmune disease, are rather scare [57]. T and B cell reactivity against the NH2-terminal portion of the BP180 ectodomain is associated with severe BP, while the central portion is more frequently recognized in patients with limited disease. In contrast, combined T and B cell response against the COOH- and NH2-terminal globular domains of BP230 were found in less than 50% [58]. The response to the Col17 ectodomain is restricted to the DQβ1*0301 allele [59, 60]. Autoreactive T cells in BP patients release a Th1/Th2 mixed cytokine profile [58, 59]. While the number of circulating CD4+CD25+FoxP3+ regulatory T cells, natural killer T cells, and natural killer cells are normal, γδ T cell numbers were reported to be reduced in BP patients [61, 62].
A plethora of data has been published about the pathogenic relevance of anti-Col17 antibodies, while only conflicting reports were available about the pathogenicity of anti-BP230 IgG. Recently, however, the injection of monoclonal BP230-specific IgG in neonatal mice induced macroscopic and microscopic blistering suggesting their pathogenic potential [63, 64].
Our knowledge about the events that lead to subepidermal blistering upon binding of anti-Col17 antibodies to their cell surface receptor on keratinocytes is derived from the observation that serum levels of Col17NC16A-specific IgG antibodies correlate with the disease activity in BP patients [65,66,67,68,69,70] as well as various experimental models. Latter models include the incubation of cultured human keratinocytes with Col17-specific IgG/IgE, the treatment of cryosections of human skin with Col17-specific IgG followed by incubation with normal human leukocytes, and, importantly, various mouse models of BP and BP-like inflammatory EBA [71,72,73,74,75,76,77] (reviewed in [78,79,80,81]).
Based on these models, the following sequence of events has appeared (Fig. 2). While most effects depend on the Fc-portion of autoantibodies, also, very early in the diseases course, Fc-independent effects have been described including the release of IL-6 and IL-8 from keratinocytes following binding of anti-Col17 IgG or IgE [72, 82] as well as internalization of Col17 and weakening of keratinocyte attachment in response to anti-Col 17 IgG [83,84,85,86,87].

Sequence of events leading to dermal-epidermal separation and potentially proresolving pathways in bullous pemphigoid (BP). Binding of autoantibodies (red) against type XVII collagen (Col17, yellow) initiates Fc-independent events, e.g. the release of IL-8 from basal keratinocytes (1). Complement is activated at the dermal-epidermal junction (DEJ) and C5a released (2). Mast cells degranulate after binding of C5A or anti-Col17 IgE (3) and inflammatory cells attracted by C5a appear in the upper dermis (4, 5). Their secretion of additional inflammatory mediators such as IL-17A, LTB4, and to a lesser extent, of IL-1β further increases and maintains the inflammatory reaction. Subsequently, neutrophils and eosinophils line along the DEJ (6) and release reactive oxygen species and specific proteases that ultimately induce dermal-epidermal separation (7). In animal models of BP and BP-like epidermolysis bullosa acquisita, the anti-inflammatory effect of regulatory T cells (Treg), C5aR2, and IL-6 (via IL-1 receptor antagonist, IL-1RA, and tissue inhibitor of metalloproteinase-1, Timp-1) was shown and point to potentially proresolving mechanisms in BP. Modified from [4] and [3]
The earliest Fc-dependent effect of autoantibodies is most likely the activation of complement at the dermal-epidermal junction, an event observed in the skin of nearly all BP patients. Complement activation leads to the influx of inflammatory cells such as neutrophils, eosinophils, and macrophages [75, 88,89,90]. Complement-derived anaphylatoxins such as C5a have strong chemotactic effects on these cells. Here, both the classical and the alternative pathway of complement activation are important and most effects are most likely exerted via the C5aR1 on leucocytes [88, 89, 91]. In addition, the degranulation of mast cells, one of the earliest events observed in BP lesions, may be exerted via C5aR1 on mast cells [92]. An even more striking usage of C5aR1 was observed in mouse models of BP-like inflammatory EBA and anti-laminin 332 mucous membrane pemphigoid [93,94,95,96].
In addition to mast cells [90, 92], neutrophils [97,98,99,100,101], eosinophils [102], and macrophages [90] were shown to contribute to subsequent tissue destruction. So far, only few individual inflammatory mediators with a striking effect on the autoantibody-mediated tissue destruction have been identified in addition to C5a, including LTB4 and IL-17A.
Leukotriene B4 (LTB4) is another chemoattractant critically involved in the regulation of neutrophil influx into the skin. Thus, deficiency in 5-lipoxygenase, the rate-limiting enzyme in the biosynthesis of LTB4, or in the LTB4 receptor BLT1 confers dramatic resistance to the recruitment of neutrophils into the skin and, consequently, to the emergence of skin lesions in both the antibody transfer BP-like EBA and BP mouse model [103, 104]. Furthermore, scavenging of LTB4 by the drugs Coversin and PAS-L-Coversin significantly attenuates disease [105]. In line with this critical role of LTB4 in the first stages of lesion development, LTB4 levels in the start increasing briefly after the injection of anti-Col7 antibodies into the skin [103]. Tissue resident cells, such as macrophages, are presumably the initial cellular sources of LTB4. This notion, however, still requires clarification. Once recruited into the skin, neutrophils themselves are an abundant source of LTB4 and amplify their recruitment into the skin in a manner similar to what was previously demonstrated for autoantibody-induced inflammation in other organs [104, 106,107,108].
CD4-positive cells were identified as the major source of IL-17A both in the blood and early skin lesions of BP patients compared to age- and sex-matched controls. mRNA analysis of early skin lesions revealed IL17A and related cytokines and chemokines to be significantly upregulated. IL17A-deficient mice were greatly protected by the otherwise pathogenic effect of anti-Col17 IgG compared to wild-type animals, and anti-Col17 IgG-injected mice developed significantly fewer clinical lesions when treated with an anti-IL17 A antibody compared to isotype control antibody-treated mice [109]. In addition, Antonicelli and coworkers showed that (i) IL-17 serum levels are lower in patients in remission compared to the time when treatment was initiated, (ii) IL-17A is involved in the formation of neutrophil extracellular traps in the BP skin lesions, and (iii) IL-17 induce the release of neutrophil elastase and matrix metalloproteinase-9 from normal human polymorphonuclear cells [110,111,112].
