
Abstract
The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor historically studied for its role in environmental chemical-mediated toxicity and carcinogenicity. In the last 5 years, however, it has become clear that the AhR, presumably activated by endogenous ligand(s), plays an important role in immune system development and function. Other articles in this edition summarize AhR function during T cell and antigen-presenting cell development and function, including the effects of AhR activation on dendritic cell function, T cell skewing, inflammation, and autoimmune disease. Here, we focus on AhR expression and function during B cell differentiation. Studies exploiting immunosuppressive environmental chemicals to probe the role of the AhR in humoral immunity are also reviewed to illustrate the multiple levels at which a “nominally activated” AhR could control B cell differentiation from the hematopoietic stem cell through the pro-B cell, mature B cell, and antibody-secreting plasma cell stages. Finally, a putative role for the AhR in the basic biology of B cell malignancies, many of which have been associated with exposure to environmental AhR ligands, is discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The study of the aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor, has come a long way. For many years, analysis of AhR function and activity was the purview solely of toxicologists interested in understanding how environmental chemicals are “sensed” by biological organisms. With regard to the immune system in particular, immunotoxicologists focused on a set of environmentally common, immunosuppressive chemicals including dioxins, most notably 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), polychlorinated biphenyls (PCBs), and polycyclic aromatic hydrocarbons (PAHs). These studies, in essence, exploited environmental chemicals as probes of biological systems to understand how the AhR functions and to begin to reveal for what purpose this evolutionarily conserved receptor/transcription factor exists. These studies were of enormous value since they provided a scaffold on which to build theories of the “nominal” function of the AhR. They also suggested that, at various points in B cell development and differentiation, B cells themselves, or stromal cells on which B cells depend for developmental signals, express AhR and serve as the immediate targets of endogenous or exogenous AhR ligands. Indeed, analysis of gene expression profiles in a panel of purified, developmentally defined normal murine B cells [1] demonstrates a hierarchy of AhR expression during B cell development (Fig. 1). Bone marrow pro- and pre-B cells express little or no AhR mRNA. In contrast, splenic transitional B cells, representing cells recently activated during clonal selection, have elevated AhR levels. Follicular, marginal zone, or germinal center B cells and plasmablasts express modest but variable AhR levels. Interestingly, plasma cells express high AhR levels, suggesting a role for the AhR in plasma cell development and/or function. This issue will be readdressed later in this manuscript.

Relative AhR mRNA expression in purified subpopulations of murine B cells. Microarray data were generated by Green et al. [1] from murine B cells sorted by flow cytometry based on B developmental stage-specific surface antigens. Expression levels of the AhR transcript within the listed differentiation stages was extracted from [1] and the corresponding distributions summarized and displayed as 'box-and-whiskers' plots (with the bottom and the top of the box corresponding to the first and third quartiles, the thick band inside the box indicating the median, and the end of the 'whiskers' extending to 1.5 times the interquartile range in both directions)
Here, we summarize representative studies that illustrate the multiple levels at which the AhR may contribute to B cell development and function. We begin with early studies that employed either in vivo models or complex in vitro systems consisting of cocultures of B cells and cells from the lymphoid microenvironment. We continue with models using B cell lines or purified B cells to assess the role of the AhR in activated B cell and plasma cell differentiation and in antibody production and conclude with a discussion of the likely role of the AhR in B cell malignancies.
Environmental chemicals as probes for AhR control of B cell development and function in vivo or in complex organ culture systems
Early studies that evaluated the mechanisms by which dioxins, PAHs, and PCBs mediate immunosuppression demonstrated that these environmental AhR ligands suppress immunity by compromising virtually every stage of lymphocyte development, activation, and effector function, implying that the AhR plays an important role at several levels of B cell differentiation. For example, halogenated aromatic hydrocarbons were shown to suppress B and T cell development in primary lymphoid organs and to compromise antibody responses in vivo and in organ cultures [2–9]. TCDD, the quintessential high-affinity AhR ligand, was shown to be particularly immunosuppressive, significantly inhibiting lymphocyte development in vivo at doses in the nanograms per kilogram range [10]. For example, administration of 10–100 ng/kg TCDD resulted in a significant increase in mortality after influenza virus infection [11–14] and weakened memory responses [15]. At slightly higher doses (100–1,000 ng/kg), TCDD induced thymic atrophy [16–18], reduced resistance to parasites [19], and suppressed humoral responses in an AhR-dependent fashion [9, 20, 21]. The ability of AhR binding, but not nonbinding TCDD congeners, to affect the suppression of B cell responses and the relative resistance of lymphocytes generated from mice expressing low-affinity AhR (AhRd) to TCDD [19] confirmed that TCDD-induced inhibition of humoral responses is AhR mediated.
PAHs and PCBs were also shown to suppress B cell responses in vivo, although likely through somewhat different mechanisms than TCDD. For example, the prototypic PAHs and AhR ligands, 7,12-dimethylbenz[a]anthracene and benzo[a]pyrene (B[a]P), suppressed in vivo humoral immune responses, and this immunosuppression was blocked by the addition of the partial AhR agonist/AhR inhibitor α-naphthoflavone [22, 23]. PAHs also decreased splenic B lymphocyte numbers and reduced the number of antigen-specific effector B cells [24–26].
Similarly, the halogenated hydrocarbons, 3,3′,4,4′-tetrachlorobiphenyl and 2,3,3′,4,4′,5-hexachlorobiphenyl, suppressed antibody responses to challenge with lipopolysaccharide (LPS) or sheep red blood cells [27, 28]. At least some of this apparent suppression of B cell differentiation induced by PAHs and PCBs was dependent on the degree to which the AhR ligand could be metabolized [22, 23, 29]. This conclusion still has relevance to AhR function, since the activation of the AhR regulates the transcription of a battery of P450 genes (CYP1A1, CYP1A2, and CYP1B1) critical to oxidative, phase I metabolism of environmental or endogenous compounds [30, 31]. Furthermore, the fact that all of these outcomes were AhR dependent suggested that the AhR is expressed in multiple components of the immune system and, therefore, is likely to play a role in the development of mature B cell responses.
Since developing biological systems are more sensitive to environmental stressors than mature systems, one might predict that developing B cells would be more sensitive to AhR ligands than mature, antibody-secreting B lineage cells. Model systems of B cell development involving cultures of bone marrow cells containing both B lineage cells and bone marrow stromal cells (e.g., Whitlock/Witte cultures) were used to test this hypothesis. Since bone marrow B cells are poised to undergo clonal deletion in response to self-antigen [32, 33], it was predicted that these cells would have a low threshold of apoptosis induction in response to AhR ligands. Indeed, in long-term Whitlock/Witte cultures of primary bone marrow cells, relatively low PAH doses (10 nM) rapidly induced apoptosis in B220+/CD43−/sIg− pre-B cells (B cell fraction D) or in a bone marrow–stromal cell-dependent B220+/CD43+ primary pro/pre-B cell line (BU-11) (B cell fraction B/C) [4, 5]. That the AhR was required for PAH-mediated apoptosis induction was supported by the ability of α-naphthoflavone or galangin, two partial AhR agonists (effective antagonists in the presence of a higher-affinity AhR ligand), to block apoptosis [4, 34]. Studies on apoptosis signaling pathways indicated: (1) PAH-induced downregulation of the anti-apoptotic NF-κB subunits Rel A and c-Rel as well as the anti-apoptotic NF-κB gene target c-myc [35, 36], (2) robust induction of the intrinsic apoptosis pathway involving cytochrome c release from mitochondria but not a mitochondrial membrane depolarization, (3) activation of APAF1 and formation of the apoptosome, (4) triggering of a caspase-8-dependent positive feedback loop, and (5) activation of executioner caspase-3 [37–39]. Interestingly, physical contact between the culture dish-adherent bone marrow stromal cells and stromal cell-adherent pro/pre-B cells was required for PAH-induced apoptosis [38], and treatment of pro/pre-B cells in the absence of stromal cells but in the presence of supportive IL-7 failed to induce B cell death [5, 40]. These results indicated that pro/pre-B cell apoptosis is the result of a “gain of function” as opposed to, for example, the loss of the production by stromal cells of cytokines critical to B cell survival. Surprisingly, murine bone marrow stromal cells, but not pro- or pre-B cells, were shown to express AhR [4, 5]. These data are consistent with microarray results in which little or no AhR mRNA was detected at the pro- or pre-B cell stage (Fig. 1). Furthermore, PAHs induced bone marrow B cell apoptosis in cocultures of primary stromal cells from AhR+/+ but not from AhR−/− mice [40]. These results indicate that stromal cells are a direct target of PAH and suggest that stromal cells deliver a cell contact-dependent “death signal” to adjacent pro/pre-B cells. Since bone marrow stromal cells express a functional AhR and since several hematopoietic cell types depend on these cells for growth and differentiation signals, it seems likely that AhR activity in the bone marrow microenvironment contributes, in an as yet undetermined manner, to the development of several hematopoietic lineages. AhR expression in stromal cells also suggests the possibility that aberrant AhR activation in the bone marrow microenvironment, for example by environmental ligands, contributes to the development of B cell malignancies that originate in the bone marrow, e.g., multiple myeloma, a disease already associated with exposure to environmental AhR ligands (see the succeeding paragraphs).
