
Abstract
Purpose
Aberrant alterations of ERBB receptor tyrosine kinases lead to tumorigenesis. Single agent therapy targeting EGFR or HER2 has shown clinical successes, but drug resistance often develops due to aberrant or compensatory mechanisms. Herein, we sought to determine the feasibility and safety of neratinib and trametinib in patients with EGFR mutation/amplification, HER2 mutation/amplification, HER3/4 mutation and KRAS mutation.
Methods
Patients with actionable somatic mutations or amplifications in ERBB genes or actionable KRAS mutations were enrolled to receive neratinib and trametinib in this phase I dose escalation trial. The primary endpoint was determination of the maximum tolerated dose (MTD) and dose-limiting toxicity (DLT). Secondary endpoints included pharmacokinetic analysis and preliminary anti-tumor efficacy.
Results
Twenty patients were enrolled with a median age of 50.5 years and a median of 3 lines of prior therapy. Grade 3 treatment-related toxicities included: diarrhea (25%), vomiting (10%), nausea (5%), fatigue (5%) and malaise (5%). The MTD was dose level (DL) minus 1 (neratinib 160 mg daily with trametinib 1 mg, 5 days on and 2 days off) given 2 DLTs of grade 3 diarrhea in DL1 (neratinib 160 mg daily with trametinib 1 mg daily). The treatment-related toxicities of DL1 included: diarrhea (100%), nausea (55.6%) and rash (55.6%). Pharmacokinetic data showed trametinib clearance was significantly reduced leading to high drug exposures of trametinib. Two patients achieved stable disease (SD) ≥ 4 months.
Conclusion
Neratinib and trametinib combination was toxic and had limited clinical efficacy. This may be due to suboptimal drug dosing given drug–drug interactions.
Trial registration ID: NCT03065387.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The epidermal growth factor family of trans-membrane receptors (ERBB family) members are effective mediators of normal cell growth and development [1]. The ERBB family of transmembrane receptor tyrosine kinases (RTKs) is comprised of four closely related receptors: epidermal growth factor receptor (EGFR; HER1), human epidermal growth factor receptor 2 (HER2), HER3 and HER4, which homo- or heterodimerize to sequentially relay signals primarily through the phosphoinositide 3-kinase (PI3K)/AKT/ mammalian target of rapamycin (mTOR) and mitogen-activated protein kinase (MAPK) pathways [2]. Aberrant expression of EGFR, HER2 and HER3, whether induced by gene amplification, gene mutation or protein overexpression, is linked to the development of many epithelial cancers [2,3,4,5,6,7,8,9,10,11,12,13].
Neratinib, a potent oral, irreversible, pan-ERBB inhibitor targets EGFR, HER2, and HER4 at the intracellular tyrosine kinase domains [14]. Neratinib reduces EGFR and ERBB2 autophosphorylation, and their downstream signaling, and inhibits the growth of EGFR- and HER2-dependent cell lines [14, 15]. Despite compelling clinical benefit from neratinib in patients with the appropriate genetic alterations, patients frequently develop resistance resulting in cancer progression and/or relapse [4, 10].
The MAPK pathway, the prominent downstream effector of ERBB signaling, is comprised of rat sarcoma virus (RAS), rapidly accelerated fibrosarcoma (RAF), mitogen-activated protein kinase kinase (MEK) and extracellular signal-regulated kinase (ERK). This pathway is involved in proliferation, survival and differentiation and as a result, deregulation of this pathway contributes to cancer [4, 16, 17]. Preclinical studies have shown that in HER2-overexpressing cells, combining MEK inhibition with neratinib reduced phosphorylated ERK more than either single agent [18]. Further, combination therapy suppressed tumor growth and reduced expression of the Forkhead box transcription factor M1 (FOXM1) in HER2-overexpressing breast cancers resistant to trastuzumab and lapatinib, and suppressed tumor growth and increased progression free survival in patient-derived xenografts of breast, colorectal and esophageal cancers with HER2 mutations [18, 19].