C5a and LTB4 may thus induce the influx of inflammatory cells in the upper dermis, while IL-17 may orchestrate the inflammatory reaction in the skin that finally leads to blister formation.
In early BP lesions, neutrophils and eosinophils are found to line up along the dermal-epidermal junction. Reactive oxygen species and specific proteases such as matrix metalloproteinase-9 and neutrophil elastase were shown to be released form infiltrating leucocytes and lead to dermal-epidermal splitting [71, 113, 114]. Although the proteolytic activity most likely not specifically targets individual basement membrane proteins, matrix metalloproteinase-9 and neutrophil elastase were found in blister fluid and lesional biopsies of BP patients and were capable to degrade Col17 [115,116,117]. In fact, the importance of individual proteases was quite well studied in the neonatal mouse model of BP [98,99,100,101, 118, 119]. It appears that in the early stages of blistering, matrix metalloproteinase-9 is mainly activated by plasmin, which is formed by activation of plasminogen by tissue plasminogen activator and/or urokinase plasminogen activator. Plasmin and the mast cell-specific serine protease-4 can activate matrix metalloproteinase-9 which then inactivates α1-proteinase inhibitor, the physiological inhibitor of neutrophil elastase. The unrestrained activity of neutrophil elastase is then responsible for the degradation of structural proteins of the dermal-epidermal junction including Col17 [98,99,100,101, 118, 119]. This cascade of events is further amplified and perpetuated by the activation of the coagulation cascade by eosinophils, which further promotes the recruitment of eosinophils into the dermis [44, 120, 121].
In summary, some aspects of BP physiology, such as the sequence of events leading to blistering, including the requirement of autoantibodies and the infiltration of inflammatory cells, have been relatively well defined. Further studies will focus on the trigger factors that induce the generation of anti-Col17 and anti-BP230 antibodies in BP and on the identification of pharmacological inhibitors of key inflammatory mediators and pathways.
Resolution
Some mediators have been described that are present in the blood and/or skin of BP patients and were shown to exert anti-inflammatory properties when their functional relevance was explored in mouse models of BP or BP-like EBA. Below we summarize the current knowledge about the so far identified anti-inflammatory factors in BP including C5aR2, IL-6, and IL-10, and discuss them as effector molecules in the resolution of proresolving potential.
Complement activation
Both, the alternative and, to even a larger extent, the classical pathway were shown to be important for blister formation. In the neonatal mouse model of BP, activation of the classical pathway was even reported to be a requisite for dermal-epidermal separation in this model. Mice deficient in C5 or C1q did not develop blisters upon injection of anti-Col17 IgG due to the lack of neutrophil recruitment to the skin [88, 89, 122]. The quasi dogma of complement dependency of BP was later questioned when the passive transfer of F(ab’)2 fragments of human Col17-specific IgG or anti-Col17 IgG4 led to skin fragility when injected in Col17-humanized mice. In the same model, the induction of skin fragility upon injection of Col17-specific IgG in C3-deficient mice pointed in the same direction [122,123,124]. In the antibody transfer model of BP employing adult mice, the injection of anti-Col17 IgG in C5-deficient mice halved the extent of skin lesions as compared to wild-type mice independent of the IgG dose [91]. In line, when C5aR1-deficient mice were injected with anti-Col17 IgG, significantly less skin lesions arose compared to wild-type animals; however, C5aR1-deficient mice still developed about two third of the extent of lesions induced in wild-type mice [91]. These results indicate that although complement activation is an important factor to recruit leucocytes to the upper dermis, BP lesions may develop independently of complement activation.
Interestingly, when Col17-specific IgG was injected in C5aR2-deficient mice, these mice developed significantly more skin lesions compared to wild-type animals. While C5aR1 clearly is a proinflammatory mediator, the role of C5aR2 in inflammation appears to be multifaceted and may depend on the individual disease and the stage of inflammation [125]. In various disorders, such as sepsis, immune-complex-mediated lung injury, and allergic contact dermatitis, like in BP, C5aR2 has a protective role [125]. In line, migration of bone marrow-derived C5aR1-deficient neutrophils towards recombinant C5a was significantly lower compared to neutrophils from C57BL/6 wild-type mice, while migration of neutrophils from C5aR2-deficient animals was similar to neutrophils from wild-type mice [91]. Current research within the Clinical Research Unit 303 Pemphigoid Diseases aims at further delineating the anti-inflammatory and potentially proresolving role of C5aR2 in BP.
Interleukin-6
IL-6 is a pleiotropic cytokine that has been identified as key proinflammatory mediator in several diseases including rheumatoid arthritis, Castleman’s disease, Takayasu arteritis, and giant cell arteritis [126]. However, proinflammatory effects of IL-6 have been described, e.g. in animal models of endotoxic lung disease and endotoxemia [127]. The classical signalling pathway is initiated by binding of IL-6 to IL-6R and a second transmembrane protein, gp130, which serves as a signal transducer of IL-6. IL-6 can also bind to soluble IL-6Rs (sIL-6Rs forming an IL-6-sIL-6R complex that can bind to membrane-bound gp130 on cells that do not express IL-6R on the surface, a process known as trans-signalling) [126, 128]. In a mouse model of BP-like EBA, we have shown that blockade of IL-6 led to significantly increased skin lesions via classical IL-6 signalling most likely by the inhibition of IL-1R antagonist [129]. More specifically, while patients as well as anti-Col7 IgG-injected mice revealed elevated levels of IL-6 in blister fluids (BP patients only), sera and skin, IL-6-deficient mice or mice treated with an blocking anti-IL-6 antibody developed significantly higher disease activity compared to wild-type animals [129, 130]. In contrast, treatment with recombinant IL-6 dose-dependently impaired the induction of experimental BP-like EBA and led to increased expression of IL-1R antagonist in skin and serum [129]. Co-injection of mice with anti-Col7 IgG and the IL-1R antagonist anakinra significantly reduced the induction of skin lesions [129]. Latter data were later corroborated by the finding that after disease induction by the immunization of susceptible SJL/J mice with recombinant Col7, anakinra prevented disease progression compared to phosphate buffer saline (PBS)-treated mice [131]. In line, IL-1R-deficient or IL-1β-deficient mice were significantly less susceptible to the skin lesion-inducing effect of anti-Col7 IgG [131]. In addition to interfering with IL-1 homeostasis, IL-6 may exert its anti-inflammatory role in experimental BP-like EBA by the observed increased dermal expression of tissue inhibitor of metalloproteinase-1 (Timp-1; a physiological inhibitor of metalloproteinase) in IL-6-treated mice [129]. Metalloproteinase-9 has previously been shown to be essential for blister formation in the neonatal mouse model of BP (see above) [100].