The failure of poorly metabolized AhR ligands to induce pre-B cell apoptosis [41], and a requirement for AhR-dependent CYP1B1 expression in bone marrow stromal cells for PAH-induced bone marrow B cell apoptosis [42, 43], indicated that AhR activation alone is not sufficient to induce B cell apoptosis and suggested that AhR-regulated PAH metabolism is required for the induction of an apoptosis signal. Indeed, the addition of PAH metabolites to bone marrow cultures obviated the need for AhR+ stromal cells for apoptosis induction in bone marrow B cells [41, 43]. Interestingly, stromal cell-derived PAH metabolites were shown to be transferred from the stromal cells to stromal cell-adherent bone marrow B cells by a unique mechanism, i.e., exchange of membranes between the two cell types (trogocytosis) [38]. The predicted sensitivity of developing B cells was supported by the failure of bone marrow stromal cells to exchange membranes with or to induce apoptosis in mature B cells or T cells, neither of which undergo apoptosis in response to PAH treatment even in the presence of stromal cells [38]. Collectively, these studies emphasize the dependence of early B cells on their stromal microenvironment and indicate that the AhR may control early B cell development indirectly by altering the bone marrow milieu. The demonstration of aberrations in bone marrow B cell development in AhR−/− mice is consistent with this model [8].
A note of caution is required in interpreting experiments with exogenous sources of AhR ligands. It is well established that different AhR ligands induce different outcomes in a tissue-specific and context-specific manner. Outcomes with TCDD may or may not exactly replicate outcomes with endogenous AhR ligands. Therefore, studies utilizing any surrogate AhR ligand can only demonstrate the presence of a functional AhR and suggest, but not prove, the nature of the AhR response to other ligands, including the response to endogenous ligands made either by the B lineage cell itself or its microenvironment.
Environmental chemicals as probes for AhR control of B cell development and function in clonal or purified B cell model systems
While the studies described previously were important for assessing AhR-mediated events in vivo or in systems designed to model interactions between developing B cells and their microenvironment, they did not determine if AhR ligands directly affect AhR+ mature B cells and, by inference, if the AhR plays a significant role in intracellular B cell signaling. In this vein, purified peripheral human B cells were isolated to assess AhR expression levels and to determine if mature B cells are affected by AhR engagement. Consistent with microarray data from purified murine B cell populations (Fig. 1), resting human B cells were shown to express relatively low AhR levels [44]. However, activation with CpG or CD40 ligand, surrogates for stimuli invoked during innate and adaptive immune responses, respectively, profoundly upregulated AhR mRNA and protein [44]. IL-4 treatment alone induced AhR expression in both murine and human B cells through a STA6-dependent pathway [45]. LPS or PMA + ionophore activation of murine splenic B cells similarly increased AhR and ARNT expression [46] and rendered activated B cells more sensitive to AhR ligands than resting B cells [19, 47, 48]. AhR nuclear translocation, constitutive DNA binding, and induction of the AhR target gene CYP1A1 in CpG-stimulated or CD40 ligand-stimulated human B cells [44], in PMA + ionophore-induced, and in IL-4-activated murine splenic B cells [47] suggested the presence of endogenous AhR ligands that drive AhR signaling in cultures of activated B cells. Inhibition of AhR activity by ectopic expression of an AhR repressor-encoding gene (AhRR) inhibited the proliferation of CpG-activated or CD40 ligand-activated human B cells (unpublished), suggesting that the AhR contributes to activated B cell proliferation. These results are consistent with those obtained in AhR−/− mice in which deficiencies in the accumulation of mature splenic lymphocytes, as well as peritoneal CD5+ B-1 cells, were noted [49]. A role for the AhR in B cell growth is consistent with studies performed with other cell types in which constitutively active AhR was shown to regulate the cell cycle [50–52]. In this context, “constitutively active” is operationally defined as AhR continuously activated by endogenous ligand(s). Dimerization of the AhR with Rb, E2F [53–56], Rel A [57, 58], or Rel B [59] suggests some mechanisms through which the AhR could influence B cell growth. These findings also suggest a possible role for the AhR in regulating apoptosis in B cells. Indeed, at least one study demonstrated that transformed human B lymphoma cells undergo apoptosis on exposure to PAH [60]. Other studies have implicated the AhR in the control of apoptosis in other cell types [61–64].
Microarray studies demonstrate relatively high AhR levels in murine plasma cells (Fig. 1). In order to determine if the AhR plays a role in the differentiation of human B cells into plasma cells, we developed an in vitro system in which up to 40 % of CD40 ligand-activated, AhRhigh human B cells could be induced to differentiate into plasma cells in the absence of feeder cells [65]. Since differentiation could be induced even when cell growth was blocked with low-level irradiation, the effects of AhR activation on differentiation and cell growth could be separated. In this system, AhR hyperactivation with B[a]P, a prototypic environmental PAH/AhR ligand, significantly blocked CD40 ligand-driven and cytokine-driven differentiation into CD138+ plasma cells in the presence or absence of cell growth without affecting cell viability [65]. As with the cocultures of bone marrow stromal cells and pre- or pro/pre-B cells, AhR-regulated metabolism of the parent PAH was required for the inhibition of plasma cell formation.
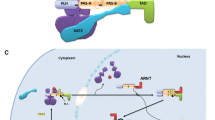
The contribution of the AhR to antibody secretion was studied extensively in a CD5+ murine B cell lymphoma line (CH12.LX). In early studies, it was shown that hyperactivation of the AhR with TCDD alters the ability of these AhR+ cells to produce antibody in response to LPS by binding to and inhibiting the transcription of the 3′ alpha immunoglobulin heavy chain gene [66–71]. Inhibition of antibody production was not seen in AhR− BCL-1 B cells [72]. AP1 (c-Jun), a component of the LPS-activated TLR-4 signaling pathway [73], also appeared to be targeted since its expression and function were downregulated by TCDD in AhR+ CH12.LX, but not in AhR− BCL-1, cells [72]. Follow-up studies further implicated the AhR in B cell differentiation by demonstrating that hyperactivated AhR decreases Blimp-1 expression and binding to the PAX5 gene promoter [66]. The decrease in Prdm1 (Blimp-1) transcription appears to be tied to the aforementioned decrease in AP1 since TCDD treatment decreased AP1 binding to Prdm1 [66]. Prdm1 transrepression may also be linked to an AhR-dependent transactivation of Bach2, a Prdm1 repressor [74]. That is, TCDD-activated AhR binds to a cognate site in the first intron of Bach2, increasing Bach2 expression and binding to the Maf elements in Prdm1 [74]. This circuit of transcription factors controlling B/plasma cell differentiation is summarized in Fig. 2.

