
Abstract
The ability of neutrophils and other leucocyte members of the innate immune system to expel their DNA into the extracellular environment in a controlled manner in order to trap and kill pathogenic microorganisms lead to a paradigm shift in our understanding of host microbe interactions. Surprisingly, the neutrophil extracellular trap (NET) cast by neutrophils is very wide and extends to the entrapment of viruses as well as multicellular eukaryotic parasites. Not unexpectedly, it has emerged that pathogenic microorganisms can employ a wide array of strategies to avoid ensnarement, including expression of DNAse enzymes that destroy the lattice backbone of NETs. Alternatively, they may use molecular mimicry to avoid detection or trigger events leading to the expression of immune modulatory cytokines such as IL-10, which dampen the NETotic response of neutrophils. In addition, the host microenvironment may contribute to the innate immune response by the production of lectin-like molecules that bind to bacteria and promote their entrapment on NETs. An example of this is the production of surfactant protein D by the lung epithelium. In addition, pregnancy provides a different challenge, as the mother needs to mount an effective response against pathogens, without harming her unborn child. An examination of these decoy and host response mechanisms may open the path for new therapies to treat pathologies mediated by overt NETosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neutrophil granulocytes, also frequently termed polymorphonuclear neutrophils (PMN), are the most abundant members of the leucocyte population, comprising between 50 and 70 % of total white blood cells in normal healthy individuals [1, 2]. PMNs are a vital component of the innate immune system and form the first line of defense against invading pathogenic microorganisms [1, 2]. Traditionally, this interaction has been considered to involve phagocytosis and lytic destruction of pathogens by toxic neutrophil granular proteins and reactive oxygen species (ROS) in the phagolysosome or the release of these agents directly into the extracellular environment by degranulation [1, 2].
This paradigm was altered by the discovery that PMNs could release their genomic DNA into the extracellular environment in the form of neutrophil extracellular traps (NETs) that could ensnare invading microorganisms [3]. Analysis of these novel NET structures showed that they were decorated by proteins from all three PMN granule groups (azurophilic, secondary, and tertiary), including neutrophil elastase (NE), myeloperoxidase (MPO), lactoferrin, and gelatinase [3]. It was proposed that by being tightly associated with the DNA lattice structure of NETs, the presence of bactericidal granular proteins in these molecular traps would be able to kill pathogenic invaders in a highly efficient manner with minimal damage to the surrounding tissue [3].
In the interim, it has emerged that the interaction between hostile microorganisms and neutrophil NETs is more complex than originally thought. In this review, we will highlight some of these developments.
Interaction with bacteria and fungi
In their seminal publication, Brinkmann et al. demonstrated that NETs could be induced by both Gram-positive (Staphylococcus aureus) and Gram-negative (Salmonella typhimurium and Shigella flexneri) bacteria and displayed signs of incapacitating the virulence of these agents by the action of NET-associated extracellular proteases [3].
This finding was rapidly extended to show that neutrophil NETs could also be induced by eukaryotic pathogens such as the ascomycetous yeast, Candida albicans [4]. In this study, it was demonstrated that NETs interact both with the single cell yeast form as well as the multicellular hyphal form and incapacitate these via action of granular components. The generation of NETs was more pronounced in cases in which the yeast cells, either yeast or hyphal form, were opsonized. Opsonization was, however, not a prerequisite for NETosis.
The fungicidal activity of NETs was subsequently shown to be largely due to the presence of calprotectin, an antimicrobial heterodimer with calcium- and zinc-binding properties [5]. The absence of calprotectin in S100A9 knockout mice rendered these animals highly susceptible to infections with C. albicans.
Evidence that NETs may play a role in the human innate immune response was provided by patients with chronic granulomatous disease (CGD) [6]. In such patients, NETosisis abrogated due to a deficiency in the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase genes that prevent efficient ROS generation. This genetic deficiency renders this patient group particularly prone to pulmonary infections with Aspergillus fungi.
To correct for this deficiency, a gene therapy approach was investigated by which the defective gp91phox gene was replaced by a functional gene encoded from a retroviral vector. By this means, it was possible to cure a child from refractory aspergillosis and to enable a return to a normal life free from the restrictions of sterile confinement [6]. Although functional NADPH oxidase was detectable in only 16 % of PMN for a period of 3 months postgene therapy, this low level was sufficient for efficient clearing of the fungal infection. Furthermore, in in vitro examinations of the gene-modified cells, it was demonstrated that they underwent NETosis following interaction with A. nidulans isolated from the patient’s lungs and that such NETs effectively inhibited fungal growth.
Direct real-time in vivo evidence of the interaction between neutrophil NETs and invading fungal pathogens was obtained in murine lungs using two-photon microscopy [7]. In this study, it was shown that neutrophils generate NETs in response to interaction with both Aspergillus fumigatus morphotypes, namely conidia and hyphae. NETs detected in the lungs of infected animals formed with rapid kinetics, occurring within 3–4 h of PMN exposure to the fungi. A discrepant finding of this study was that NETs did not appear to be the major contributor to the killing of A. fumigatus, unlike what was previously clearly demonstrated to be the case for C. albicans, but may, in this instance, contribute to fungal demise by binding of secreted proteins or membrane structures and thereby preventing fungal spread.
Mechanisms to evade NET entrapment by bacteria or fungi
Similar to “spy vs. spy” scenarios, it is obvious that in biology, attacker and evader will develop different strategies to counteract each other. In the case of NETs, several systems appear to be employed by bacteria or fungi to render them ineffective. Not unexpectedly, these strategies rely on the use of nucleases that attack the DNA backbone of the NET lattice structure [8, 9].
The first report of such an evasive strategy was made in strains of Group A streptococcus, which, by the expression of a DNAse molecule Sda1, were determined to be more virulent than strains lacking this enzyme. In addition, the inhibition of this enzyme reduced infectivity and promoted clearance by PMN [10] (Fig. 1a).

Mechanisms of defense against bacteria and fungi and strategies of evasion of NET entrapment. NETs can be induced by both Gram-positive or Gram-negative bacteria (a). Evasion of the antimicrobial activity of NETs may include the use of nucleases that attack the DNA backbone of the NETs' lattice structure (a) or elements of molecular mimicry that stimulate the production of the potent immune-modulating cytokines (b). NE neutrophil elastase, PMN polymorphonuclear neutrophil, MPO myeloperoxidase, LL37 cathelicidin, CG cathepsin G, IL10 interleukin 10, TGFβ transforming growth factor beta
In a similar manner, endonuclease expression by Streptococcus pneumoniae [11] or S. aureus [12] was shown to permit these organisms to evade ensnarement by NETs, thereby boosting their virulence (Fig. 1a). Consequently, these nucleases are currently being evaluated as targets for novel antibacterial therapies [13].