Tregs and Interleukin-10
On the cellular level, Tregs have been highlighted to promote the timely resolution of skin lesions in the antibody transfer BP-like EBA mouse model. Thus, depletion of Tregs in DEREG significantly aggravated and prolonged disease [132]. Supporting also a similar role of Tregs in the human situation, Tregs are also present in lesional skin of BP patients. Notably, their density in lesional BP skin is, however, significant lower than in psoriasis and atopic dermatitis suggesting a relative deficiency in Tregs in BP [133], which may contribute to the persistence of skin inflammation in pemphigoid diseases. The mechanisms Tregs apply to counteract skin inflammation are still largely elusive, but there is first evidence for a significant role of IL-10 in this process. Thus, the induction of IL-10+ plasma cells efficiently suppressed skin inflammation in the antibody transfer BP-like EBA mouse model, among other, by inducing the release of IL-10 from Tregs in the skin [134]. Intriguingly, a series of in vitro experiments suggested that IL-10 may suppress disease in pemphigoid diseases by directly inhibiting the effect of C5a on neutrophils [134].
Proresolving lipid mediators
Multiple lines of evidence predominantly generated in mouse models of acute peritonitis, pouchitis, ischemia, or colitis point at a central role of proresolving lipid mediators as orchestrators of resolution [135,136,137]. The validity of this principle for the resolution of autoantibody-induced tissue inflammation is, however, still uncertain [135], and the role of proresolving lipid mediators in pemphigoid diseases has not been investigated either. However, profiling the lipidome in emerging skin lesions in the antibody transfer mouse model of BP-like EBA, we detected 12/15-lipoxygenase-derived proresolving lipid mediators [103], suggesting that 12/15-lipoxygenase is already active in the initiation phase of skin lesions and might counteract their emergence from the very beginning. Whether the enzyme is still active during the resolution of skin lesions is currently under investigation.
Concluding remarks
In recent years, a number of anti-inflammatory pathways counteracting skin inflammation in pemphigoid diseases have been uncovered. These pathways may also play a role in the resolution of skin inflammation in pemphigoid diseases which still requires detailed investigation. With single skin lesions in BP continuously emerging and completely resolving, BP appears as excellent model to dissect the mechanisms of resolution in autoantibody-induced tissue inflammation and injury. In that line, the disease may potentially also respond particularly well to proresolving therapeutic strategies, and, with pemphigoid disease patients often succumbing to the side effects of immunosuppressive drugs, these diseases may be among those benefiting the most from the introduction of proresolving therapies into treatment regimens.
References
Ludwig RJ, Vanhoorelbeke K, Leypoldt F, Kaya Z, Bieber K, McLachlan SM et al (2017) Mechanisms of autoantibody-induced pathology. Front Immunol 8:603
Amber KT, Murrell DF, Schmidt E, Joly P, Borradori L (2018) Autoimmune subepidermal bullous diseases of the skin and mucosae: clinical features, diagnosis, and management. Clin Rev Allergy Immunol 54(1):26–51
Schmidt E, Groves R (2016) Immunobullous diseases. In: Griffith C, Barker J, Chalmers BT, Creamer D (eds) Rook’s textbook of dermatology, part 3, chapter 50, vol 50, 9th edn. Wiley-Blackwell, Chichester, pp 1–56
Schmidt E, Zillikens D (2013) Pemphigoid diseases. Lancet. 381(9863):320–332
Hubner F, Recke A, Zillikens D, Linder R, Schmidt E (2016) Prevalence and age distribution of pemphigus and pemphigoid diseases in Germany. J Invest Dermatol 136(12):2495–2498
Bertram F, Brocker EB, Zillikens D, Schmidt E (2009) Prospective analysis of the incidence of autoimmune bullous disorders in Lower Franconia, Germany. J Dtsch Dermatol Ges 7(5):434–440
Gudi VS, White MI, Cruickshank N, Herriot R, Edwards SL, Nimmo F et al (2005) Annual incidence and mortality of bullous pemphigoid in the Grampian Region of North-east Scotland. Br J Dermatol 153(2):424–427
Marazza G, Pham HC, Scharer L, Pedrazzetti PP, Hunziker T, Trueb RM et al (2009) Incidence of bullous pemphigoid and pemphigus in Switzerland: a 2-year prospective study. Br J Dermatol 161(4):861–868
Brick KE, Weaver CH, Lohse CM, Pittelkow MR, Lehman JS, Camilleri MJ et al (2014) Incidence of bullous pemphigoid and mortality of patients with bullous pemphigoid in Olmsted County, Minnesota, 1960 through 2009. J Am Acad Dermatol 71(1):92–99
Joly P, Baricault S, Sparsa A, Bernard P, Bedane C, Duvert-Lehembre S et al (2012) Incidence and mortality of bullous pemphigoid in France. J Invest Dermatol 132(8):1998–2004
Langan SM, Smeeth L, Hubbard R, Fleming KM, Smith CJ, West J (2008) Bullous pemphigoid and pemphigus vulgaris—incidence and mortality in the UK: population based cohort study. BMJ 337:a180
Thorslund K, Seifert O, Nilzen K, Gronhagen C (2017) Incidence of bullous pemphigoid in Sweden 2005-2012: a nationwide population-based cohort study of 3761 patients. Arch Dermatol Res 309(9):721–727