AhR interactions with B cell differentiation decisions. A “bi-stable circuit” controls the decision of activated B cells to differentiate into antibody-secreting plasma cells. The consensus “all-or-none” switching pathway involves five interacting transcription factors: Prdm (encoding Blimp-1), AP1, Bach2, Pax5 (BSAP), Bcl6, and XBP1. The AhR has been shown to suppress the transcription of AP1, Prdm1, and IgM while enhancing the transcription of Bach2

AhR mRNA expression varies among five human B lineage cancers. Relative levels of AhR expression in five human B lineage cancers were assessed by analysis of microarray data obtained from 1,036 human cancer cell lines (http://www.broadinstitute.org/ccle/home). Each box plot reports the distribution of the AhR transcript within the samples belonging to the corresponding B lineage cancer type
These comprehensive studies illustrate the intersection of AhR signaling with several interconnecting pathways of B cell differentiation and effector function. Apropos of this, it has been postulated [75] that the AhR could alter B cell function by inhibiting antibody production to a level dictated by the degree of AhR activation and/or by interference with the “bi-stable switch circuit” involving Bcl6, Blimp-1, PAX5, and Bach2 and known to control an all-or-none cellular decision to differentiate from activated B cell into antibody-secreting plasma cell [76, 77] (Fig. 2). The demonstration that the AhR directly binds to the 3′ alpha Ig heavy chain promoter [66–71] suggests a mechanism for the former possibility, while AhR control of AP1 [72], Prdm1 [66], and Bach2 transcription [74] is consistent with the latter. To distinguish between these two, non-mutually exclusive models, Zhang et al. performed an elegant study in which the propensity for LPS to induce antibody production and/or an all-or-none commitment to generating plasma cells in the presence or absence of TCDD was evaluated [75]. Using a combination of computational biology and flow cytometric analysis of LPS-induced antibody production and B cell terminal differentiation, it was concluded that TCDD-induced suppression of the IgM response occurs as a binary function, i.e., TCDD reduces the number of IgM-secreting cells in a dose-dependent manner in an “all-or-none” response rather than by proportionally decreasing the amount of antibody produced by any given plasma cell [75].
A more global analysis of potential targets of the AhR in TCDD-treated, LPS-activated CH12LX B lymphoma cells revealed a number of potential interactions between the AhR and genes critical to B cell development and function [78]. In this study, 1,893 regions, 1,035 of which mapped to within 10 kb of a known gene, exhibited increased AhR binding after TCDD treatment. These identified regions were then compared to a gene expression profile in which 422 genes exhibited increased expression 8 h after AhR activation. Seventy-eight of the upregulated genes were also contained in the set of 1,035 regions immunoprecipitated with the AhR, suggesting direct interactions between the AhR and these genes. Several of these genes are known to be critical to B cell development or function and contain multiple consensus AhR binding sites (5′-TNGCGTG-3′) within 3,000 bp upstream and 299 bp downstream of their respective gene start sites. (AhR binding sites are known to be located further upstream than is generally considered to be part of a prototypical gene promoter [79, 80]). For example, c-myc (seven consensus AhREs) plays a critical role in normal B cell growth [81] and has previously been shown to be directly regulated by the AhR in human mammary tumor cells [57, 82]. Runx2 and Runx3 (eight and seven AhREs, respectively), although most frequently associated with osteoblast [83] and T cell development [84], respectively, also appear to be involved in memory B cell formation [85]. XBP1 (four AhREs), repressed by Pax5 (nine AhREs), plays a critical role in plasma cell differentiation [86]. Finally, SOCS3 (five AhREs) controls the response of B cells and plasma cells to STAT-3-dependent cytokines, including IL-21 and IL-6, and is critical to the formation of germinal centers [87]. Therefore, there is the potential for the AhR, activated by as yet unidentified endogenous ligands, to influence the transcription of several master regulators of B cell development.
AhR control of hematopoietic stem cell (HSC) development
The differentiation of HSCs into all eight blood cell lineages is a tightly regulated process [88] that changes in subtle but important ways during life [89–92]. Disruption of this regulation has a profound downstream effect on multiple hematopoietic cell types, including B cells, leading to mixed lineage leukemias [93], lymphomas [94], stem cell exhaustion [95], and other blood cell disorders [96, 97]. Therefore, any role that the AhR plays in HSC differentiation will have a bearing on B cell development. Several studies indicate that murine and human HSC express modest AhR levels [98–100] and recent breakthrough studies indicate that the AhR plays a critical role in HSC growth and differentiation [95, 101–105]. For example, in vivo AhR modulation disrupts HSC growth, senescence, and migration [95, 101–103, 106, 107]. Of note is that most of these studies used environmental AhR ligands as probes to establish the nominal function of the AhR in HSCs [101, 102, 104, 106, 107]. Furthermore, AhR−/− mice exhibit an increased number of bone marrow HSCs [95] and pro/pre-B cells [8]. Perhaps most dramatically, AhR inhibition promotes the expansion of purified, human HSC, suggesting the use of AhR inhibitors as a clinical method for expanding HSC populations prior to stem cell transplant [105].
The AhR in B cell malignancies
The studies summarized previously demonstrate that the AhR plays an integral role in B cell development, likely through controlling cell growth and apoptosis. In other types of cells, the AhR regulates cell migration potentially through Slug, Vav3, TGF-β, and/or c-Jun [108–113]. Since dysregulated cell growth, apoptosis, and migration are hallmarks of cancer, it could reasonably be hypothesized that aberrant AhR expression or activity could contribute to B cell malignancies. One prediction from this hypothesis would be an association between exposure to environmental AhR ligands and the risk of B lymphomas, leukemias, or multiple myeloma. Consistent with this prediction, exposure to TCDD, halogenated hydrocarbon-containing organochlorine pesticides, or PCBs significantly increases the risk of non-Hodgkin’s lymphoma (NHL) [114–116]. In perhaps the best documented study of TCDD exposure which occurred following an explosion at an herbicide manufacturing facility in Seveso, Italy, a significant increase in the risk of NHL was documented in inhabitants of the surrounding communities (relative odds ratio = 4.45) [117]. In addition, human AhR polymorphisms have been linked to the risk of NHL following exposure to organochlorines, some of which are AhR ligands [118]. Similarly, the risk of multiple myeloma in the TCDD-exposed Seveso population was significantly elevated, with a relative risk of 3.07 [117, 119]. Although controversial, a link between multiple myeloma risk in Vietnam War veterans exposed to the TCDD-contaminated defoliant, Agent Orange, has been suggested by a National Academy of Sciences review committee (http://books.nap.edu/openbook.php?record_id=13166). In interpreting these studies, it is important to note that TCDD, which exhibits a biological half-life of 7–11 years in humans, is a known human carcinogen but is not genotoxic, i.e., it does not directly induce mutations. Thus, its carcinogenicity likely reflects, at least in part, persistent AhR signaling.
In a fashion analogous to the analysis of immune modulation by environmental AhR ligands to elucidate AhR function during normal B cell development, analysis of cancer induction with environmental AhR ligands pointed to a general role for the AhR in malignant B cell transformation. That the AhR plays a role in B cell malignancy regardless of cancer etiology was supported by many studies demonstrating elevated AhR levels and “constitutive” activity in a variety of cancer cell lines including lymphomas, myelomas, and T cell leukemias [50, 82, 120–126].
Microarray analysis of 1,036 human cancer cell lines generated at the Broad Institute, i.e., the Cancer Cell Line Encyclopedia (http://www.broadinstitute.org/ccle/home) (Fig. 3), revealed a hierarchy of AhR expression in which low levels of AhR were expressed in diffuse large B cell lymphomas, unspecified B cell lymphomas, and myelomas and notably higher levels in Hodgkin’s lymphomas and chronic lymphocytic leukemias (Fig. 4). Interestingly, expression of three (CYP1B1, TIPARP, and AhRR) of four (CYP1B1, TIPARP, AhRR, and NQO1) well-established AhR target genes chosen at random appear to track with AhR expression in the B lineage cancer subtypes, suggesting, but not proving, constitutive AhR activity in these tumor lines as previously documented in other tumor types [50, 82, 120–126].