Recently, evidence of more complex evasive strategies employing elements of molecular mimicry has emerged. In the instance of Pseudomonas aeruginosa, it appears that this opportunistic pathogen exploits the acquisition of sialic acid motifs form host cells [14]. Sialic acids, also termed Sias, are unusual monosaccharides characterized by a shared nine-carbon backbone [15]. They are widely expressed on the cell surface of most mammalian cells. In order to evade detection and destruction by the host immune system, a number of bacteria have been shown to acquire and coat themselves with host Sias [16]. In this manner, such pathogens will not only immunologically appear as “self” rather than as “foreign,” but by binding to specific Sias, lectin-like receptors (siglecs) on host cells dampen the host immune response against them [16].
By absorbing host serum Sias, P. aeruginosa was able to bind directly to PMN via siglec-9 and, thereby, stimulate the production of the potent immune-modulating cytokines IL-10 and TGF-β. Furthermore, this interaction attenuated the generation of ROS and the release of neutrophil elastase (NE) by PMN engaged in this manner. Since the generation of ROS is a pivotal step required for NETosis, it comes as no surprise that PMN exhibited a reduced ability to generate NETs when confronted with Sias-coated P. aeruginosa bacteria [14] (Fig. 1b). It is not clear if the IL-10 present in this system is produced by neutrophils or by bystander cells, as it has been suggested that human neutrophils have an inactive IL-10 gene locus [17].
Fungi have also developed analogous evasive countermeasure. In the interaction between PMN and with Aspergillus species, it was determined that the presence of the hydrophobin RodA protein on the cell surface of conidia leads to a reduction in NETosis [7]. The presence of this hydrophobic protein that coats resting conidia, but not hyphae, has previously been shown to largely negate an antifungal innate immune response [7] (Fig. 1b). A summary of the reported mechanisms of pathogen evasion from NETs is given in Table 1.
Tuberculosis: interaction between macrophages and neutrophils
The elimination of invasive pathogenic microorganisms involves a delicately orchestrated interplay between them and among members of the innate and the adaptive immune systems [18, 19]. A prime example of this system breaking down, permitting the virulent bacterium to thrive and eventually kill the host, is provided by Mycobacterium tuberculosis [20].
Even though the tubercle bacillus was described by Robert Koch back in 1882, tuberculosis (TB) remains a leading cause of worldwide mortality (1.4 million deaths in 2011) and morbidity (8.7 million new infections in 2011), second only to human immunodeficiency virus (HIV) [20]. Despite this large health care burden, surprisingly little has been undertaken to investigate the potential role of neutrophils or their NETs in combating this insidious infection [21].
This may be largely due to the intrinsic properties of PMN, in that they are short lived and cannot be cryopreserved, necessitating the constant use of fresh primary cells, which restrict their availability and utilization [21]. Furthermore, macrophages are considered to be the prime target of this bacterium and to be crucial for its removal and containment in granulomas.
Nonetheless, the role played by PMNs in the initial combating and subsequent harboring of the M. tuberculosis bacilli is gaining recognition, since a large influx of PMNs is evident at the site of infection and PMNs may account for the predominant phagocytic cells infected by M. tuberculosis bacilli [22]. In addition, a transcriptomic analysis of human blood samples from TB patients suggests that tuberculosis is associated with a neutrophil-driven interferon-induced profile [23]. Furthermore, PMNs are required for granuloma formation in chronic infection. Experiments in mice suggest that the recruitment of PMNs to the site of infection reduces the number of infective M. tuberculosis bacilli from the lungs and prevents their spread to secondary organs, such as the spleen [21]. This is consistent with reports in humans, where the risk of infection is inversely proportional to peripheral PMN cell counts [21].
That M. tuberculosis bacilli induce NETosis has become evident from the study by Ramos-Kichik et al. [24]. In examining the influence of two strains of M. tuberculosis with high or low (M. tuberculosis H37Rv and M. canetti) virulence on isolated PMNs in vitro, it was observed that both strains lead to the formation of leucocyte clusters or aggregates [24]. Remarkably, both M. tuberculosis strains induced the formation of neutrophil NETs, as detected by scanning and transmission electron microscopy. This feature was confirmed by fluorescence immunohistochemical staining for DNA, histones, and neutrophil elastase [24]. NETosis was found to be associated with the demise of PMNs and the release of intracellular constituents (LDH), as originally reported by Brinkmann et al. [3]. No significant difference in the degree of NETosis induction by either the virulent N37Rv or the nonvirulent M. canetti strains could be discerned. Of particular interest was the observation that, although the PMN NETs were induced by M. tuberculosis bacilli and could ensnare these microorganisms, they were incapable of killing these bacilli. This was in strong contrast to what was observed in the case of Listeria monocytogenes under the same conditions, in which the entrapment by NETs lead to a significant reduction in bacterial survival and infectivity. This effect was not due to the M. tuberculosis bacilli rendering the NETs inert, as the addition of such NETs to L. monocytogenes cultures was capable of killing the latter. This effect was abolished if the DNA lattice of the NETs was destroyed by addition of nuclease. Consequently, the inability of NETs to kill M. tuberculosis bacilli must be a reflection upon the unique nature of the cell wall of these bacilli, its high lipid content rendering them impervious to the action of immune cell attack, be it by PMNs or macrophages.
The interplay between M. tuberculosis bacilli and members of the innate immune system may, however, be more complex than previously thought, a change in concept supported by a spate of recent publications [18, 19, 25]. In this context, it has been observed that macrophages are able to ingest neutrophil azurophil granular proteins and use them to kill invasive mycobacteria [26, 27]. In the first of these reports, Steinwede et al. examined the role of cathepsin G (CG) and neutrophil elastase (NE) in the alveolar clearance of mycobacterial infections, in particular M. bovis bacillus Calmette-Guérin (M. bovis-BCG). They observed that genetically altered knockout mice for CG, and especially CG/NE, exhibited a reduced capacity for clearing bacterial infections, resulting in increased bronchoalveolar pathology. This could be overcome by aerosol therapy with CG/NE-loaded liposomes. Of interest is that these enzymes were taken up by alveolar macrophages and used by these to expedite bacterial elimination.
In a subsequent study, Jena et al. observed that macrophages can endocytose neutrophil-derived azurophilic enzymes and employ these for bacillus destruction [27]. They included M. smegmatis, M. bovis-BCG as well as the virulent M. tuberculosis H37Rv strain. Their data indicate that the acquisition of azurophilic enzymes and their presence within macrophages leads to the disintegration of the bacterial cell membrane and to bacterial cell lysis (Fig. 2).

Neutrophils and macrophages cooperate in order to confront mycobacterial infections. Mycobacterium bacilli induce leucocyte clustering or aggregation and, subsequently, NETosis, but not killing of these pathogens. Macrophages are able to ingest neutrophil azurophil granular proteins and dispatch invasive mycobacteria. The generation of ETs (extracellular traps) by macrophages is dependent on the presence of IFN-γ and the mycobacterial ESX-1 system, which mediate phagosomal rupture and subsequent bacterial escape
As these two studies report on very similar phenomena, this would suggest that the uptake of PMN-derived granules by macrophages could be a relatively common cooperative mechanism utilized by the innate immune system to eliminate certain bacteria. It also opens a potential pathway for novel therapeutic strategies.