Lever WF (1953) Pemphigus. Medicine. 32:1–123
Joly P, Roujeau JC, Benichou J, Picard C, Dreno B, Delaporte E et al (2002) A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med 346(5):321–327
Feliciani C, Joly P, Jonkman MF, Zambruno G, Zillikens D, Ioannides D et al (2015) Management of bullous pemphigoid: the European Dermatology Forum consensus in collaboration with the European Academy of Dermatology and Venereology. Br J Dermatol 172(4):867–877
Bagci IS, Horvath ON, Ruzicka T, Sardy M (2017) Bullous pemphigoid. Autoimmun Rev 16(5):445–455
Delaporte E, Dubost-Brama A, Ghohestani R, Nicolas JF, Neyrinck JL, Bergoend H et al (1996) IgE autoantibodies directed against the major bullous pemphigoid antigen in patients with a severe form of pemphigoid. J Immunol 157(8):3642–3647
Hashimoto T, Ohzono A, Teye K, Numata S, Hiroyasu S, Tsuruta D et al (2017) Detection of IgE autoantibodies to BP180 and BP230 and their relationship to clinical features in bullous pemphigoid. Br J Dermatol 177(1):141–151
Ishiura N, Fujimoto M, Watanabe R, Nakashima H, Kuwano Y, Yazawa N et al (2008) Serum levels of IgE anti-BP180 and anti-BP230 autoantibodies in patients with bullous pemphigoid. J Dermatol Sci 49(2):153–161
Bastuji-Garin S, Joly P, Lemordant P, Sparsa A, Bedane C, Delaporte E et al (2011) Risk factors for bullous pemphigoid in the elderly: a prospective case-control study. J Invest Dermatol 131(3):637–643
Langan SM, Groves RW, West J (2010) The relationship between neurological disease and bullous pemphigoid: a population-based case-control study. J Invest Dermatol 131(3):631–636
Taghipour K, Chi CC, Vincent A, Groves RW, Venning V, Wojnarowska F (2010) The association of bullous pemphigoid with cerebrovascular disease and dementia: a case-control study. Arch Dermatol 146(11):1251–1254
Forsti AK, Huilaja L, Schmidt E, Tasanen K (2017) Neurological and psychiatric associations in bullous pemphigoid-more than skin deep? Exp Dermatol 26(12):1228–1234
Bastuji-Garin S, Joly P, Picard-Dahan C, Bernard P, Vaillant L, Pauwels C et al (1996) Drugs associated with bullous pemphigoid. A case-control study. Arch Dermatol 132(3):272–276
Lloyd-Lavery A, Chi CC, Wojnarowska F, Taghipour K (2013) The associations between bullous pemphigoid and drug use: a UK case-control study. JAMA Dermatol 149(1):58–62
Kridin K, Cohen AD (2018) Dipeptidyl-peptidase IV inhibitor-associated bullous pemphigoid: a systematic review and meta-analysis. J Am Acad Dermatol
Varpuluoma O, Forsti AK, Jokelainen J, Turpeinen M, Timonen M, Huilaja L et al (2018) Vildagliptin significantly increases the risk of bullous pemphigoid: a Finnish nationwide registry study. J Invest Dermatol 138(7):1659–1661
Benzaquen M, Borradori L, Berbis P, Cazzaniga S, Valero R, Richard MA, Feldmeyer L (2018) Dipeptidyl peptidase IV inhibitors, a risk factor for bullous pemphigoid: retrospective multicenter case-control study from France and Switzerland. J Am Acad Dermatol 78(6):1090–1096
Schmidt E, della Torre R, Borradori L (2011) Clinical features and practical diagnosis of bullous pemphigoid. Dermatol Clin 29(3):427–438 viii-ix
Schmidt E, Zillikens D (2010) Modern diagnosis of autoimmune blistering skin diseases. Autoimmun Rev 10(2):84–89
Schmidt E, Zillikens D (2011) The diagnosis and treatment of autoimmune blistering skin diseases. Deutsches Arzteblatt international 108(23):399–405 I-III
Blocker IM, Dahnrich C, Probst C, Komorowski L, Saschenbrecker S, Schlumberger W et al (2012) Epitope mapping of BP230 leading to a novel enzyme-linked immunosorbent assay for autoantibodies in bullous pemphigoid. Br J Dermatol 166(5):964–970
Sitaru C, Dahnrich C, Probst C, Komorowski L, Blocker I, Schmidt E et al (2007) Enzyme-linked immunosorbent assay using multimers of the 16th non-collagenous domain of the BP180 antigen for sensitive and specific detection of pemphigoid autoantibodies. Exp Dermatol 16(9):770–777
Charneux J, Lorin J, Vitry F, Antonicelli F, Reguiai Z, Barbe C et al (2011) Usefulness of BP230 and BP180-NC16a enzyme-linked immunosorbent assays in the initial diagnosis of bullous pemphigoid: a retrospective study of 138 patients. Arch Dermatol 147(3):286–291
Schmidt E, Goebeler M, Hertl M, Sardy M, Sitaru C, Eming R et al (2015) S2k guideline for the diagnosis of pemphigus vulgaris/foliaceus and bullous pemphigoid. J Dtsch Dermatol Ges 13(7):713–727
van Beek N, Zillikens D, Schmidt E (2018) Diagnosis of autoimmune bullous diseases. J Dtsch Dermatol Ges 16(9):1077–1091
Eming R, Sticherling M, Hofmann SC, Hunzelmann N, Kern JS, Kramer H et al (2015) S2k guidelines for the treatment of pemphigus vulgaris/foliaceus and bullous pemphigoid. J Dtsch Dermatol Ges 13(8):833–844
Sticherling M, Franke A, Aberer E, Glaser R, Hertl M, Pfeiffer C et al (2017) An open, multicentre, randomized clinical study in patients with bullous pemphigoid comparing methylprednisolone and azathioprine with methylprednisolone and dapsone. Br J Dermatol 177(5):1299–1305