Relative expression of AhR mRNA and AhR target genes in human tumor cell lines. Analysis of microarray data obtained from 1,036 human cancer cell lines (http://www.broadinstitute.org/ccle/home) is presented. a Data corresponding to the five lymphoid malignancies listed and five transcripts, including AhR and four of its putative targets, are displayed as a color-coded gene-by-sample heat map, with rows (genes) and columns (samples) sorted by hierarchical clustering [136]. b Microarray data for the same five lymphoid cancers were analyzed, and genes ranked by Pearson correlation between the level of AhR expression and that of four known AhR target genes, CYP1B1, NQO1, TIPARP, and AhRR. Permutation-based p values and the corresponding FDR-corrected q values are shown. c A Kolmogorov–Smirnov test was performed to assess the strength of the association between AhR and its four targets. The x-axis lists the genes in the human transcriptome sorted by their distance from AhR (from the closest, left, to the furthest, right). The position of the four AhR targets (red ticks) is significantly skewed toward the left-hand side of the list (permutation-based p value = 0.021) [137]
Although the molecular mechanisms through which constitutively active AhR may contribute to B lineage cancers is unknown, several possibilities exist. With regard to Burkitt’s lymphoma, the AhR complex directly interacts with EBNA-3, a protein required for EBV-mediated cell transformation and involved in cell growth and survival. This interaction enhances AhR nuclear translocation and reporter gene transactivation [127]. Using histiocytic lymphoma, Burkitt’s lymphoma, and NHL cell lines, Vogel et al. demonstrated that AhR hyperactivation with TCDD resulted in a loss of the apoptosis response, likely through the modulation of cyclooxygenase-2 (Cox-2) and Bcl-xL [128]. Both Cox-2 and Bcl-xL are known to also inhibit apoptosis in B chronic lymphocytic leukemias [129]. Furthermore, it was noted that TCDD promoted the development of lymphomas and Cox-2 expression in lymphoma-bearing lymph nodes [128]. With regard to TGF-β, a cytokine that inhibits lymphoma apoptosis, AhR has been shown to both suppress [130, 131] and enhance [121] TGF-β expression in a tissue-specific and/or ligand-specific fashion.
Finally, the role of the AhR in the development of B lineage malignancies may not be restricted to the transforming cell itself but may be a function of AhR-dependent events in either the malignant cell or the tumor microenvironment, specifically in AhR+ bone marrow stromal cells [4, 5, 40, 132]. For example, a constitutively active AhR increases IL-6 production in head and neck cancers [109] and modulates IL-6 production in bone marrow stromal cells [133]. IL-6, produced by the bone marrow microenvironment, is a critical cytokine in the development and maintenance of multiple myeloma [134].
Conclusions
The use of environmental AhR ligands has enabled immunotoxicologists to probe the immune system in order to identify in which cells and at what stage of their development the AhR is expressed and functional. These studies demonstrated that the AhR is variably expressed during B cell differentiation from the HSC to the antibody-secreting cell stage (Fig. 5). These results imply that the AhR is a critical mediator of B cell development and function and that environmental AhR ligands have the potential to adversely affect B lineage cells at multiple levels. The expression of a functional AhR in bone marrow stromal cells further indicates that the AhR may affect B cell development indirectly by altering the function of bone marrow stromal cells critical to B cell growth and differentiation (Fig. 5). Notably, the expression of the AhR in both HSCs and bone marrow stromal cells suggests that the AhR may have a more global effect on other hematopoietic cell lineages, all of which derive from common HSCs and which require signals provided by bone marrow stromal elements.

Role of the AhR in the development of normal and malignant B cells. AhR is expressed variably during B cell differentiation. While HSC express low AhR levels, AhR expression is lost by the pro-B cell stage. Pre-B cells are similarly AhR−. Transitional splenic B cells, which may have been recently activated by low-affinity autoantigens [138], upregulate AhR expression. Following clonal selection, resting B cells express little or no AhR. AhR is again upregulated on activation by foreign antigens with T cell help and during differentiation into plasma cells. AhR+ bone marrow stromal cells facilitate normal bone marrow B cell growth and differentiation and likely play a critical role in supporting B lineage malignancies including lymphomas and multiple myelomas
These studies also point to a possible role for the AhR in B cell malignancies, the incidence of which has risen considerably since 1975 (http://seer.cancer.gov/statfacts/html/nhl.html) [135]. That is, AhR expression in B cell malignancies or in the bone marrow microenvironment provides a mechanism through which three large classes of environmental chemicals, dioxins, planar PCBs, and PAHs, could contribute to B cell cancers. Of equal importance is the likelihood that the AhR, in the absence of environmental chemicals but, presumably, in the presence of endogenous ligands, contributes to B lineage cancers by influencing cell growth and/or survival either directly within the malignant cell or indirectly via the tumor microenvironment. If proven, this hypothesis would raise the exciting possibility that the AhR signaling pathway could be targeted for B lineage cancer therapy. Thus, studies exploiting environmental chemicals as biologic probes have not only helped to reveal the biological functions of what had previously been thought of only as an environmental chemical sensor but also now suggest a novel strategy for targeted cancer therapy.
References
Green MR, Monti S, Dalla-Favera R, Pasqualucci L, Walsh NC, Schmidt-Supprian M, Kutok JL, Rodig SJ, Neuberg DS, Rajewsky K, Golub TR, Alt FW, Shipp MA, Manis JP (2011) Signatures of murine B-cell development implicate Yy1 as a regulator of the germinal center-specific program. Proc Natl Acad Sci U S A 108(7):2873–2878. doi:10.1073/pnas.1019537108
Lai ZW, Fiore NC, Hahn PJ, Gasiewicz TA, Silverstone AE (2000) Differential effects of diethylstilbestrol and 2,3,7,8-tetrachlorodibenzo-p-dioxin on thymocyte differentiation, proliferation, and apoptosis in bcl-2 transgenic mouse fetal thymus organ culture. Toxicol Appl Pharmacol 168(1):15–24
Shepherd DM, Steppan LB, Hedstrom OR, Kerkvliet NI (2001) Anti-CD40 treatment of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-exposed C57Bl/6 mice induces activation of antigen presenting cells yet fails to overcome TCDD-induced suppression of allograft immunity. Toxicol Appl Pharmacol 170(1):10–22
Yamaguchi K, Matulka RA, Shneider AM, Toselli P, Trombino AF, Yang S, Hafer LJ, Mann KK, Tao XJ, Tilly JL, Near RI, Sherr DH (1997) Induction of PreB cell apoptosis by 7,12-dimethylbenz[a]anthracene in long-term primary murine bone marrow cultures. Toxicol Appl Pharmacol 147(2):190–203
Yamaguchi K, Near RI, Matulka RA, Shneider A, Toselli P, Trombino AF, Sherr DH (1997) Activation of the aryl hydrocarbon receptor/transcription factor and bone marrow stromal cell-dependent preB cell apoptosis. J Immunol 158(5):2165–2173