Such cooperation may include other factors such as microparticles (MP) released by infected macrophages or PMN. In the instance of macrophage-derived MPs, these appear to be proinflammatory and attract PMN and other immune effector cells to sites of application [28]. These MPs were shown to contain mycobacterial antigens and promote specific CD4+ T cell activation both in vitro and in vivo. In contrast to these proinflammatory observations, it has been suggested that ectosomes derived from M. tuberculosis bacilli-infected PMN reduced the antimycobacterial activity of accessory macrophages [29]. Clearly, these disparities will need to be resolved.
A very intriguing recent observation in this context was made by Wong and Jacobs, in which they report on the generation of ETs (extracellular traps) by macrophages heavily infected with M. tuberculosis bacilli [30]. This effect was dependent on the presence of IFN-γ and the mycobacterial ESX-1 system, which mediated phagosomal rupture and subsequent bacterial escape. The macrophage ETs were determined to be remarkably similar to neutrophil NETs by having a DNA backbone readily destroyed by DNAse action and the occurrence of citrullinated histones. Furthermore, akin to the neutrophil NETotic process, the generation of macrophage ETs was also dependent on an elastase activity, as the process could be blocked by treatment with an elastase inhibitor, AAPV. The ability to generate traps was strongly dependent on whether the primary human macrophages had been differentiated with the aid of G-CSF but not GM-CSF. In addition, priming with IFN-γ was an essential requirement for ET triggering by M. tuberculosis bacilli. This feature was observable both under physiological oxygen tensions (5–10 %) as well as under normal tissue culture conditions (20 %).
The triggering of ETs by M. tuberculosis bacilli was strongly dependent on the bacteria possessing an intact ESX-1 system. It has previously been shown that the expression of these factors leads to caspase-1-independent cell death of infected macrophages. ESX-1 also leads to the activation of the inflammasome and release of IL-1 beta [31]. The role of this proinflammatory cytokine in macrophage ETosis is currently unclear.
On the other hand, the action of IFN-γ appears to promote the aggregation of M. tuberculosis bacilli in macrophages, thereby providing the necessary impetus for ETosis. This action was also accompanied by widespread necrosis of infected macrophages. It, therefore, appears that the ESX-1 system subverts the action of IFN-γ to enhance or amplify the aggregation of bacilli, macrophage ETosis, and necrosis (Fig. 2).
Role of cathelicidin in combating microorganisms
Antimicrobial peptides play a fundamental role in the protection against microbial attack in a number of systems, evident by their widespread distribution throughout the animal and plant kingdoms [32]. In humans, two major families of antimicrobial peptides have been identified as defensins and cathelicidin. Cathelicidin family of antimicrobial polypeptides are characterized by a highly conserved region (cathelin domain) and a highly variable cathelicidin peptide domain [33]. Human Cathelicidin(LL-37) is a cationic, host defense peptide, mainly expressed by neutrophils and epithelial cells during acute inflammation. LL-37 corresponds to amino acid 134–170 of Human cationic Antimicrobial Protein 18 (hCAP-18), which is present in specific granules of human neutrophils and produced after the C terminal cleavage of hCAP 18 by caspase 3 [34].
LL-37 is known to exhibit broad-spectrum antimicrobial activity against a wide range of Gram-positive and Gram-negative bacteria species [35–37], including tuberculosis, where in combination with lipocalin 2, it restricts the growth of this microorganism in an iron-dependent manner [38]. It also exhibits inhibitory activity against certain fungi and enveloped viruses [39]. This capability can be transferred to other species, including plants, as demonstrated by a recent experiment in which expression of LL-37 in transgenic plants lead to resistance to bacterial and fungal pathogens [40]. The mechanism of bacterial killing by LL-37 is rapid and mostly involves intercalation and assembly of peptides within the bacterial membrane to disrupt membrane integrity [41]. By the use of time-lapse fluorescence microscopy, it was shown that LL-37 adopted alternate mechanisms for membrane disruption depending upon the concentration of the peptide [42]. In addition to antimicrobial activity, LL-37 has been shown to be associated with a wide range of biological activities including the inhibition of biofilm formation, chemotaxis and mast cell degranulation, induction of chemokines, enhancement of keratinocyte migration and proliferation, and enhancement of vascularization [43, 44].
Inappropriate activity by LL-37 can lead to considerable tissue damage, especially in infections of the respiratory tract by pathogens such as S. aureus, which can lead to excessive fibrinolysis [45]. In addition, aberrant expression of cathelicidin has been linked to increased susceptibility to skin infections, frequent oral bacterial infections, severe periodontal disease, and cutaneous infections [46]. For example, histological examination of a number of chronic inflammatory skin diseases such as atopic dermatitis, rosacea, and psoriasis have shown that the proinflammatory peptide is dysregulated in these disorders [39].
This facet may be due to an unexpected interaction between LL-37 and extracellular host DNA derived from dying cells [47, 48]. In these studies, it was shown that LL-37 binds very stably to DNA and then acts as a transfecting agent, dragging this macromolecular complex into adjacent cells, where it can interact with toll-like pattern recognition receptors. In psoriasis, these bystander cells happen to be plasmacytoid dendritic cells (pDCs), which can elicit a potent immune response via the production of interferons [47].
Since cathelicidin is intimately associated with NETs, its presence may contribute to inflammatory conditions associated with aberrant NETosis such as occurs in autoantibody (ANCA) small vessel vasculitis [49]. This may involve further autoinflammatory conditions such as rheumatoid arthritis [50].
Although LL-37 constitutes a formidable innate immunity defense barrier against infections, some bacterial species have developed various mechanisms to circumvent its action. An example is the oral pathogen Porphyromonas gingivalis associated with chronic periodontal disease, which can degrade LL-37 by utilizing its arginine-specific gingipains enzyme [51]. In addition, S. aureus can cleave and inactivate LL-37 by the action of its metalloproteinase called “aureolysin,” in a time- and concentration-dependent manner [52]. Analogously, M1 protein promotes the survival of Group A streptococcus through the inhibition of cathelicidin [53]. On the other hand, the action of cathelicidin may be affected by pathological conditions such as cystic fibrosis, where its interaction with glycosaminoglycans can inhibit its antimicrobial activity, a facet that can be restored by treatment with hypertonic saline [54].
LL-37 may also influence neutrophil behavior, inducing secondary necrosis in these cells, leading to the release of IL-8, IL-1R antagonist, and granules [55]. Furthermore, LL-37 seemed to enhance the production of IL-8 and ROS production. It is currently unclear if such action by LL-37 leads to the induction of NETosis. Apart from powerful chemotactic activities, which appear to be, in part, mediated via interaction with the formyl peptide receptors [56], LL-37 can also promote monocyte adhesion, thereby illustrating a new mode of neutrophil interaction with these cells [57].