Williams HC, Wojnarowska F, Kirtschig G, Mason J, Godec TR, Schmidt E et al (2017) Doxycycline versus prednisolone as an initial treatment strategy for bullous pemphigoid: a pragmatic, non-inferiority, randomised controlled trial. Lancet. 389(10079):1630–1638
Hubner F, Kasperkiewicz M, Knuth-Rehr D, Shimanovich I, Hubner J, Sufke S et al (2018) Adjuvant treatment of severe/refractory bullous pemphigoid with protein A immunoadsorption. J Dtsch Dermatol Ges 16(9):1109–1118
Kasperkiewicz M, Schulze F, Meier M, van Beek N, Nitschke M, Zillikens D et al (2014) Treatment of bullous pemphigoid with adjuvant immunoadsorption: a case series. J Am Acad Dermatol 71(5):1018–1020
Kasperkiewicz M, Shimanovich I, Ludwig RJ, Rose C, Zillikens D, Schmidt E (2011) Rituximab for treatment-refractory pemphigus and pemphigoid: a case series of 17 patients. J Am Acad Dermatol 65(3):552–558
Lamberts A, Euverman HI, Terra JB, Jonkman MF, Horvath B (2018) Effectiveness and safety of rituximab in recalcitrant pemphigoid diseases. Front Immunol 9:248
Genovese G, Di Zenzo G, Cozzani E, Berti E, Cugno M, Marzano AV (2019) New insights into the pathogenesis of bullous pemphigoid: 2019 update. Front Immunol 10:1506
Dopp R, Schmidt E, Chimanovitch I, Leverkus M, Brocker EB, Zillikens D (2000) IgG4 and IgE are the major immunoglobulins targeting the NC16A domain of BP180 in bullous pemphigoid: serum levels of these immunoglobulins reflect disease activity. J Am Acad Dermatol 42(4):577–583
Iwata Y, Komura K, Kodera M, Usuda T, Yokoyama Y, Hara T et al (2008) Correlation of IgE autoantibody to BP180 with a severe form of bullous pemphigoid. Arch Dermatol 144(1):41–48
Messingham KA, Noe MH, Chapman MA, Giudice GJ, Fairley JA (2009) A novel ELISA reveals high frequencies of BP180-specific IgE production in bullous pemphigoid. J Immunol Methods 346(1–2):18–25
Kromminga A, Scheckenbach C, Georgi M, Hagel C, Arndt R, Christophers E et al (2000) Patients with bullous pemphigoid and linear IgA disease show a dual IgA and IgG autoimmune response to BP180. J Autoimmun 15(3):293–300
Christophoridis S, Budinger L, Borradori L, Hunziker T, Merk HF, Hertl M (2000) IgG, IgA and IgE autoantibodies against the ectodomain of BP180 in patients with bullous and cicatricial pemphigoid and linear IgA bullous dermatosis. Br J Dermatol 143(2):349–355
van Beek N, Luttmann N, Huebner F, Recke A, Karl I, Schulze FS et al (2017) Correlation of serum levels of IgE autoantibodies against BP180 with bullous pemphigoid disease activity. JAMA Dermatol 153(1):30–38
van Beek N, Schulze FS, Zillikens D, Schmidt E (2016) IgE-mediated mechanisms in bullous pemphigoid and other autoimmune bullous diseases. Expert Rev Clin Immunol 12(3):267–277
Kromminga A, Sitaru C, Hagel C, Herzog S, Zillikens D (2004) Development of an ELISA for the detection of autoantibodies to BP230. Clin Immunol 111(1):146–152
Thoma-Uszynski S, Uter W, Schwietzke S, Hofmann SC, Hunziker T, Bernard P et al (2004) BP230- and BP180-specific auto-antibodies in bullous pemphigoid. J Invest Dermatol 122(6):1413–1422
Yoshida M, Hamada T, Amagai M, Hashimoto K, Uehara R, Yamaguchi K et al (2006) Enzyme-linked immunosorbent assay using bacterial recombinant proteins of human BP230 as a diagnostic tool for bullous pemphigoid. J Dermatol Sci 41(1):21–30
Tampoia M, Lattanzi V, Zucano A, Villalta D, Filotico R, Fontana A et al (2009) Evaluation of a new ELISA assay for detection of BP230 autoantibodies in bullous pemphigoid. Ann N Y Acad Sci 1173:15–20
Roussel A, Benichou J, Randriamanantany ZA, Gilbert D, Drenovska K, Houivet E et al (2011) Enzyme-linked immunosorbent assay for the combination of bullous pemphigoid antigens 1 and 2 in the diagnosis of bullous pemphigoid. Arch Dermatol 147(3):293–298
Hertl M, Eming R, Veldman C (2006) T cell control in autoimmune bullous skin disorders. J Clin Invest 116(5):1159–1166
Thoma-Uszynski S, Uter W, Schwietzke S, Schuler G, Borradori L, Hertl M (2006) Autoreactive T and B cells from bullous pemphigoid (BP) patients recognize epitopes clustered in distinct regions of BP180 and BP230. J Immunol 176(3):2015–2023
Budinger L, Borradori L, Yee C, Eming R, Ferencik S, Grosse-Wilde H et al (1998) Identification and characterization of autoreactive T cell responses to bullous pemphigoid antigen 2 in patients and healthy controls. J Clin Invest 102(12):2082–2089
Lin MS, Fu CL, Giudice GJ, Olague-Marchan M, Lazaro AM, Stastny P et al (2000) Epitopes targeted by bullous pemphigoid T lymphocytes and autoantibodies map to the same sites on the bullous pemphigoid 180 ectodomain. J Invest Dermatol 115(6):955–961
Rensing-Ehl A, Gaus B, Bruckner-Tuderman L, Martin SF (2007) Frequency, function and CLA expression of CD4+CD25+FOXP3+ regulatory T cells in bullous pemphigoid. Exp Dermatol 16(1):13–21
Oswald E, Fisch P, Jakob T, Bruckner-Tuderman L, Martin SF, Rensing-Ehl A (2009) Reduced numbers of circulating gammadelta T cells in patients with bullous pemphigoid. Exp Dermatol 18(11):991–993
Haeberle S, Wei X, Bieber K, Goletz S, Ludwig RJ, Schmidt E et al (2018) Regulatory T-cell deficiency leads to pathogenic bullous pemphigoid antigen 230 autoantibody and autoimmune bullous disease. J Allergy Clin Immunol 142:1831–1842.e7