Kerkvliet NI, Steppan LB, Brauner JA, Deyo JA, Henderson MC, Tomar RS, Buhler DR (1990) Influence of the Ah locus on the humoral immunotoxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin: evidence for Ah-receptor-dependent and Ah-receptor-independent mechanisms of immunosuppression. Toxicol Appl Pharmacol 105(1):26–36
Thurmond TS, Silverstone AE, Baggs RB, Quimby FW, Staples JE, Gasiewicz TA (1999) A chimeric aryl hydrocarbon receptor knockout mouse model indicates that aryl hydrocarbon receptor activation in hematopoietic cells contributes to the hepatic lesions induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Appl Pharmacol 158(1):33–40
Thurmond TS, Staples JE, Silverstone AE, Gasiewicz TA (2000) The aryl hydrocarbon receptor has a role in the in vivo maturation of murine bone marrow B lymphocytes and their response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Appl Pharmacol 165(3):227–236
Vorderstrasse BA, Steppan LB, Silverstone AE, Kerkvliet NI (2001) Aryl hydrocarbon receptor-deficient mice generate normal immune responses to model antigens and are resistant to TCDD-induced immune suppression. Toxicol Appl Pharmacol 171(3):157–164
Vogel C, Donat S, Dohr O, Kremer J, Esser C, Roller M, Abel J (1997) Effect of subchronic 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure on immune system and target gene responses in mice: calculation of benchmark doses for CYP1A1 and CYP1A2 related enzyme activities. Arch Toxicol 71(6):372–382
Burleson GR, Lebrec H, Yang YG, Ibanes JD, Pennington KN, Birnbaum LS (1996) Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on influenza virus host resistance in mice. Fundam Appl Toxicol 29(1):40–47
Vorderstrasse BA, Bohn AA, Lawrence BP (2003) Examining the relationship between impaired host resistance and altered immune function in mice treated with TCDD. Toxicology 188(1):15–28
Head JL, Lawrence BP (2009) The aryl hydrocarbon receptor is a modulator of anti-viral immunity. Biochem Pharmacol 77(4):642–653. doi:10.1016/j.bcp.2008.10.031
Warren TK, Mitchell KA, Lawrence BP (2000) Exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) suppresses the humoral and cell-mediated immune responses to influenza A virus without affecting cytolytic activity in the lung. Toxicol Sci 56(1):114–123
Lawrence BP, Vorderstrasse BA (2004) Activation of the aryl hydrocarbon receptor diminishes the memory response to homotypic influenza virus infection but does not impair host resistance. Toxicol Sci 79(2):304–314
Laiosa MD, Lai ZW, Thurmond TS, Fiore NC, DeRossi C, Holdener BC, Gasiewicz TA, Silverstone AE (2002) 2,3,7,8-Tetrachlorodibenzo-p-dioxin causes alterations in lymphocyte development and thymic atrophy in hemopoietic chimeras generated from mice deficient in ARNT2. Toxicol Sci 69(1):117–124
Rhile MJ, Nagarkatti M, Nagarkatti PS (1996) Role of Fas apoptosis and MHC genes in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced immunotoxicity of T cells. Toxicology 110(1–3):153–167
Blaylock BL, Holladay SD, Comment CE, Heindel JJ, Luster MI (1992) Exposure to tetrachlorodibenzo-p-dioxin (TCDD) alters fetal thymocyte maturation. Toxicol Appl Pharmacol 112:207–213
Tucker AN, Vore SJ, Luster MI (1986) Suppression of B cell differentiation by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Mol Pharmacol 29(4):372–377
Fernandez-Salguero PM, Hilbert DM, Rudikoff S, Ward JM, Gonzalez FJ (1996) Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol Appl Pharmacol 140(1):173–179
Staples JE, Murante FG, Fiore NC, Gasiewicz TA, Silverstone AE (1998) Thymic alterations induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin are strictly dependent on aryl hydrocarbon receptor activation in hemopoietic cells. J Immunol 160(8):3844–3854
Ladics GS, Kawabata TT, White KL (1991) Suppression of the in vitro humoral immune response of mouse splenocytes by 7,12-dimethylbenz[a]anthracene metabolites and inhibition of immunosuppression by a-naphthoflavone. Toxicol Appl Pharmacol 110:31–44
Ladics GS, Kawabata TT, Munson AE, White KL Jr (1992) Evaluation of murine splenic cell type metabolism of benzo[a]pyrene and functionality in vitro following repeated in vivo exposure to benzo[a]pyrene. Toxicol Appl Pharmacol 116(2):258–266
De Jong WH, Kroese ED, Vos JG, Van Loveren H (1999) Detection of immunotoxicity of benzo[a]pyrene in a subacute toxicity study after oral exposure in rats. Toxicol Sci 50(2):214–220
Ward EC, Murray MJ, Lauer LD, House RV, Irons R, Dean JH (1984) Immunosuppression following 7,12-dimethylbenz[a]anthracene exposure in B6C3F1 mice. I. Effects on humoral immunity and host resistance. Toxicol Appl Pharmacol 75(2):299–308
Holladay SD, Smith BJ (1995) Benzo[a]pyrene-induced alterations in total immune cell number and cell-surface antigen expression in the thymus, spleen and bone marrow of B6C3F1 mice. Vet Human Toxicol 37:99–104
Silkworth JB, Antrim L (1985) Relationship between Ah receptor-mediated polychlorinated biphenyl (PCB)-induced humoral immunosuppression and thymic atrophy. J Pharmacol Exp Ther 235(3):606–611
Silkworth JB, Antrim LA, Sack G (1986) Ah receptor mediated suppression of the antibody response in mice is primarily dependent on the Ah phenotype of lymphoid tissue. Toxicol Appl Pharmacol 86(3):380–390
Silkworth JB, Antrim L, Grabstein EM (1984) Correlations between polychlorinated biphenyl immunotoxicity, the aromatic hydrocarbon locus, liver microsomal enzyme induction in C57BL/6 and DBA/2 mice. Toxicol Appl Pharmacol 75:156
Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP (2000) Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol 59(1):65–85
Shimizu Y, Nakatsuru Y, Ichinose M, Takahashi Y, Kume H, Mimura J, Fujii-Kuriyama Y, Ishikawa T (2000) Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A 97(2):779–782
Ruiz-Vela A, Gonzalez de Buitrago G, Martinez AC (1999) Implication of calpain in caspase activation during B cell clonal deletion. EMBO J 18(18):4988–4998
Scott DW, Tuttle J, Livnat D, Haynes W, Cogswell JP, Keng P (1985) Lymphoma models for B-cell activation and tolerance. II. Growth inhibition by anti-mu of WEHI-231 and the selection and properties of resistant mutants. Cell Immunol 93(1):124–131
Quadri S, Qadri A, Mann KL, Sherr DH (2000) The bioflavonoid galangin blocks aryl hydrocarbon receptor (AhR) activation and polycyclic aromatic hydrocarbon-induced pre-B cell apoptosis. Mol Pharmacol 58:515–525
Mann KK, Doerre S, Schlezinger JJ, Sherr DH, Quadri S (2001) The role of NF-kappaB as a survival factor in environmental chemical-induced pre-B cell apoptosis. Mol Pharmacol 59(2):302–309
Ryu HY, Mann KK, Schlezinger JJ, Jensen B, Sherr DH (2003) Environmental chemical-induced pro/pre-B cell apoptosis: analysis of c-Myc, p27Kip1, and p21WAF1 reveals a death pathway distinct from clonal deletion. J Immunol 170(10):4897–4904
Ryu HY, Emberley JK, Schlezinger JJ, Allan LL, Na S, Sherr DH (2005) Environmental chemical-induced bone marrow B cell apoptosis: death receptor-independent activation of a caspase-3 to caspase-8 pathway. Mol Pharmacol 68(4):1087–1096
Teague JE, Ryu HY, Kirber M, Sherr DH, Schlezinger JJ (2010) Proximal events in 7,12-dimethylbenz[a]anthracene-induced, stromal cell-dependent bone marrow B cell apoptosis: stromal cell–B cell communication and apoptosis signaling. J Immunol 185(6):3369–3378
Page TJ, O’Brien S, Jefcoate CR, Czuprynski CJ (2002) 7,12-Dimethylbenz[a]anthracene induces apoptosis in murine pre-B cells through a caspase-8-dependent pathway. Mol Pharmacol 62(2):313–319