Additionally, the mutant RAS pathway amplifies the ERBB kinase activity both in vitro and in vivo [20]. Broad inhibition of kinases of the ERBB family by neratinib has been shown to suppress KRAS G12D mutant-driven lung tumors and enhance the potency of MEK inhibition by trametinib in a cre-inducible immunocompetent mouse model of autochthonous lung cancer [20]. Similarly, another pan-ERBB inhibitor, afatinib, was shown to reduce the progression of KRAS G12D driven lung cancer in preclinical mouse models [21]. Both studies have shown that cancers with KRAS mutations rely on resistance mechanisms that involve signaling through the ERBB network. These mechanisms can be effectively targeted either using a combination of neratinib and trametinib or using a single agent afatinib, irrespective of the expression or mutation of EGFR/ERBB [20, 21]. Herein, we report the feasibility and safety results of a single-center study of neratinib and trametinib in patients with EGFR mutation/amplification, HER2 mutation/amplification, HER3/4 mutation and KRAS mutation.
Methods
Study design and dosing
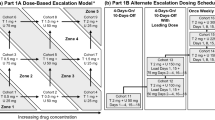
This study was part of an ongoing open-label, non-randomized, single-center, phase I dose-escalation trial, conducted in patients with advanced or metastatic cancer with EGFR mutation/amplification, HER-2 mutation/amplification, HER-3/4 mutation or KRAS mutation (NCT03065387). Neratinib was provided by Puma Biotechnology and trametinib was commercially obtained. Neratinib was orally administered in a continuous 28-day cycle and trametinib was given orally once daily or followed a 5 day on and 2 days off schedule. Compliance was determined through review of pill diaries and unused drugs returned at the end of every cycle. The study was sponsored by Puma Biotechnology and was approved by the Institutional Review Board (IRB) in accordance with the Declaration of Helsinki, Good Clinical Practice, and all federal, state and local regulatory guidelines. Consent was obtained from all patients prior to study enrollment.
The protocol followed a standard 3 + 3 design [22]. Adverse events (AE) were graded based on the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events, version 4.0 (CTCAEv4.0). Dose limiting toxicity (DLT) was defined as any treatment-related grade ≥ 3 non-hematologic toxicities (except for: grade 3 nausea and vomiting lasting < 72 h with adequate anti-emetic and supportive care; grade 3 diarrhea lasting < 48 h with optimal medical therapy; alopecia; and, electrolyte imbalances that resolved with supportive care); any grade 4 hematologic toxicity lasting more than a week; grade 3 thrombocytopenia with bleeding; neutropenic fever; all other grade 3 non-hematologic toxicity and any study drugs related severe or life-threatening conditions not defined in the CTCAEv4.0. DLT evaluable patients were defined as patient that had received at least 75% of the study drugs in the first cycle (28 days). A patient who discontinued therapy without completing the first tumor assessment (radiographic evaluation approximately 8 weeks after baseline) would be in-evaluable for response assessment. The maximum tolerated dose (MTD) was defined as the highest dose at which no more than 1 of 6 evaluable patients had a DLT.
Safety assessments
All patients were evaluated for new or worsening adverse events by investigator or qualified designee. The assessments were conducted weekly during the first cycle (28 days), and then monthly from cycle 2 onwards. Evaluation could be conducted at higher frequency if required by the patient’s clinical condition. The assessment included a complete physical examination, eastern cooperative oncology group (ECOG) assessment, and the recording of vital signs. All toxicities were carefully evaluated in terms of grading, seriousness, action taken regarding trial agents and causality to each study agent.
Study oversight
The study was conducted in compliance with the Declaration of Helsinki, Good Clinical Practice, and all relevant federal, state and local regulatory guidelines and was approved by the MD Anderson cancer center Institutional Review Board (IRB). To ensure adherence to the study procedures and patient safety, the study was monitored by Investigational New Drug (IND) office at MD Anderson Cancer Center. All treatment-related toxicities experienced by the patients, including any cases of early termination, were reported to and reviewed by the IND office before approval of enrollment to subsequent cohorts.
Eligibility criteria
Key inclusion criteria included patients with advanced solid tumors (not hematologic malignancy), either relapsed after standard therapy or without standard therapy available. Patients must have had one of the following pre-identified somatic molecular aberrations predicted to be activating or pathogenic as performed in the Clinical Laboratory Improvement Amendments (CLIA) environment and suitable for enrollment: EGFR mutation/amplification, HER-2 mutation/amplification, HER-3/4 mutation or KRAS mutation; age 18 years or older; measurable disease by Response Evaluation Criteria in Solid Tumors (RECIST) v1.1; ECOG status ≤ 1; with adequate organ functions including absolute neutrophils > 1500 cells/uL, platelets ≥ 100,000/uL, hemoglobin ≥ 9 g/dL, total bilirubin ≤ 1.5 × upper limit of normal (ULN), serum creatinine < 1.5 × ULN and alanine transaminase (ALT) ≤ 2.5 × ULN (≤ 5 × ULN if with liver metastases); patients that are of child-bearing potential must agree to use adequate contraception. Key exclusion criteria included concurrent chemotherapy treatment; uncontrolled illness such as active infection requiring intravenous (IV) antibiotics; any clinically significant heart conditions or gastrointestinal abnormalities that may alter absorption or inability to swallow pills; skin rash > grade 1; albumin < 3 gm/dL; or, history of retinal disorder, dry eye syndrome or blurry vision not evaluated and cleared by ophthalmology prior to starting treatment.