The induction of IL-8 by LL-37 may be pivotal in the combat of respiratory infections. Of particular interest is that this is not restricted to neutrophils but may include airway smooth muscle cells [58].
Role of immunothrombosis in combating microbial infections
In a recent review by Engelmann and Massberg [59], recapitulating the recent progress in the field of hemostasis and thrombosis activated by circulating microorganisms, the concept of immunothrombosis was coined. Immunothrombosis is defined as an innate immune response led by specific cells and molecules, which interact to form thrombi inside the blood vessels. When uncontrolled, it represents a major biological process fostering thrombosis-associated pathologies.
NETs appear to play a central role in immunothrombosis, supporting the above-described process through several means. NETs can directly activate factor XII (the contact pathway of coagulation), probably due to their electrochemical properties [60]. It has been shown that NETs bind directly to von Willebrand factor (vWF) and enhance the process of platelet recruitment [61]. Moreover, histones H3 and H4, present in the NETs, can trigger the activation of platelets [62] and stimulate platelet adhesion [63]. Other indispensable components of NETs, such as the proteolytic antimicrobial enzymes NE and MPO, can actively regulate the clotting cascade, on the one hand, by cleaving or oxidizing major natural anticoagulant substances, including tissue factor pathway inhibitor (TFPI) [64] and thrombomodulin [65], which propagates coagulation. On the other hand, in vitro studies have shown that the aforementioned proteases can also degrade fibrin [66], and since they are present on NETs, they could be potentially involved in the resolution of the clots [67, 68]. In all cases, treatment with DNase1 prevents thrombus formation, underscoring the importance of NETs for the process. Additionally, NETs can bind to TF and promote the activation of the extrinsic pathway of coagulation [69]. In vitro, NETs stimulate fibrin formation and deposition, and fibrin colocalizes with NETs in blood clots [67, 70].
In this context, the engaged neutrophils and monocytes are most probably stimulated by activated platelets via PRRs in order to form NETs [60, 71]. In turn, activated platelets not only stimulate NET formation but also the NETs that are generated trigger de novo platelet activation, red cell accumulation, and thrombosis [72, 73].
In the framework of the organism's defense tactics against pathogens, immunothrombosis is thought to perform a broad series of physiological functions. First, it facilitates capture and tanglement of blood-borne pathogens, limiting their spread by retaining the microorganisms within the fibrin clot. In this way, tissue invasion by pathogens is prevented through microthrombus formation [68]. Further, these intravascular clots generate a separate compartment that focuses all antimicrobial strategies to distinct loci favoring pathogen killing. This involves both innate immune cells and the antimicrobial substances generated during their activation [74, 75]. Finally, fibrinogen or fibrin accumulation and deposition in the small vessels enhances the additional recruitment of reactive cells to the damaged site of tissue infection, supporting the pathogen recognition and directing the immune response [76]. The basic features of immunothrombosis are depicted in Fig. 3.

Immunothrombosis supports the innate immune defense against pathogens. Immunothrombosis involves an intricate interplay between complement, coagulation, and innate immune effector cells. By assembly of a thrombus around the pathogenic microorganisms, their systemic spread is prevented and the damage to the host is minimized. PRR pattern recognition receptor, TLR toll-like receptor, TF tissue factor, vWF von Willebrand factor, NE neutrophil elastase, PMN polymorphonuclear neutrophil. Adapted and modified from Engelmann et al. [59]
The broad range of sophisticated strategies that pathogenic microorganisms have evolved in in order to subvert, additionally, immunothrombosis, apart from the herein described NETosis, underlines the crucial role of this process against their propagation [75, 77–80]. These strategies are also summarized in Table 1.
Viral interactions with neutrophil NETs
The interaction of neutrophils and viruses is less well characterized than those with bacteria or fungi, especially with regard to NETosis. Indeed, it was unclear if PMN NETs were beneficial or detrimental in cases of viral infections, as PMNs have been associated with increased pulmonary damage in murine influenza model systems [81–83].
This enigma was clearly addressed in a recent publication within travital microscopy to track the murine PMN response to infection with an oncolytic poxvirus in vivo [84]. In their experiments, the authors determined that PMN were recruited to the liver following administration with poly I:C (polyinosinic:poliycitidylic acid), a double-strand nucleotide viral analog, lypopolysaccheride (LPS), or challenge with the pox virus [84]. The activation of PMNs by poly I:C or pox virus involved toll-like receptor 3 (TLR3) of the innate immune pattern recognition receptors (PRR). This treatment increased the expression of the cell surface integrin molecule CD11b, which facilitates attachment of PMN on host endothelium. Furthermore, viral challenge induced thrombocytopenia, platelet accumulation in the liver, and generation of PMN NETs. In this manner, it is possible that infection with pox virus mobilizes an immune–thrombotic response [59], which would serve to immobilize the virus for efficient clearance by macrophages. The generation of NETs appears to be useful in the clearance of virus, as their induction by LPS treatment promoted viral clearance, whereas their dissolution by DNAse treatment facilitated viral persistence [84].
Viral modulation of NETosis: feline leukemia virus
Viral infections appear to be able to modulate the host NETotic response. The first indication of such an occurrence was in cases with feline leukemia virus (FeLV) [85]. Feline infections with this gammaretrovirus generally result in a mild viremia with high titers of neutralizing antibodies, while about a third of cases fail to mount an effective humoral response and succumb to complications arising from virally induced immunosuppression. For this purpose, FeLV has been suggested to serve as a model system for human infections with HIV (human immunodeficiency virus-1) [86].
Since immunosuppression in FeLV cases has been associated with reduced PMN function, Wardini et al. addressed this issue in more detail, including an analysis of the host NETotic response [85]. Their results indicated that feline PMNs underwent NETosis when exposed to Leishmania promastigotes. The degree of NETosis was elevated in asymptomatic FeLV-infected cats and was dramatically elevated in symptomatic FeLV-infected cats. These data suggest that FeLV alters the NETotic response of PMNs and that the measure of this index can be used as a prognostic marker of disease progression [85]. It is currently unclear how virally enhanced NETosis can lead to immunosuppression, but it is possible that overt stimulation of the NETotic response may lead to PMN depletion and exhaustion of the underlying progenitors.
Downmodulation of NETosis by HIV: paving the way for secondary infections?
In a recent report by Saitoh et al., it was shown that HIV triggers NETosis via a process that involved the recognition of viral nucleic acids via engagement with TLR7 and TLR8 [87, 88]. A unique feature of this study is that by the use of super-resolution structured illumination microscopy (SR-SIM) [89], they were able to directly visualize individual HIV virions trapped in the NET lattice. They were, furthermore, able to determine that PMN NETs not only immobilized HIV virions but were able to reduce their infectivity via the action of MPO (myeloperoxidase) and α-defensin. This appears to involve intact NETs, as it was reduced if the DNA lattice backbone was digested by DNAse treatment. These data, therefore, suggest that PMN NETs could play an important part in the initial innate response to HIV infection by facilitating viral trapping, inactivation, and clearance.