Near RI, Matulka RA, Mann KK, Gogate SU, Trombino AF, Sherr DH (1999) Regulation of preB cell apoptosis by aryl hydrocarbon receptor/transcription factor-expressing stromal/adherent cells. Proc Soc Exp Biol Med 221(3):242–252
Mann KK, Matulka RA, Hahn ME, Trombino AF, Lawrence BP, Kerkvliet NI, Sherr DH (1999) The role of polycyclic aromatic hydrocarbon metabolism in dimethylbenz[a]anthracene-induced pre-B lymphocyte apoptosis. Toxicol Appl Pharmacol 161(1):10–22. doi:10.1006/taap.1999.8778
Heidel SM, Czuprynski CJ, Jefcoate CR (1998) Bone marrow stromal cells constitutively express high levels of cytochrome P4501B1 that metabolize 7,12-dimethylbenz[a]anthracene. Mol Pharmacol 54(6):1000–1006
Heidel SM, Holston K, Buters JT, Gonzalez FJ, Jefcoate CR, Czupyrynski CJ (1999) Bone marrow stromal cell cytochrome P4501B1 is required for pre-B cell apoptosis induced by 7,12-dimethylbenz[a]anthracene. Mol Pharmacol 56(6):1317–1323
Allan LL, Sherr DH (2005) Constitutive activation and environmental chemical induction of the aryl hydrocarbon receptor/transcription factor in activated human B lymphocytes. Mol Pharmacol 67(5):1740–1750. doi:10.1124/mol.104.009100
Tanaka G, Kanaji S, Hirano A, Arima K, Shinagawa A, Goda C, Yasunaga S, Ikizawa K, Yanagihara Y, Kubo M, Kuriyama-Fujii Y, Sugita Y, Inokuchi A, Izuhara K (2005) Induction and activation of the aryl hydrocarbon receptor by IL-4 in B cells. Int Immunol 17(6):797–805
Marcus RS, Holsapple MP, Kaminski NE (1998) Lipopolysaccharide activation of murine splenocytes and splenic B cells increased the expression of aryl hydrocarbon receptor and aryl hydrocarbon receptor nuclear translocator. J Pharmacol Exp Ther 287(3):1113–1118
Crawford RB, Holsapple MP, Kaminski NE (1997) Leukocyte activation induces aryl hydrocarbon receptor up-regulation, DNA binding, and increased Cyp1a1 expression in the absence of exogenous ligand. Mol Pharmacol 52(6):921–927
Wood SC, Jeong HG, Morris DL, Holsapple MP (1993) Direct effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on human tonsillar lymphocytes. Toxicology 81(2):131–143
Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, Nebert DW, Rudikoff S, Ward JM, Gonzalez FJ (1995) Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 268(5211):722–726
Schlezinger JJ, Liu D, Farago M, Seldin DC, Belguise K, Sonenshein GE, Sherr DH (2006) A role for the aryl hydrocarbon receptor in mammary gland tumorigenesis. Biol Chem 387(9):1175–1187. doi:10.1515/BC.2006.145
Ma Q, Whitlock J (1996) The aromatic hydrocarbon receptor modulates the Hepa 1c1c7 cell cycle and differentiated state independently of dioxin. Mol Cell Biol 16:2144–2150
Abdelrahim M, Smith R 3rd, Safe S (2003) Aryl hydrocarbon receptor gene silencing with small inhibitory RNA differentially modulates Ah-responsiveness in MCF-7 and HepG2 cancer cells. Mol Pharmacol 63(6):1373–1381
Puga A, Marlowe J, Barnes S, Chang CY, Maier A, Tan Z, Kerzee JK, Chang X, Strobeck M, Knudsen ES (2002) Role of the aryl hydrocarbon receptor in cell cycle regulation. Toxicology 181–182:171–177
Marlowe JL, Knudsen ES, Schwemberger S, Puga A (2004) The aryl hydrocarbon receptor displaces p300 from E2F-dependent promoters and represses S-phase specific gene expression. J Biol Chem 279:29013–29022
Marlowe JL, Fan Y, Chang X, Peng L, Knudsen ES, Xia Y, Puga A (2008) The aryl hydrocarbon receptor binds to E2F1 and inhibits E2F1-induced apoptosis. Mol Biol Cell 19(8):3263–3271
Puga A, Barnes SJ, Dalton TP, Chang C-Y, Knudsen ES, Maier MA (2000) Aromatic hydrocarbon receptor interaction with the retinoblastoma protein potentiates repression of E2F-dependent transcription and cell cycle arrest. J Biol Chem 275:2943–2950
Kim DW, Gazourian L, Quadri SA, Romieu-Mourez R, Sherr DH, Sonenshein GE (2000) The RelA NF-kB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene 19(48):5498–5506
Tian Y, Ke S, Denison MS, Rabson AB, Gallo MA (1999) Ah receptor and NF-kappaB interactions, a potential mechanism for dioxin toxicity. J Biol Chem 274(1):510–515
Vogel CF, Li W, Wu D, Miller JK, Sweeney C, Lazennec G, Fujisawa Y, Matsumura F (2011) Interaction of aryl hydrocarbon receptor and NF-kappaB subunit RelB in breast cancer is associated with interleukin-8 overexpression. Arch Biochem Biophys 512(1):78–86. doi:10.1016/j.abb.2011.05.011
Salas VM, Burchiel SW (1998) Apoptosis in Daudi human B cells in response to benzo[a]pyrene and benzo[a]pyrene-7,8-dihydrodiol. Toxicol Appl Pharmacol 151(2):367–376. doi:10.1006/taap.1998.8455
Matikainen TM, Moriyama T, Morita Y, Perez GI, Korsmeyer SJ, Sherr DH, Tilly JL (2002) Ligand activation of the aromatic hydrocarbon receptor transcription factor drives Bax-dependent apoptosis in developing fetal ovarian germ cells. Endocrinology 143(2):615–620
Robles R, Morita Y, Mann KK, Perez GI, Yang S, Matikainen T, Sherr DH, Tilly JL (2000) The aryl hydrocarbon receptor, a basic helix–loop–helix transcription factor of the PAS gene family, is required for normal ovarian germ cell dynamics in the mouse. Endocrinology 141(1):450–453
Caruso JA, Mathieu PA, Joiakim A, Leeson B, Kessel D, Sloane BF, Reiners JJ Jr (2004) Differential susceptibilities of murine hepatoma 1c1c7 and Tao cells to the lysosomal photosensitizer NPe6: influence of aryl hydrocarbon receptor on lysosomal fragility and protease contents. Mol Pharmacol 65(4):1016–1028
Caruso JA, Mathieu PA, Joiakim A, Zhang H, Reiners JJ Jr (2006) Aryl hydrocarbon receptor modulation of TNFalpha-induced apoptosis and lysosomal disruption in a hepatoma model that is caspase-8 independent. J Biol Chem 281:10954–10967
Allan LL, Sherr DH (2010) Disruption of human plasma cell differentiation by an environmental polycyclic aromatic hydrocarbon: a mechanistic immunotoxicological study. Environ Health 9:15. doi:10.1186/1476-069X-9-15
Schneider D, Manzan MA, Yoo BS, Crawford RB, Kaminski N (2009) Involvement of Blimp-1 and AP-1 dysregulation in the 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated suppression of the IgM response by B cells. Toxicol Sci 108(2):377–388
Sulentic CE, Holsapple MP, Kaminski NE (1998) Aryl hydrocarbon receptor-dependent suppression by 2,3,7, 8-tetrachlorodibenzo-p-dioxin of IgM secretion in activated B cells. Mol Pharmacol 53(4):623–629
Sulentic CE, Holsapple MP, Kaminski NE (2000) Putative link between transcriptional regulation of IgM expression by 2,3,7,8-tetrachlorodibenzo-p-dioxin and the aryl hydrocarbon receptor/dioxin-responsive enhancer signaling pathway. J Pharmacol Exp Ther 295(2):705–716
Sulentic CE, Kang JS, Na YJ, Kaminski NE (2004) Interactions at a dioxin responsive element (DRE) and an overlapping kappaB site within the hs4 domain of the 3′alpha immunoglobulin heavy chain enhancer. Toxicology 200(2–3):235–246. doi:10.1016/j.tox.2004.03.015
Sulentic CE, Zhang W, Na YJ, Kaminski NE (2004) 2,3,7,8-Tetrachlorodibenzo-p-dioxin, an exogenous modulator of the 3′alpha immunoglobulin heavy chain enhancer in the CH12.LX mouse cell line. J Pharmacol Exp Ther 309(1):71–78. doi:10.1124/jpet.103.059493
Fernando TM, Ochs SD, Liu J, Chambers-Turner RC, Sulentic CE (2012) 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces transcriptional activity of the human polymorphic hs1,2 enhancer of the 3′Igh regulatory region. J Immunol 188(7):3294–3306. doi:10.4049/jimmunol.1101111