Genomic eligibility
All genomic alterations in eligible genes were reviewed by the MD Anderson Precision Oncology Decision Support (PODS) prior to patient enrollment. Alterations were researched within the published literature for any known effect on function, stability, expression, or therapeutic sensitivity. Alterations were then classified for their functional significance and variant-level actionability, as previously described [23].
Assessment of tumor response
Baseline radiographic imaging (e.g., computed tomography (CT) scan or magnetic resonance imaging (MRI)) was performed within four weeks of the start of treatment. Tumor measurements were performed on patients at baseline and at the end of every two cycles (three cycles after 24 weeks). Measurable target lesions were evaluated for response using RECIST v1.1. For purposes of this report, prolonged stable disease (SD) was defined as lasting ≥ 16 weeks.
Pharmacokinetics
Pharmacokinetic (PK) analysis for trametinib and neratinib was performed on cycle 1, day 15 with blood samples for neratinib and trametinib collected at pre-dose, 1, 3, 4, 6, 8- and 24-h post-dose. Trametinib and neratinib plasma concentrations were quantified using a validated tandem LC/MS assay as described [24, 25]. PK parameters for trametinib and neratinib were determined using noncompartmental analysis methods using Phoenix WinNonlin 8.1 (Certara USA Inc, Princeton, NJ, USA).
Results
Patient characteristics
Between the period of November 2017 and March 2020, a total of 20 patients with advanced solid tumor were enrolled to receive combination therapy with neratinib and trametinib. The demographic and clinical characteristics of the patients are summarized in Table 1. The median age of patients was 50.5 years (range 26–71 years) with the majority being female (60%). The median number of prior systemic therapies was 3 (range 1–11). Five (25%) patients had received prior therapy with either EGFR (n = 1) or HER-2 targeted therapies (n = 4). The most common malignancies were colorectal cancer (n = 10, 50%), ovarian cancer (n = 3, 15%) and esophageal cancer (n = 2, 10%).
Table 1 summarizes the pre-identified somatic molecular alterations that were deemed activating or pathogenic and enrolled in the study, including KRAS mutations (n = 14; 70%) and pan-ERBB alterations (n = 15, 75%). ERBB2 amplification (n = 7, 35%) was the most common alteration in enrolled patients with pan-ERBB alterations followed by ERBB2 mutation (n = 4, 20%). Co-occurring alterations were reported in three patients including an ovarian patient with somatic EGFR G724S and ERBB2 Y590C mutation; a colorectal patient with HER2 amplification and ERBB2 D769Y mutation; and a colorectal patient with co-existing EGFR amplification by next generation sequencing (NGS) and EGFR L861R mutation. Among the ERBB2 amplified patients, 5 of seven patients had ERBB2 amplification by fluorescence in situ hybridization (FISH) and 2 patients had amplification confirmed by next generation sequencing (NGS).
Toxicity assessment and adverse event
Twenty patients received study drugs and were evaluable for safety. A total of 9 patients were treated on dose level 1, with continuous oral administration of 160 mg neratinib in combination with 1 mg of trametinib daily. Three of 9 patients treated on dose level 1 discontinued treatment prematurely and failed to complete 75% of study drug due to clinical progression (n = 1), consent withdrawal after 7 doses (n = 1) and hospitalization due to anemia unrelated to study drugs (n = 1). Six of 9 patients completed at least 75% of study drug during first cycle and were evaluable for DLT. Grade 3 diarrhea were reported as DLT in 2 patients at dose level 1. Therefore, dose level minus 1 (160 mg neratinib daily with 1 mg trametinib 5 days on and 2 days off) was selected as maximum tolerated dose (MTD) per protocol guidance. Eleven patients were treated at dose level minus 1 with 8 patients completing at least 75% of the study drug during the first cycle. Three patients failed to complete 75% of study drug in cycle one due to hospital admission for disease-related conditions (n = 2) and withdrawal of consent to seek other treatment options (n = 1).