Unfortunately, HIV is a devious pathogen and will use a whole range of mechanisms to modulate and manage the host immune system in order to foster its propagation. One such means is by binding to Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN), also termed CD209. This mannose-binding C-type lectin receptor can serve as an anchor for HIV virions on dendritic cells (DC) and, thereby, promote the efficient infection of CD4+ T cells that engage with this antigen-presenting cell [90].
By examining this interaction, it was determined that the binding of HIV virions to isolated DCs leads to the production of a factor that inhibited PMN NETosis. This feature was dependent on the gp120 membrane glycoprotein of HIV, as it could be mimicked by addition of recombinant gp120 to DCs. (Fig. 4). Further analysis indicated that the factor released by gp120/CD-SIGN-primed DCs was the immune-modulating cytokine IL-10. In this manner, addition of recombinant IL-10 to PMN cultures lead to a reduction in their NETotic capability and prevented these cells from effectively engaging and debilitating HIV.

HIV downmodulates NETosis via dendritic cells. HIV triggers NETosis via a process that involves TLR7 and TLR8, trapping individual HIV virions in the NET lattice. Viral binding to CD209 anchors the virions on dendritic cells (DC), thereby promoting the efficient infection of CD4+ T cells. The gp120/CD209-primed DC cells produce the immune-modulating cytokine IL-10, leading to a reduction in NETotic capability, thereby preventing PMN from effectively engaging and eliminating HIV
Individuals with HIV are highly prone to secondary infections, especially those of the pulmonary system with pathogens such as M. tuberculosis or S. pneumoniae, which result in high degrees of mortality. Consequently, these findings have considerable implications for anti-HIV therapeutic strategies in order to counter AIDS-associated neutropenia and to ensure an effective innate immune response. In this context, it is worth noting that coinfection with M. tuberculosis bacilli can accelerate AIDS progression, since this bacterium can promote the replication of HIV and infection of CD4+ T cells [91].
Malaria: a possible autoimmune component?
A number of previous studies have shown that PMNs can generate NETs against multicellular eukaryotic parasites, including the use of Leishmania promastigotes in the study of NETosis in FeLV-infected cats [85, 92]. In human pathology, among the most important parasites are Plasmodium falciparum and Plasmodium vivax, the agents responsible for malaria with lethal outcome [93].
In pediatric patients infected with P. falciparum, Baker et al. made the striking observation that NETting PMNs could be readily detected directly in blood smears [94]. An examination of total PMNs in the smears indicated that P. falciparum infection was associated with a high proportion of segmented PMN, which decreased following treatment with Fansidar® (sulphadoxine–pyrimethamine). Of interest is that drug therapy leads to an increase in immature PMNs (metamyelocytes) and a significant decrease in bands. The degree of NETosis did not appear to be significantly influenced by therapy, although there was an increase in the number of cases observed post therapy (100 vs. 86 %). NETosis did, however, appear to correlate with plasma TNFα levels, indicating that a strong proinflammatory component was driving this response.
Of additional interest is that the occurrence of NETs appeared to correlate with the presence of antinuclear antibodies (ANA), with high levels of antibodies reactive with dsDNA, predictive of autoimmunity. Such autoantibodies could contribute to other malaria-related features such as renal dysfunction and uremia. These data may provide additional insight into the possible link between lupus susceptibility genetic loci and resistance to malaria [95], or into the possible mechanism of action of antimalarial drugs such as chloroquine and hydroxychloroquine used for the treatment of autoimmune conditions [96].
Influence of the host: pulmonary system
The ability of PMNs to form NETs is not only dependent on the external stimulus but also depends on the physiological status of the PMNs and the host microenvironment. One of the best characterized host microenvironments is the pulmonary system.
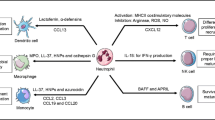
The alveolar epithelium synthesizes proteins such as the surfactant protein A and D (SP-A, SP-D), members of the collectin family of PPRs, which bind carbohydrate moieties on pulmonary pathogens such as M. tuberculosis or S. pneumonia as well as fungi. Absence of these factors in genetically manipulated knockout mice diminished their capability to clear P. aeruginosa infections [97]. That these factors can interact with members of the innate immune system to promote an antibacterial response is underscored by the recent finding that SP-D can bind pathogens as well as NETs in order to augment NET-mediated bacterial trapping and eradication [98] (Fig. 5).

NETosis—modulation by the respiratory system. The response of the respiratory system to infection is complex involving chemokine recruitment of neutrophils and monocytes into the lumen of the alveoli. The NETotic release of cytotoxic DNA–protein complexes with NE, MPO, LL37, and other neutrophil proteases increases mucus viscosity and contributes to lung epithelial damage, thereby perpetuating a vicious cycle of injury and inflammation. Innate immune surfactant proteins (also termed collectins) contribute in maintaining the lungs' homeostasis with minimal inflammation. NETs are removed by DNAse degradation and macrophage ingestion. A delicate equilibrium between NETosis and NET clearance is fundamental for the successful combating of infectious agents with minimum tissue damage. NE neutrophil elastase, MPO myeloperoxidase, LL37 cathelicidin, SP-A surfactant protein A, SP-D surfactant protein D, PMN polymorphonuclear neutrophil, Mφ macrophages. Adapted and modified from Chen et al. [81]
On the other hand, numerous observations suggest that aberrant NETosis can lead to damage of the fragile alveoli [81]. Such NETosis-associated damage may be triggered by host trauma in conditions such as transfusion-related acute lung injury (TRALI) following blood transfusions [99, 100] or be the result of viral infection [82]. Indeed, under such conditions of influenza infection, NETs appear to be incapable of clearing any secondary infections with pneumococci but rather contribute to increasingly compromised lung function [101].
It has, furthermore, been shown that an underlying disturbance of the alveolar tissue, as in the instance in cystic fibrosis (CF), can trigger NETosis, thereby contributing to the bronchial pool of cell-free DNA [102]. In order to clear this thick and sticky sputum, exogenous DNAse is used as a therapeutic agent [102]. In examining the contribution of PMN NETs to this phenomenon, it was observed that neutrophil elastase (NE) promoted the solubilisation of CF sputum by assisting with the degradation of histones.
This PMN-derived enzyme, however, does not only play a role in the clearance of excess NET debris but also, in combination with protease 3 and cathepsin G, may play a significant role in the damage caused to the underlying alveolar epithelium [103]. In a further study using atomic force and scanning electron microscopy, it was determined that the major portion of DNA in CF sputum resulted from excessive NETosis [104]. Consequently, it will be necessary to refocus therapeutic strategies to tackle NETs as a whole rather than focus on the sole dissolution of individual DNA fibers.