Muramatsu K, Ujiie H, Kobayashi I, Nishie W, Izumi K, Ito T et al (2018) Regulatory T-cell dysfunction induces autoantibodies to bullous pemphigoid antigens in mice and human subjects. J Allergy Clin Immunol 142(6):1818–30 e6
Schmidt E, Obe K, Brocker EB, Zillikens D (2000) Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Arch Dermatol 136(2):174–178
Tsuji-Abe Y, Akiyama M, Yamanaka Y, Kikuchi T, Sato-Matsumura KC, Shimizu H (2005) Correlation of clinical severity and ELISA indices for the NC16A domain of BP180 measured using BP180 ELISA kit in bullous pemphigoid. J Dermatol Sci 37(3):145–149
Kobayashi M, Amagai M, Kuroda-Kinoshita K, Hashimoto T, Shirakata Y, Hashimoto K et al (2002) BP180 ELISA using bacterial recombinant NC16a protein as a diagnostic and monitoring tool for bullous pemphigoid. J Dermatol Sci 30(3):224–232
Feng S, Wu Q, Jin P, Lin L, Zhou W, Sang H et al (2008) Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Int J Dermatol 47(3):225–228
Di Zenzo G, Thoma-Uszynski S, Fontao L, Calabresi V, Hofmann SC, Hellmark T et al (2008) Multicenter prospective study of the humoral autoimmune response in bullous pemphigoid. Clin Immunol 128(3):415–426
Amo Y, Ohkawa T, Tatsuta M, Hamada Y, Fujimura T, Katsuoka K et al (2001) Clinical significance of enzyme-linked immunosorbent assay for the detection of circulating anti-BP180 autoantibodies in patients with bullous pemphigoid. J Dermatol Sci 26(1):14–18
Sitaru C, Schmidt E, Petermann S, Munteanu LS, Brocker EB, Zillikens D (2002) Autoantibodies to bullous pemphigoid antigen 180 induce dermal-epidermal separation in cryosections of human skin. J Invest Dermatol 118(4):664–671
Messingham KN, Srikantha R, DeGueme AM, Fairley JA (2011) FcR-independent effects of IgE and IgG autoantibodies in bullous pemphigoid. J Immunol 187(1):553–560
Liu Z, Diaz LA, Troy JL, Taylor AF, Emery DJ, Fairley JA et al (1993) A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest 92(5):2480–2488
Nishie W, Sawamura D, Goto M, Ito K, Shibaki A, McMillan JR et al (2007) Humanization of autoantigen. Nat Med 13(3):378–383
Schulze FS, Beckmann T, Nimmerjahn F, Ishiko A, Collin M, Kohl J et al (2014) Fcgamma receptors III and IV mediate tissue destruction in a novel adult mouse model of bullous pemphigoid. Am J Pathol 184(8):2185–2196
Sitaru C, Chiriac MT, Mihai S, Buning J, Gebert A, Ishiko A et al (2006) Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol 177(5):3461–3468
Sitaru C, Mihai S, Otto C, Chiriac MT, Hausser I, Dotterweich B et al (2005) Induction of dermal-epidermal separation in mice by passive transfer of antibodies specific to type VII collagen. J Clin Invest 115(4):870–878
Kasperkiewicz M, Sadik CD, Bieber K, Ibrahim SM, Manz RA, Schmidt E et al (2016) Epidermolysis bullosa Acquisita: from pathophysiology to novel therapeutic options. J Invest Dermatol 136(1):24–33
Ludwig RJ (2013) Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol 2013:812029
Bieber K, Koga H, Nishie W (2017) In vitro and in vivo models to investigate the pathomechanisms and novel treatments for pemphigoid diseases. Exp Dermatol 26:1163–1170
Liu Y, Li L, Xia Y (2017) BP180 is critical in the autoimmunity of bullous pemphigoid. Front Immunol 8:1752
Schmidt E, Reimer S, Kruse N, Jainta S, Brocker EB, Marinkovich MP et al (2000) Autoantibodies to BP180 associated with bullous pemphigoid release interleukin-6 and interleukin-8 from cultured human keratinocytes. J Invest Dermatol 115(5):842–848
Iwata H, Kamio N, Aoyama Y, Yamamoto Y, Hirako Y, Owaribe K et al (2009) IgG from patients with bullous pemphigoid depletes cultured keratinocytes of the 180-kDa bullous pemphigoid antigen (type XVII collagen) and weakens cell attachment. J Invest Dermatol 129(4):919–926
Hiroyasu S, Ozawa T, Kobayashi H, Ishii M, Aoyama Y, Kitajima Y et al (2013) Bullous pemphigoid IgG induces BP180 internalization via a macropinocytic pathway. Am J Pathol 182(3):828–840
Kitajima Y, Nojiri M, Yamada T, Hirako Y, Owaribe K (1998) Internalization of the 180 kDa bullous pemphigoid antigen as immune complexes in basal keratinocytes: an important early event in blister formation in bullous pemphigoid. Br J Dermatol 138(1):71–76
Wada M, Nishie W, Ujiie H, Izumi K, Iwata H, Natsuga K, Nakamura H, Kitagawa Y, Shimizu H (2016) Epitope-dependent pathogenicity of antibodies targeting a major bullous pemphigoid autoantigen collagen XVII/BP180. J Invest Dermatol 136(5):938–946
Iwata H, Kamaguchi M, Ujiie H, Nishimura M, Izumi K, Natsuga K et al (2016) Macropinocytosis of type XVII collagen induced by bullous pemphigoid IgG is regulated via protein kinase C. Lab Investig 96(12):1301–1310
Liu Z, Giudice GJ, Swartz SJ, Fairley JA, Till GO, Troy JL et al (1995) The role of complement in experimental bullous pemphigoid. J Clin Invest 95(4):1539–1544
Nelson KC, Zhao M, Schroeder PR, Li N, Wetsel RA, Diaz LA et al (2006) Role of different pathways of the complement cascade in experimental bullous pemphigoid. J Clin Invest 116(11):2892–2900