Suh J, Jeon YJ, Kim HM, Kang JS, Kaminski NE, Yang KH (2002) Aryl hydrocarbon receptor-dependent inhibition of AP-1 activity by 2,3,7,8-tetrachlorodibenzo-p-dioxin in activated B cells. Toxicol Appl Pharmacol 181(2):116–123
Genestier L, Taillardet M, Mondiere P, Gheit H, Bella C, Defrance T (2007) TLR agonists selectively promote terminal plasma cell differentiation of B cell subsets specialized in thymus-independent responses. J Immunol 178(12):7779–7786
De Abrew KN, Phadnis AS, Crawford RB, Kaminski NE, Thomas RS (2011) Regulation of Bach2 by the aryl hydrocarbon receptor as a mechanism for suppression of B-cell differentiation by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Appl Pharmacol 252(2):150–158. doi:10.1016/j.taap.2011.01.020
Zhang Q, Kline DE, Bhattacharya S, Crawford RB, Conolly RB, Thomas RS, Andersen ME, Kaminski NE (2013) All-or-none suppression of B cell terminal differentiation by environmental contaminant 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Appl Pharmacol 268(1):17–26. doi:10.1016/j.taap.2013.01.015
Delogu A, Schebesta A, Sun Q, Aschenbrenner K, Perlot T, Busslinger M (2006) Gene repression by Pax5 in B cells is essential for blood cell homeostasis and is reversed in plasma cells. Immunity 24(3):269–281. doi:10.1016/j.immuni.2006.01.012
Mora-Lopez F, Reales E, Brieva JA, Campos-Caro A (2007) Human BSAP and BLIMP1 conform an autoregulatory feedback loop. Blood 110(9):3150–3157. doi:10.1182/blood-2007-05-092262
De Abrew KN, Kaminski NE, Thomas RS (2010) An integrated genomic analysis of aryl hydrocarbon receptor-mediated inhibition of B-cell differentiation. Toxicol Sci 118(2):454–469. doi:10.1093/toxsci/kfq265
Watson AJ, Hankinson O (1992) Dioxin- and Ah receptor-dependent protein binding to xenobiotic responsive elements and G-rich DNA studied by in vivo footprinting. J Biol Chem 267(10):6874–6878
Zhang L, Savas U, Alexander DL, Jefcoate CR (1998) Characterization of the mouse Cyp1B1 gene. Identification of an enhancer region that directs aryl hydrocarbon receptor-mediated constitutive and induced expression. J Biol Chem 273(9):5174–5183
Klapproth K, Wirth T (2010) Advances in the understanding of MYC-induced lymphomagenesis. Br J Haematol 149(4):484–497. doi:10.1111/j.1365-2141.2010.08159.x
Yang X, Liu D, Murray TJ, Mitchell GC, Hesterman EV, Karchner SI, Merson RR, Hahn ME, Sherr DH (2005) The aryl hydrocarbon receptor constitutively represses c-myc transcription in human mammary tumor cells. Oncogene 24(53):7869–7881
Nishimura R, Hata K, Matsubara T, Wakabayashi M, Yoneda T (2012) Regulation of bone and cartilage development by network between BMP signalling and transcription factors. J Biochem 151(3):247–254. doi:10.1093/jb/mvs004
Smeets RL, Fleuren WW, He X, Vink PM, Wijnands F, Gorecka M, Klop H, Bauerschmidt S, Garritsen A, Koenen HJ, Joosten I, Boots AM, Alkema W (2012) Molecular pathway profiling of T lymphocyte signal transduction pathways; Th1 and Th2 genomic fingerprints are defined by TCR and CD28-mediated signaling. BMC Immunol 13:12. doi:10.1186/1471-2172-13-12
Ehrhardt GR, Hijikata A, Kitamura H, Ohara O, Wang JY, Cooper MD (2008) Discriminating gene expression profiles of memory B cell subpopulations. J Exp Med 205(8):1807–1817. doi:10.1084/jem.20072682
Calame KL, Lin KI, Tunyaplin C (2003) Regulatory mechanisms that determine the development and function of plasma cells. Annu Rev Immunol 21:205–230
Jones SA, White CA, Robb L, Alexander WS, Tarlinton DM (2011) SOCS3 deletion in B cells alters cytokine responses and germinal center output. J Immunol 187(12):6318–6326. doi:10.4049/jimmunol.1102057
Orkin SH, Zon LI (2008) Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132(4):631–644. doi:10.1016/j.cell.2008.01.025
Chambers SM, Goodell MA (2007) Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev 3(3):201–211
Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA (2007) Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol 5(8):e201. doi:10.1371/journal.pbio.0050201
Morrison SJ, Wandycz AM, Akashi K, Globerson A, Weissman IL (1996) The aging of hematopoietic stem cells. Nat Med 2(9):1011–1016
Christensen JL, Wright DE, Wagers AJ, Weissman IL (2004) Circulation and chemotaxis of fetal hematopoietic stem cells. PLoS Biol 2(3):E75. doi:10.1371/journal.pbio.0020075
Jude CD, Climer L, Xu D, Artinger E, Fisher JK, Ernst P (2007) Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell 1(3):324–337. doi:10.1016/j.stem.2007.05.019
Delgado MD, Leon J (2010) Myc roles in hematopoiesis and leukemia. Genes Cancer 1(6):605–616. doi:10.1177/1947601910377495
Singh KP, Garrett RW, Casado FL, Gasiewicz TA (2011) Aryl hydrocarbon receptor-null allele mice have hematopoietic stem/progenitor cells with abnormal characteristics and functions. Stem Cells Dev 20(5):769–784. doi:10.1089/scd.2010.0333
Van Zant G, Liang Y (2003) The role of stem cells in aging. Exp Hematol 31(8):659–672
Bell DR, Van Zant G (2004) Stem cells, aging, and cancer: inevitabilities and outcomes. Oncogene 23(43):7290–7296. doi:10.1038/sj.onc.1207949
Lindsey S, Papoutsakis ET (2011) The aryl hydrocarbon receptor (AHR) transcription factor regulates megakaryocytic polyploidization. Br J Haematol 152(4):469–484. doi:10.1111/j.1365-2141.2010.08548.x
van Grevenynghe J, Bernard M, Langouet S, Le Berre C, Fest T, Fardel O (2005) Human CD34-positive hematopoietic stem cells constitute targets for carcinogenic polycyclic aromatic hydrocarbons. J Pharmacol Exp Ther 314(2):693–702. doi:10.1124/jpet.105.084780
Ivanova NB, Dimos JT, Schaniel C, Hackney JA, Moore KA, Lemischka IR (2002) A stem cell molecular signature. Science 298(5593):601–604. doi:10.1126/science.1073823
Casado FL, Singh KP, Gasiewicz TA (2010) The aryl hydrocarbon receptor: regulation of hematopoiesis and involvement in the progression of blood diseases. Blood Cells Mol Dis 44(4):199–206. doi:10.1016/j.bcmd.2010.01.005
Casado FL, Singh KP, Gasiewicz TA (2011) Aryl hydrocarbon receptor activation in hematopoietic stem/progenitor cells alters cell function and pathway-specific gene modulation reflecting changes in cellular trafficking and migration. Mol Pharmacol 80(4):673–682. doi:10.1124/mol.111.071381
Gasiewicz TA, Singh KP, Casado FL (2010) The aryl hydrocarbon receptor has an important role in the regulation of hematopoiesis: implications for benzene-induced hematopoietic toxicity. Chem Biol Interact 184(1–2):246–251. doi:10.1016/j.cbi.2009.10.019
Murante FG, Gasiewicz TA (2000) Hemopoietic progenitor cells are sensitive targets of 2,3,7,8-tetrachlorodibenzo-p-dioxin in C57BL/6J mice. Toxicol Sci 54(2):374–383
Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, Sutton SE, Walker JR, Flaveny CA, Perdew GH, Denison MS, Schultz PG, Cooke MP (2010) Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 329(5997):1345–1348. doi:10.1126/science.1191536
Singh KP, Wyman A, Casado FL, Garrett RW, Gasiewicz TA (2009) Treatment of mice with the Ah receptor agonist and human carcinogen dioxin results in altered numbers and function of hematopoietic stem cells. Carcinogenesis 30(1):11–19. doi:10.1093/carcin/bgn224
Singh KP, Casado FL, Opanashuk LA, Gasiewicz TA (2009) The aryl hydrocarbon receptor has a normal function in the regulation of hematopoietic and other stem/progenitor cell populations. Biochem Pharmacol 77(4):577–587. doi:10.1016/j.bcp.2008.10.001