The occurrence of this highly gelatinous mucous in the lungs may, however, render this microenvironment more amenable to infections, such as those with P. aeruginosa, which frequently occur in patients with CF [105]. In this scenario, it appears that the highly dense DNA-based biofilm permits P. aeruginosa to adopt a NET resistance phenotype, thereby evading efficacious removal by this arm of the innate immune system. This phenomenon appears to be specific for P. aeruginosa isolates from affected CF lungs, as PMN isolated from CF patients were quite capable of generating NETs against normal P. aeruginosa isolates and killing these efficiently [105].
Influence of the host: pregnancy
Pregnancy poses a unique immune challenge as the mother has to maintain an effective immune response against pathogens deleterious to her own health and yet not mount a response that be deleterious to her unborn child [106]. Although most studies have largely focussed on how the maternal T cell response is modulated to avoid rejection of the semiallogeneic components of her fetus via regulatory T cells [107] or a shift in T cell cytokine profiles (Th1 vs. Th2) [108], it has become evident that pregnancy involves considerable changes in the activity of the innate immune system, specifically in that of neutrophils [109]. In general, pregnancy is associated with neutrophil leucoytosis, with numbers increasing from the second month of pregnancy and plateauing at term. Neutrophil action in the reproductive cycle is quite diverse and may range from the trapping and elimination of excess spermatozoa directly in the reproductive tract during fertilization, the rendering of the myometrium more receptive to implantation, or the contribution to the etiology of preeclampsia via overt NETosis [109, 110].
The modulation of the immune system, both innate and adaptive, during pregnancy may, however, be more extensive that initially thought. In this manner, due to elevated oestrogen levels, the upper respiratory tract in pregnancy is associated with increased mucous secretion, capillary congestion, and fragility [81]. This may render pregnant women more susceptible to airborne noxins or pathogens. Such respiratory infections can have severe consequences for the fetus, as they can lead to secondary placental infections, which may even result in stillbirth or septic abortion [111].
While it is well appreciated that prenatal exposure to maternal smoking or airborne particulate matter is associated with higher rates of pediatric asthma [112], new data indicate that these events may debilitate the neonatal immune response [113], rendering the developing child more susceptible to infections [114].
With regard to the pathogenic agents covered in this review in the context of neutrophil interactions, two points are worth noting:
-
1.
Low maternal and neonatal neutrophil counts are associated with an increased risk of HIV transmission in utero in African populations [115]. This suggests that anti-HIV therapy in women with low neutrophil counts may need to be started earlier in pregnancy to prevent viral transmission to the fetus.
-
2.
Pregnant women are more susceptible to infections with influenza, resulting in high degrees of maternal or fetal morbidity and mortality [116].
-
3.
In affected populations, pregnant women generally have a higher risk for malaria than nonpregnant women [117]. Since the malaria parasite is sequestered to the placenta, this leads to severe complications such as fetal growth restriction and preterm delivery [117, 118].
It is not clear why pregnant women should be more susceptible to malaria infection, especially if they are primigravid, but it may be linked to a reduction in neutrophil counts [119] and dysfunction of residual neutrophils due to impaired NADPH oxidase activity [120].
These findings suggest that pregnant women should be regarded as a separate entity at high risk for infection with a diverse array of pathogens. This may result from an altered immune response and, consequently, implies that pregnant women will require tailored therapeutic strategies.
Discussion and conclusions
In this review, the emphasis has been placed on mechanisms used by pathogenic microorganisms to evade entrapment and killing by PMN NETs. These range from the expression of DNAse molecules that degrade the nucleic acid lattice backbone [10] to the induction of immune-modulatory molecules such as IL-10 that dampen the NETotic response of PMN. The latter is employed by both HIV [88] or Sias-coated P. aeruginosa bacteria [14]. In the context of HIV, it is possible that IL-10-mediated dampening of the innate immune response may contribute to the secondary infection of affected individuals with tuberculosis or pneumococci.
By studying such interactions in more detail, it may not only be possible to devise strategies to optimize the host immune response but also develop therapies to counter pathologies associated with overt NETosis. In this manner, by antagonising the effect of IL-10 in cases with AIDS, it may be possible to reduce concomitant secondary infections. On the other hand, the use of IL-10 may be of interest in autoimmune conditions associated with increased NETosis such as systemic lupus erythematosus [121, 122].
In this regard, it is ironic that the first therapy used to alleviate and facilitate clearing of the dense mucus, which largely consists of NETs, from the lungs of CF patients was by the application of rhDNAse [123], thereby mimicking a strategy first explored by streptococcal bacteria [10, 11].
References
Borregaard N (2010) Neutrophils, from marrow to microbes. Immunity 33:657–670
Mantovani A et al (2011) Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 11:519–531
Brinkmann V et al (2004) Neutrophil extracellular traps kill bacteria. Science 303:1532–1535
Urban CF et al (2006) Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol 8:668–676
Urban CF et al (2009) Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defence against Candida albicans. PLoS pathogens 5:e1000639
Bianchi M et al (2009) Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 114:2619–2622
Bruns S et al (2010) Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS pathogens 6:e1000873
Medina E (2009) Neutrophil extracellular traps: a strategic tactic to defeat pathogens with potential consequences for the host. Journal of innate immunity 1:176–180
Ermert D et al (2009) Fungal and bacterial killing by neutrophils. Methods in molecular biology 470:293–312
Buchanan JT et al (2006) DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Current biology : CB 16:396–400
Beiter K et al (2006) An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Current biology : CB 16:401–407
Berends ET et al (2010) Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. Journal of innate immunity 2:576–586
Peterson EJ et al (2013) Inhibitors of Streptococcus pneumoniae surface endonuclease EndA discovered by high-throughput screening using a PicoGreen fluorescence assay. J Biomol Screen 18:247–257
Khatua B et al (2012) Sialoglycoproteins adsorbed by Pseudomonas aeruginosa facilitate their survival by impeding neutrophil extracellular trap through siglec-9. J Leukoc Biol 91:641–655
Varki A, Gagneux P (2012) Multifarious roles of sialic acids in immunity. Ann N Y Acad Sci 1253:16–36