Chen R, Fairley JA, Zhao ML, Giudice GJ, Zillikens D, Diaz LA et al (2002) Macrophages, but not T and B lymphocytes, are critical for subepidermal blister formation in experimental bullous pemphigoid: macrophage-mediated neutrophil infiltration depends on mast cell activation. J Immunol 169(7):3987–3992
Karsten CM, Beckmann T, Holtsche MM, Tillmann J, Tofern S, Schulze FS et al (2018) Tissue destruction in bullous pemphigoid can be complement independent and may be mitigated by C5aR2. Front Immunol 9:488
Heimbach L, Li Z, Berkowitz P, Zhao M, Li N, Rubenstein DS et al (2011) The C5a receptor on mast cells is critical for the autoimmune skin-blistering disease bullous pemphigoid. J Biol Chem 286(17):15003–15009
Karsten CM, Pandey MK, Figge J, Kilchenstein R, Taylor PR, Rosas M et al (2012) Anti-inflammatory activity of IgG1 mediated by Fc galactosylation and association of FcgammaRIIB and dectin-1. Nat Med 18(9):1401–1406
Mihai S, Chiriac MT, Takahashi K, Thurman JM, Holers VM, Zillikens D et al (2007) The alternative pathway of complement activation is critical for blister induction in experimental epidermolysis bullosa acquisita. J Immunol 178(10):6514–6521
Mihai S, Hirose M, Wang Y, Thurman JM, Holers VM, Morgan BP et al (2018) Specific inhibition of complement activation significantly ameliorates autoimmune blistering disease in mice. Front Immunol 9:535
Heppe EN, Tofern S, Schulze FS, Ishiko A, Shimizu A, Sina C et al (2017) Experimental laminin 332 mucous membrane pemphigoid critically involves C5aR1 and reflects clinical and Immunopathological characteristics of the human disease. J Invest Dermatol 137(8):1709–1718
Liu Z, Giudice GJ, Zhou X, Swartz SJ, Troy JL, Fairley JA et al (1997) A major role for neutrophils in experimental bullous pemphigoid. J Clin Invest 100(5):1256–1263
Liu Z, Li N, Diaz LA, Shipley M, Senior RM, Werb Z (2005) Synergy between a plasminogen cascade and MMP-9 in autoimmune disease. J Clin Invest 115(4):879–887
Liu Z, Shapiro SD, Zhou X, Twining SS, Senior RM, Giudice GJ et al (2000) A critical role for neutrophil elastase in experimental bullous pemphigoid. J Clin Invest 105(1):113–123
Liu Z, Shipley JM, Vu TH, Zhou X, Diaz LA, Werb Z et al (1998) Gelatinase B-deficient mice are resistant to experimental bullous pemphigoid. J Exp Med 188(3):475–482
Liu Z, Zhou X, Shapiro SD, Shipley JM, Twining SS, Diaz LA et al (2000) The serpin alpha1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell. 102(5):647–655
Lin L, Hwang BJ, Culton DA, Li N, Burette S, Koller BH et al (2018) Eosinophils mediate tissue injury in the autoimmune skin disease bullous pemphigoid. J Invest Dermatol 138(5):1032–1043
Sezin T, Krajewski M, Wutkowski A, Mousavi S, Chakievska L, Bieber K et al (2017) The leukotriene B4 and its receptor BLT1 act as critical drivers of neutrophil recruitment in murine bullous pemphigoid-like epidermolysis bullosa acquisita. J Invest Dermatol 137(5):1104–1113
Sadik CD, Miyabe Y, Sezin T, Luster AD (2018) The critical role of C5a as an initiator of neutrophil-mediated autoimmune inflammation of the joint and skin. Semin Immunol 37:21–29
Sezin T, Murthy S, Attah C, Seutter M, Holtsche MM, Hammers CM et al (2019) Dual inhibition of complement factor 5 and leukotriene B4 synergistically suppresses murine pemphigoid disease. JCI Insight 4(15)
Sadik CD, Kim ND, Iwakura Y, Luster AD (2012) Neutrophils orchestrate their own recruitment in murine arthritis through C5aR and FcgammaR signaling. Proc Natl Acad Sci U S A 109(46):E3177–E3185
Sadik CD, Kim ND, Luster AD (2011) Neutrophils cascading their way to inflammation. Trends Immunol 32(10):452–460
Sadik CD, Luster AD (2012) Lipid-cytokine-chemokine cascades orchestrate leukocyte recruitment in inflammation. J Leukoc Biol 91(2):207–215
Chakievska L, Holtsche MM, Kunstner A, Goletz S, Petersen BS, Thaci D et al (2019) IL-17A is functionally relevant and a potential therapeutic target in bullous pemphigoid. J Autoimmun 96:104–112
Le Jan S, Plee J, Vallerand D, Dupont A, Delanez E, Durlach A et al (2014) Innate immune cell-produced IL-17 sustains inflammation in bullous pemphigoid. J Invest Dermatol 134(12):2908–2917
Plee J, Le Jan S, Giustiniani J, Barbe C, Joly P, Bedane C et al (2015) Integrating longitudinal serum IL-17 and IL-23 follow-up, along with autoantibodies variation, contributes to predict bullous pemphigoid outcome. Sci Rep 5:18001
Giusti D, Bini E, Terryn C, Didier K, Le Jan S, Gatouillat G et al (2019) NET formation in bullous pemphigoid patients with relapse is modulated by IL-17 and IL-23 interplay. Front Immunol 10:701
Shimanovich I, Mihai S, Oostingh GJ, Ilenchuk TT, Brocker EB, Opdenakker G et al (2004) Granulocyte-derived elastase and gelatinase B are required for dermal-epidermal separation induced by autoantibodies from patients with epidermolysis bullosa acquisita and bullous pemphigoid. J Pathol 204(5):519–527
Yu X, Holdorf K, Kasper B, Zillikens D, Ludwig RJ, Petersen F (2010) FcgammaRIIA and FcgammaRIIIB are required for autoantibody-induced tissue damage in experimental human models of bullous pemphigoid. J Invest Dermatol 130(12):2841–2844