Safety assessments are summarized in Table 2. Treatment-related adverse events (TRAEs) were observed in 95% of patients on study. Seven patients (35%) experienced at least 1 or more grade 3 adverse events that were attributed to study drugs. No patients experienced grade 4 or higher TRAEs. There was a higher frequency and grading of TRAEs reported for neratinib as compared to trametinib. Nineteen of 20 patients (95%) had TRAEs that were attributed to neratinib and 80% of patients had TRAEs that were attributed to trametinib. After the completion of cycle 1, three patients discontinued study treatment in subsequent cycles secondary to toxicity. This includes 2 patients coming off trial due to diarrhea attributed to both study drugs during cycles 4 and 7, respectively, and the third patient withdrew consent due to nausea and vomiting attributed to both study drugs during cycle 2.
The TRAEs of neratinib and trametinib are shown in Table 3. Diarrhea (n = 19, 95%) was the most common TRAE followed by rash (n = 13, 65%), nausea (n = 10, 50%), vomiting (n = 6, 30%), fatigue (n = 4, 20%), mucositis (n = 4, 20%) and anorexia (n = 3, 15%). Notably, diarrhea also accounted for the most frequent grade 3 AE, observed in 5 patients (25%). Six serious adverse event (SAE) reports of diarrhea, 1 event of vomiting and 1 event of nausea, that were at least possibly related to study drugs, were reported on study (Supp. Table 1).
To evaluate whether combination therapy of neratinib with trametinib increased toxicity, we compared the TRAEs of all grades (frequency > 10%) between the monotherapies of neratinib or trametinib reported in previous studies, and the TRAEs observed in our combination therapy study. The single agent neratinib taken at 180 mg, the most comparable dose level, has similar toxicity profile and frequency as combination therapy [27]. Trametinib monotherapy (2 mg) has overlapping TRAEs with combination therapy, albeit at a lower frequency. Supp. Table 3 shows the frequency of grade 3 TRAEs for monotherapies of trametinib or neratinib, and for combination therapy. Combination therapy had increased toxicity with increased instances of grade 3 AEs of diarrhea, nausea and vomiting compared to either single agent neratinib or trametinib.
Pharmacokinetic (PK) analysis
PK parameters were assessed for neratinib and trametinib from available samples obtained from 8 patients that were dosed at dose level minus 1 (160 mg neratinib daily with 1 mg trametinib 5 days on and 2 days off). The PK parameters are summarized in Table 4 and plasma concentration profiles in Fig. 2. After oral administration, trametinib concentration peaked at 5.6 h with Cmax of 38.1 ± 12.3 ng/mL and trough concentration (C24) was 27.8 ± 12.7 ng/ml. The mean area under the concentration–time curve (AUC) was 732 ± 339 ng*h/mL, mean half-life was 52 ± 38 h, and the oral clearance was 1.55 ± 0.63 L/h. For neratinib, the mean AUC, half-life and oral clearance were 1491 ± 1189 ng·h/mL, 18 ± 6.15 h and 76.7 ± 16.9 L/h, respectively. The mean time to maximum concentration was about 6 h post dose administration with Cmax concentration of 102 ± 79.99 ng/mL. Neratinib trough concentration (C24) was 43.99 ± 33.19 ng/mL.

Waterfall plot and swimmer plot depicting response by patient a Patients/cohorts are represented by vertical bars on the X-axis. The best RECIST response (%) is depicted on the Y-axis. Seventeen patients were measurable by RECIST 1.1. Four of 17 patients were assigned a value of + 21% for clinical progression (*) and 2 were assigned a value of + 21% for new lesion or PD in non-target lesions (^). The upper dotted line indicates progressive disease by RECIST 1.1 (+ 20% from baseline). The lower dotted line indicates partial response RECIST 1.1 (-30% from baseline). b Swimmer plot analysis of 20 patients treated on study. Each lane presents a patient. X-axis represent time from treatment start (months). Y-axis represents patient number. SD, stable disease, PD, progression disease

Plasma concentration profiles of (a) trametinib and (b) neratinib. Mean (standard of deviation) plasma concentrations over time on cycle 1, day 15 (n = 8) after co-administration of trametinib 1 mg on 5 days on, 2 days off schedule and neratinib 160 mg daily
Antitumor activity
Among the 20 treated patients, 17 (85%) were evaluable for efficacy due to having RECISTv1.1 measurable disease and having completed post-baseline tumor assessment by radiographic imaging studies or physician-determined clinical progression. Two patients withdrew from the study during the first cycle to seek other treatment options. One patient was deemed non-evaluable for follow-up tumor re-assessment due to lack of contrast in target lesions to accurately assess the response.