Carlin AF et al (2009) Molecular mimicry of host sialylated glycans allows a bacterial pathogen to engage neutrophil Siglec-9 and dampen the innate immune response. Blood 113:3333–3336
Tamassia N et al (2013) Cutting edge: an inactive chromatin configuration at the IL-10 locus in human neutrophils. J Immunol 190:1921–1925
Huynh KK et al (2011) A delicate dance: host response to mycobacteria. Curr Opin Immunol 23:464–472
Torrado E et al (2011) Cellular response to mycobacteria: balancing protection and pathology. Trends in immunology 32:66–72
Zumla A et al (2013) Tuberculosis. N Engl J Med 368:745–755
Lowe DM et al (2012) Neutrophils in tuberculosis: friend or foe? Trends in immunology 33:14–25
Eum SY et al (2010) Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest 137:122–128
Berry MP et al (2010) An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466:973–977
Ramos-Kichik V et al (2009) Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis 89:29–37
Doherty TM (2012) Immunotherapy for TB. Immunotherapy 4:629–647
Steinwede K et al (2012) Cathepsin G and neutrophil elastase contribute to lung-protective immunity against mycobacterial infections in mice. J Immunol 188:4476–4487
Jena P et al (2012) Azurophil granule proteins constitute the major mycobactericidal proteins in human neutrophils and enhance the killing of mycobacteria in macrophages. PLoS One 7:e50345
Walters SB et al (2013) Microparticles from mycobacteria-infected macrophages promote inflammation and cellular migration. J Immunol 190:669–677
Duarte TA et al (2012) Mycobacterium tuberculosis-induced neutrophil ectosomes decrease macrophage activation. Tuberculosis 92:218–225
Wong, K.W. and Jacobs, W.R., Jr. (2013) Mycobacterium tuberculosis exploits human interferon-gamma to stimulate macrophage extracellular trap formation and necrosis. The Journal of infectious diseases
Feltcher ME et al (2010) Protein export systems of Mycobacterium tuberculosis: novel targets for drug development? Future Microbiol 5:1581–1597
Yount NY, Yeaman MR (2013) Peptide antimicrobials: cell wall as a bacterial target. Ann N Y Acad Sci 1277:127–138
Zanetti M (2004) Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol 75:39–48
Gudmundsson GH et al (1996) The human gene FALL39 and processing of the cathelin precursor to the antibacterial peptide LL-37 in granulocytes. European journal of biochemistry / FEBS 238:325–332
Dorschner RA et al (2001) Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A Streptococcus. J Investig Dermatol 117:91–97
Thennarasu S et al (2010) Antimicrobial and membrane disrupting activities of a peptide derived from the human cathelicidin antimicrobial peptide LL37. Biophys J 98:248–257
Turner J et al (1998) Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob Agents Chemother 42:2206–2214
Martineau AR et al (2007) Neutrophil-mediated innate immune resistance to mycobacteria. J Clin Invest 117:1988–1994
Reinholz M et al (2012) Cathelicidin LL-37: an antimicrobial peptide with a role in inflammatory skin disease. Ann Dermatol 24:126–135
Jung YJ et al (2012) Enhanced resistance to bacterial and fungal pathogens by overexpression of a human cathelicidin antimicrobial peptide (hCAP18/LL-37) in Chinese cabbage. Plant biotechnology reports 6:39–46
Nizet V, Gallo RL (2003) Cathelicidins and innate defence against invasive bacterial infection. Scand J Infect Dis 35:670–676
Barns, K.J. and Weisshaar, J.C. (2013) Real-time attack of LL-37 on single Bacillus subtilis cells. Biochimica et biophysica acta
Nijnik A, Hancock RE (2009) The roles of cathelicidin LL-37 in immune defences and novel clinical applications. Curr Opin Hematol 16:41–47
Niyonsaba F et al (2007) Antimicrobial peptides human beta-defensins stimulate epidermal keratinocyte migration, proliferation and production of proinflammatory cytokines and chemokines. J Investig Dermatol 127:594–604
Braff MH et al (2007) Staphylococcus aureus exploits cathelicidin antimicrobial peptides produced during early pneumonia to promote staphylokinase-dependent fibrinolysis. J Infect Dis 195:1365–1372
Choi KY, Mookherjee N (2012) Multiple immune-modulatory functions of cathelicidin host defence peptides. Front Immunol 3:149
Lande R et al (2007) Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449:564–569
Baumgarth N, Bevins CL (2007) Autoimmune disease: skin deep but complex. Nature 449:551–553
Kessenbrock K et al (2009) Netting neutrophils in autoimmune small-vessel vasculitis. Nature medicine 15:623–625
Hoffmann, M.H., et al. (2012) The cathelicidins LL-37 and rCRAMP are associated with pathogenic events of arthritis in humans and rats. Annals of the rheumatic diseases
Gutner M et al (2009) Saliva enables the antimicrobial activity of LL-37 in the presence of proteases of Porphyromonas gingivalis. Infect Immun 77:5558–5563
Sieprawska-Lupa M et al (2004) Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob Agents Chemother 48:4673–4679
Lauth X et al (2009) M1 protein allows Group A streptococcal survival in phagocyte extracellular traps through cathelicidin inhibition. Journal of innate immunity 1:202–214
Bergsson G et al (2009) LL-37 complexation with glycosaminoglycans in cystic fibrosis lungs inhibits antimicrobial activity, which can be restored by hypertonic saline. J Immunol 183:543–551
Zheng Y et al (2007) Cathelicidin LL-37 induces the generation of reactive oxygen species and release of human alpha-defensins from neutrophils. Br J Dermatol 157:1124–1131
Tjabringa GS et al (2006) Human cathelicidin LL-37 is a chemoattractant for eosinophils and neutrophils that acts via formyl-peptide receptors. International archives of allergy and immunology 140:103–112
Wantha S et al (2013) Neutrophil-derived cathelicidin promotes adhesion of classical monocytes. Circ Res 112:792–801
Zuyderduyn S et al (2006) The antimicrobial peptide LL-37 enhances IL-8 release by human airway smooth muscle cells. The Journal of allergy and clinical immunology 117:1328–1335
Engelmann B, Massberg S (2013) Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol 13:34–45
Semeraro F et al (2011) Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 118:1952–1961
Brill A et al (2012) Neutrophil extracellular traps promote deep vein thrombosis in mice. Journal of thrombosis and haemostasis : JTH 10:136–144
Das R et al (2007) Histone H2B as a functionally important plasminogen receptor on macrophages. Blood 110:3763–3772
Fuchs TA et al (2010) Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 107:15880–15885
Higuchi DA et al (1992) The effect of leukocyte elastase on tissue factor pathway inhibitor. Blood 79:1712–1719
Takano S et al (1990) Plasma thrombomodulin in health and diseases. Blood 76:2024–2029
Plow EF (1980) The major fibrinolytic proteases of human leukocytes. Biochim Biophys Acta 630:47–56
von Bruhl ML et al (2012) Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. The Journal of experimental medicine 209:819–835
Massberg S et al (2010) Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med 16:887–896
Kambas K et al (2012) The emerging role of neutrophils in thrombosis—the journey of TF through NETs. Front Immunol 3:385
Kambas K et al (2012) Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PLoS One 7:e45427
Xu J et al (2011) Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol 187:2626–2631
Clark SR et al (2007) Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nature medicine 13:463–469