Stahle-Backdahl M, Inoue M, Guidice GJ, Parks WC (1994) 92-kD gelatinase is produced by eosinophils at the site of blister formation in bullous pemphigoid and cleaves the extracellular domain of recombinant 180-kD bullous pemphigoid autoantigen. J Clin Invest 93(5):2022–2030
Niimi Y, Pawankar R, Kawana S (2006) Increased expression of matrix metalloproteinase-2, matrix metalloproteinase-9 and matrix metalloproteinase-13 in lesional skin of bullous pemphigoid. Int Arch Allergy Immunol 139(2):104–113
Verraes S, Hornebeck W, Polette M, Borradori L, Bernard P (2001) Respective contribution of neutrophil elastase and matrix metalloproteinase 9 in the degradation of BP180 (type XVII collagen) in human bullous pemphigoid. J Invest Dermatol 117(5):1091–1096
Lin L, Bankaitis E, Heimbach L, Li N, Abrink M, Pejler G et al (2011) Dual targets for mouse mast cell protease-4 in mediating tissue damage in experimental bullous pemphigoid. J Biol Chem 286(43):37358–37367
Lin L, Betsuyaku T, Heimbach L, Li N, Rubenstein D, Shapiro SD et al (2012) Neutrophil elastase cleaves the murine hemidesmosomal protein BP180/type XVII collagen and generates degradation products that modulate experimental bullous pemphigoid. Matrix Biol 31(1):38–44
Marzano AV, Tedeschi A, Berti E, Fanoni D, Crosti C, Cugno M (2011) Activation of coagulation in bullous pemphigoid and other eosinophil-related inflammatory skin diseases. Clin Exp Immunol 165(1):44–50
Cugno M, Marzano AV, Bucciarelli P, Balice Y, Cianchini G, Quaglino P et al (2016) Increased risk of venous thromboembolism in patients with bullous pemphigoid. The INVENTEP (INcidence of VENous ThromboEmbolism in bullous Pemphigoid) study. Thromb Haemost 115(1):193–199
Iwata H, Ujiie H (2017) Complement-independent blistering mechanisms in bullous pemphigoid. Exp Dermatol 26(12):1235–1239
Natsuga K, Nishie W, Shinkuma S, Ujiie H, Nishimura M, Sawamura D et al (2012) Antibodies to pathogenic epitopes on type XVII collagen cause skin fragility in a complement-dependent and -independent manner. J Immunol 188(11):5792–5799
Ujiie H, Sasaoka T, Izumi K, Nishie W, Shinkuma S, Natsuga K et al (2014) Bullous pemphigoid autoantibodies directly induce blister formation without complement activation. J Immunol 193(9):4415–4428
Li XX, Lee JD, Kemper C, Woodruff TM (2019) The complement receptor C5aR2: a powerful modulator of innate and adaptive immunity. J Immunol 202(12):3339–3348
Kang S, Tanaka T, Narazaki M, Kishimoto T (2019) Targeting Interleukin-6 signaling in clinic. Immunity. 50(4):1007–1023
Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei XF et al (1998) IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest 101(2):311–320
Murakami M, Kamimura D, Hirano T (2019) Pleiotropy and specificity: insights from the interleukin 6 family of cytokines. Immunity. 50(4):812–831
Samavedam UK, Kalies K, Scheller J, Sadeghi H, Gupta Y, Jonkman MF et al (2012) Recombinant IL-6 treatment protects mice from organ specific autoimmune disease by IL-6 classical signalling-dependent IL-1ra induction. J Autoimmun 40:74–85
Schmidt E, Bastian B, Dummer R, Tony HP, Brocker EB, Zillikens D (1996) Detection of elevated levels of IL-4, IL-6, and IL-10 in blister fluid of bullous pemphigoid. Arch Dermatol Res 288(7):353–357
Sadeghi H, Lockmann A, Hund AC, Samavedam UK, Pipi E, Vafia K et al (2015) Caspase-1-independent IL-1 release mediates blister formation in autoantibody-induced tissue injury through modulation of endothelial adhesion molecules. J Immunol 194(8):3656–3663
Bieber K, Sun S, Witte M, Kasprick A, Beltsiou F, Behnen M et al (2017) Regulatory T cells suppress inflammation and blistering in pemphigoid diseases. Front Immunol 8:1628
Antiga E, Quaglino P, Volpi W, Pierini I, Del Bianco E, Bianchi B et al (2014) Regulatory T cells in skin lesions and blood of patients with bullous pemphigoid. J Eur Acad Dermatol Venereol 28(2):222–230
Kulkarni U, Karsten CM, Kohler T, Hammerschmidt S, Bommert K, Tiburzy B et al (2016) IL-10 mediates plasmacytosis-associated immunodeficiency by inhibiting complement-mediated neutrophil migration. J Allergy Clin Immunol 137(5):1487–97 e6
Sugimoto MA, Vago JP, Perretti M, Teixeira MM (2019) Mediators of the resolution of the inflammatory response. Trends Immunol 40(3):212–227
Headland SE, Norling LV (2015) The resolution of inflammation: principles and challenges. Semin Immunol 27(3):149–160
Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH et al (2005) Molecular circuits of resolution: formation and actions of resolvins and protectins. J Immunol 174(7):4345–4355
Funding
This work was supported by the Schleswig-Holstein Cluster of Excellence Precision Medicine in Chronic Inflammation (EXC 2167-390884018) and the Clinical Research Unit 303 Pemphigoid Diseases (Sa1960/5-1, SCHM1686/7-2).
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on Resolution of Inflammation in Chronic Diseases - Guest Editor: Markus Neurath
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sadik, C.D., Schmidt, E. Resolution in bullous pemphigoid. Semin Immunopathol 41, 645–654 (2019). https://doi.org/10.1007/s00281-019-00759-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-019-00759-y