Carvajal-Gonzalez JM, Mulero-Navarro S, Roman AC, Sauzeau V, Merino JM, Bustelo XR, Fernandez-Salguero PM (2009) The dioxin receptor regulates the constitutive expression of the vav3 proto-oncogene and modulates cell shape and adhesion. Mol Biol Cell 20(6):1715–1727. doi:10.1091/mbc.E08-05-0451
DiNatale BC, Schroeder JC, Perdew GH (2011) Ah receptor antagonism inhibits constitutive and cytokine inducible IL6 production in head and neck tumor cell lines. Mol Carcinog 50(3):173–183. doi:10.1002/mc.20702
Barouki R, Coumoul X, Fernandez-Salguero PM (2007) The aryl hydrocarbon receptor, more than a xenobiotic-interacting protein. FEBS Lett 581(19):3608–3615
Fernandez-Salguero PM (2010) A remarkable new target gene for the dioxin receptor: the Vav3 proto-oncogene links AhR to adhesion and migration. Cell Adh Migr 4(2):172–175
Ikuta T, Kawajiri K (2006) Zinc finger transcription factor Slug is a novel target gene of aryl hydrocarbon receptor. Exp Cell Res 312(18):3585–3594
Peng TL, Chen J, Mao W, Song X, Chen MH (2009) Aryl hydrocarbon receptor pathway activation enhances gastric cancer cell invasiveness likely through a c-Jun-dependent induction of matrix metalloproteinase-9. BMC Cell Biol 10:27. doi:10.1186/1471-2121-10-27
Quintana PJ, Delfino RJ, Korrick S, Ziogas A, Kutz FW, Jones EL, Laden F, Garshick E (2004) Adipose tissue levels of organochlorine pesticides and polychlorinated biphenyls and risk of non-Hodgkin’s lymphoma. Environ Heal Perspect 112(8):854–861
Hardell L, Eriksson M, Nordstrom M (2002) Exposure to pesticides as risk factor for non-Hodgkin’s lymphoma and hairy cell leukemia: pooled analysis of two Swedish case–control studies. Leuk Lymphoma 43(5):1043–1049
Viel JF, Arveux P, Baverel J, Cahn JY (2000) Soft-tissue sarcoma and non-Hodgkin’s lymphoma clusters around a municipal solid waste incinerator with high dioxin emission levels. Am J Epidemiol 152(1):13–19
Consonni D, Pesatori AC, Zocchetti C, Sindaco R, D’Oro LC, Rubagotti M, Bertazzi PA (2008) Mortality in a population exposed to dioxin after the Seveso, Italy, accident in 1976: 25 years of follow-up. Am J Epidemiol 167(7):847–858. doi:10.1093/aje/kwm371
Ng CH, Janoo-Gilani R, Sipahimalani P, Gallagher RP, Gascoyne RD, Connors JM, Weber JP, Lai AS, Leach S, Le ND, Brooks-Wilson AR, Spinelli JJ (2010) Interaction between organochlorines and the AHR gene, and risk of non-Hodgkin lymphoma. Cancer Causes Control 21(1):11–22. doi:10.1007/s10552-009-9429-5
Pesatori AC, Consonni D, Rubagotti M, Grillo P, Bertazzi PA (2009) Cancer incidence in the population exposed to dioxin after the “Seveso accident”: twenty years of follow-up. Environ Health 8:39. doi:10.1186/1476-069X-8-39
Barhoover MA, Hall JM, Greenlee WF, Thomas RS (2010) Aryl hydrocarbon receptor regulates cell cycle progression in human breast cancer cells via a functional interaction with cyclin-dependent kinase 4. Mol Pharmacol 77(2):195–201. doi:10.1124/mol.109.059675
Gramatzki D, Pantazis G, Schittenhelm J, Tabatabai G, Kohle C, Wick W, Schwarz M, Weller M, Tritschler I (2009) Aryl hydrocarbon receptor inhibition downregulates the TGF-beta/Smad pathway in human glioblastoma cells. Oncogene 28(28):2593–2605. doi:10.1038/onc.2009.104
Moennikes O, Loeppen S, Buchmann A, Andersson P, Ittrich C, Poellinger L, Schwarz M (2004) A constitutively active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res 64(14):4707–4710
Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, Jugold M, Guillemin GJ, Miller CL, Lutz C, Radlwimmer B, Lehmann I, von Deimling A, Wick W, Platten M (2011) An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478(7368):197–203. doi:10.1038/nature10491
Yang X, Solomon S, Fraser LR, Trombino AF, Liu D, Sonenshein GE, Hestermann EV, Sherr DH (2008) Constitutive regulation of CYP1B1 by the aryl hydrocarbon receptor (AhR) in pre-malignant and malignant mammary tissue. J Cell Biochem 104(2):402–417. doi:10.1002/jcb.21630
Belguise K, Guo S, Yang S, Rogers AE, Seldin DC, Sherr DH, Sonenshein GE (2007) Green tea polyphenols reverse cooperation between c-Rel and CK2 that induces the aryl hydrocarbon receptor, slug, and an invasive phenotype. Cancer Res 67(24):11742–11750. doi:10.1158/0008-5472.CAN-07-2730
Brooks J, Eltom SE (2011) Malignant transformation of mammary epithelial cells by ectopic overexpression of the aryl hydrocarbon receptor. Curr Cancer Drug Targets 11(5):654–669
Kashuba EV, Gradin K, Isaguliants M, Szekely L, Poellinger L, Klein G, Kazlauskas A (2006) Regulation of transactivation function of the aryl hydrocarbon receptor by the Epstein–Barr virus-encoded EBNA-3 protein. J Biol Chem 281(2):1215–1223. doi:10.1074/jbc.M509036200
Vogel CF, Li W, Sciullo E, Newman J, Hammock B, Reader JR, Tuscano J, Matsumura F (2007) Pathogenesis of aryl hydrocarbon receptor-mediated development of lymphoma is associated with increased cyclooxygenase-2 expression. Am J Pathol 171(5):1538–1548. doi:10.2353/ajpath.2007.070406
Secchiero P, Barbarotto E, Gonelli A, Tiribelli M, Zerbinati C, Celeghini C, Agostinelli C, Pileri SA, Zauli G (2005) Potential pathogenetic implications of cyclooxygenase-2 overexpression in B chronic lymphoid leukemia cells. Am J Pathol 167(6):1599–1607. doi:10.1016/S0002-9440(10)61244-8
Chang X, Fan Y, Karyala S, Schwemberger S, Tomlinson CR, Sartor MA, Puga A (2007) Ligand-independent regulation of transforming growth factor beta1 expression and cell cycle progression by the aryl hydrocarbon receptor. Mol Cell Biol 27(17):6127–6139
Haarmann-Stemmann T, Bothe H, Abel J (2009) Growth factors, cytokines and their receptors as downstream targets of arylhydrocarbon receptor (AhR) signaling pathways. Biochem Pharmacol 77(4):508–520. doi:10.1016/j.bcp.2008.09.013
Allan LL, Mann KK, Matulka RA, Ryu HY, Schlezinger JJ, Sherr DH (2003) Bone marrow stromal–B cell interactions in polycyclic aromatic hydrocarbon-induced pro/pre-B cell apoptosis. Toxicol Sci 76(2):357–365. doi:10.1093/toxsci/kfg239
Jensen BA, Leeman RJ, Schlezinger JJ, Sherr DH (2003) Aryl hydrocarbon receptor (AhR) agonists suppress interleukin-6 expression by bone marrow stromal cells: an immunotoxicology study. Environ Health 2(1):16. doi:10.1186/1476-069X-2-16
Perez LE, Parquet N, Shain K, Nimmanapalli R, Alsina M, Anasetti C, Dalton W (2008) Bone marrow stroma confers resistance to Apo2 ligand/TRAIL in multiple myeloma in part by regulating c-FLIP. J Immunol 180(3):1545–1555
Clarke CA, Glaser SL (2002) Changing incidence of non-Hodgkin lymphomas in the United States. Cancer 94(7):2015–2023. doi:10.1002/cncr.10403
Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 95(25):14863–14868
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102(43):15545–15550. doi:10.1073/pnas.0506580102
Carsetti R, Kohler G, Lamers MC (1995) Transitional B cells are the target of negative selection in the B cell compartment. J Exp Med 181(6):2129–2140
Acknowledgments
This work was supported by P01-ES11624, the Art beCAUSE Breast Cancer Foundation, P42ES007381, and an Evans Center Interdisciplinary Biomedical Research Affinity Research Collaborative Award.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on Roles of Aryl Hydrocarbon Receptor in Controlling Immunity-Guest Editors: C. Pot, V. Kuchroo and F. Quintaña
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Sherr, D.H., Monti, S. The role of the aryl hydrocarbon receptor in normal and malignant B cell development. Semin Immunopathol 35, 705–716 (2013). https://doi.org/10.1007/s00281-013-0390-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-013-0390-8