73 Caudrillier, A., et al. (2012) Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest
Frick IM et al (2006) The contact system—a novel branch of innate immunity generating antibacterial peptides. EMBO J 25:5569–5578
Yeaman MR (2010) Platelets in defence against bacterial pathogens. Cellular and molecular life sciences : CMLS 67:525–544
Szaba FM, Smiley ST (2002) Roles for thrombin and fibrin(ogen) in cytokine/chemokine production and macrophage adhesion in vivo. Blood 99:1053–1059
Bergmann S, Hammerschmidt S (2007) Fibrinolysis and host response in bacterial infections. Thromb Haemost 98:512–520
Sun H et al (2004) Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science 305:1283–1286
Guggenberger C et al (2012) Two distinct coagulase-dependent barriers protect Staphylococcus aureus from neutrophils in a three dimensional in vitro infection model. PLoS Pathog 8:e1002434
Cheng AG et al (2010) Contribution of coagulases towards Staphylococcus aureus disease and protective immunity. PLoS Pathog 6:e1001036
Cheng OZ, Palaniyar N (2013) NET balancing: a problem in inflammatory lung diseases. Front Immunol 4:1
Narasaraju T et al (2011) Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol 179:199–210
Hemmers S et al (2011) PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS One 6:e22043
Jenne CN et al (2013) Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell host & microbe 13:169–180
Wardini AB et al (2010) Characterization of neutrophil extracellular traps in cats naturally infected with feline leukemia virus. J Gen Virol 91:259–264
Jarrett O (1996) The relevance of feline retroviruses to the development of vaccines against HIV. AIDS research and human retroviruses 12:385–387
Jenne CN, Kubes P (2012) NETs tangle with HIV. Cell host & microbe 12:5–7
Saitoh T et al (2012) Neutrophil extracellular traps mediate a host defence response to human immunodeficiency virus-1. Cell host & microbe 12:109–116
Gustafsson MG et al (2008) Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys J 94:4957–4970
da Silva RC et al (2011) Role of DC-SIGN and L-SIGN receptors in HIV-1 vertical transmission. Hum Immunol 72:305–311
Goletti D et al (1996) Effect of Mycobacterium tuberculosis on HIV replication. Role of immune activation. J Immunol 157:1271–1278
Papayannopoulos V, Zychlinsky A (2009) NETs: a new strategy for using old weapons. Trends in immunology 30:513–521
Battle KE et al (2012) The global public health significance of Plasmodium vivax. Adv Parasitol 80:1–111
Baker VS et al (2008) Cytokine-associated neutrophil extracellular traps and antinuclear antibodies in Plasmodium falciparum infected children under six years of age. Malar J 7:41
Waisberg M et al (2011) Genetic susceptibility to systemic lupus erythematosus protects against cerebral malaria in mice. Proc Natl Acad Sci U S A 108:1122–1127
Ben-Zvi I et al (2012) Hydroxychloroquine: from malaria to autoimmunity. Clinical reviews in allergy & immunology 42:145–153
Giannoni E et al (2006) Surfactant proteins A and D enhance pulmonary clearance of Pseudomonas aeruginosa. American journal of respiratory cell and molecular biology 34:704–710
Douda DN et al (2011) Innate immune collectin surfactant protein D simultaneously binds both neutrophil extracellular traps and carbohydrate ligands and promotes bacterial trapping. J Immunol 187:1856–1865
Thomas GM et al (2012) Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood 119:6335–6343
Caudrillier A et al (2012) Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest 122:2661–2671
Narayana Moorthy A et al (2013) In vivo and in vitro studies on the roles of neutrophil extracellular traps during secondary pneumococcal pneumonia after primary pulmonary influenza infection. Front Immunol 4:56
Papayannopoulos V et al (2011) Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy. PLoS One 6:e28526
Dubois AV et al (2012) Influence of DNA on the activities and inhibition of neutrophil serine proteases in cystic fibrosis sputum. American journal of respiratory cell and molecular biology 47:80–86
Manzenreiter R et al (2012) Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 11:84–92
Young RL et al (2011) Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa: evidence of acquired resistance within the CF airway, independent of CFTR. PLoS One 6:e23637
Aagaard-Tillery KM et al (2006) Immunology of normal pregnancy. Semin Fetal Neonatal Med 11:279–295
Erlebacher A (2013) Mechanisms of T cell tolerance towards the allogeneic fetus. Nat Rev Immunol 13:23–33
Sykes L et al (2012) The Th1:th2 dichotomy of pregnancy and preterm labour. Mediat Inflamm 2012:967629
Hahn S et al (2012) Neutrophil NETs in reproduction: from infertility to preeclampsia and the possibility of fetal loss. Front Immunol 3:362
Gupta A et al (2006) Occurrence of neutrophil extracellular DNA traps (NETs) in pre-eclampsia: a link with elevated levels of cell-free DNA? Ann N Y Acad Sci 1075:118–122
Sandu C et al (2013) Hematogenous placental infection in acute respiratory infections. Romanian journal of morphology and embryology = Revue roumaine de morphologie et embryologie 54:157–161
Proietti E et al (2013) Air pollution during pregnancy and neonatal outcome: a review. Journal of aerosol medicine and pulmonary drug delivery 26:9–23
Singh SP et al (2011) Prenatal secondhand cigarette smoke promotes Th2 polarization and impairs goblet cell differentiation and airway mucus formation. J Immunol 187:4542–4552
Claude JA et al (2012) Perinatal exposure to environmental tobacco smoke (ETS) enhances susceptibility to viral and secondary bacterial infections. International journal of environmental research and public health 9:3954–3964
Kourtis AP et al (2012) Neutrophil count in African mothers and newborns and HIV transmission risk. N Engl J Med 367:2260–2262
Cantu J, Tita AT (2013) Management of influenza in pregnancy. Am J Perinatol 30:99–103
Takem EN, D'Alessandro U (2013) Malaria in pregnancy. Mediterranean journal of hematology and infectious diseases 5:e2013010
Rogerson SJ et al (2007) Malaria in pregnancy: linking immunity and pathogenesis to prevention. AmJTrop Med Hyg 77:14–22
Davison BB et al (2005) Alterations in the profile of blood cell types during malaria in previously unexposed primigravid monkeys. J Infect Dis 191:1940–1952
Cunnington AJ et al (2012) Prolonged neutrophil dysfunction after Plasmodium falciparum malaria is related to hemolysis and heme oxygenase-1 induction. J Immunol 189:5336–5346
Garcia-Romo, G.S., et al. (2011) Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Science translational medicine 3, 73ra20
Lande, R., et al. (2011) Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA–peptide complexes in systemic lupus erythematosus. Science translational medicine 3, 73ra19
Suri R (2005) The use of human deoxyribonuclease (rhDNase) in the management of cystic fibrosis. BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy 19:135–144
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on Neutrophils - Guest Editors: Paul Hasler and Sinuhe Hahn
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Hahn, S., Giaglis, S., Chowdury, C.S. et al. Modulation of neutrophil NETosis: interplay between infectious agents and underlying host physiology. Semin Immunopathol 35, 439–453 (2013). https://doi.org/10.1007/s00281-013-0380-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-013-0380-x