Figure 1a is a waterfall plot depicting best response of these 17 patients. SD ≥ 4 months was observed in 2 patients treated at dose level 1 for a clinical benefit rate of 10% (2/20 patients). These patients included an ovarian cancer patient with KRAS G12D and EGFR G724S mutations with SD (- 9%) for 7 months and a salivary gland cancer patient with EGFR amplification by IHC with SD (+ 3%) for 5 months. Both patients eventually withdrew consent from study due to treatment-related diarrhea. Figure 1b shows the treatment duration of patients by swimmer plot. The median treatment duration for all patients was 1.8 months (95% CI 1.69 to 2.40 months). Four patients had stable disease by RECIST1.1, however, 2 patients failed to reach prolonged stable disease which we defined as greater than or equal to 4 months duration. As of data cut for analysis (March 1, 2020), only 1 patient from study remained alive and all patients were off study, including 15 patients (75%) due to progressive disease (6 clinical PD and 9 PD by RECIST) and 5 withdrew consent from study (3 patients due to toxicity and 2 patients decided to seek other treatment options).
Discussion
Aberration in the function or expression of ERBB receptor tyrosine kinases contributes to tumorigenesis. EGFR or HER2 targeting agents are widely used and have shown substantial clinical efficacy. However, drug resistance often arises from aberrant or compensatory mechanism from downstream signaling proteins. Neratinib and trametinib have demonstrated clinical benefits when used as monotherapy or in combination with chemotherapy [26,27,28,29,30,31]. Furthermore, preclinical data have revealed synergistic effects of combination therapy with neratinib and trametinib including enhanced tumor inhibition [18].
Here, we are the first to examine the safety, toxicity and preliminary anti-tumor efficacy in patients with advanced solid tumors treated with neratinib and trametinib. Unfortunately, continuous daily dosing of neratinib and trametinib was poorly tolerated. Two patients experienced DLTs of diarrhea at the initial dose level resulting in dose de-escalation to dose level minus 1 which was neratinib at a dose of 160 mg once daily in combination with trametinib at a dose of 1 mg once daily on a 5 days on and 2 days off schedule. This cohort was expanded to include a minimum of six patients treated and evaluable for DLT to further evaluate safety and tolerability. In total, 8 patients were DLT evaluable, and dose level minus one was determined to be the MTD. The TRAEs observed in this study are consistent with previous studies and were mainly gastrointestinal toxicities such as diarrhea, nausea and vomiting [27, 32]. Monotherapy with neratinib at 180 mg daily displayed a similar toxicity profile and frequency with combination therapy on this study. Supp. Table 2 summarizes the TRAEs of all grades with neratinib or trametinib, and in combination. However, combination therapy revealed increased instances of grade 3 AEs including diarrhea, nausea, and vomiting as compared to either single agent neratinib or trametinib. This is likely due to overlapping toxicities causing additive side effects (Supp. Table 3). A previous study with dabrafenib and trametinib combination therapy has shown improvement in treatment tolerance when patients transitioned from continuous to intermittent dosing schedule [33]. Further exploration with intermittent dosing schedules of neratinib in combination with trametinib should be considered for future studies to help alleviate overlapping toxicity.
In our PK cohort of 8 patients treated at dose level minus 1 (160 mg neratinib daily with 1 mg trametinib 5 days on and 2 days off), trametinib pharmacokinetics, when co-administered with neratinib, appears to have a much lower oral clearance (1.55 L/h) leading to higher exposure (AUC0-24) compared to published single agent data results of 3.81 L/h [32, 34]. In fact, in our cohort, the mean trametinib exposure was 732 ng*h/mL ranging from 197 to 1054 ng*h/mL which was closer to the single agent dosing of trametinib at ≥ 2.5 mg [32]. Furthermore, at day 15, the accumulation ratios (AR) in our patient cohort are 2–3 times those reported by Infante et al. [32].
Studies of trametinib and neratinib have shown that each agent has different metabolism pathways. Neratinib is metabolized via cytochrome P450-3A4 (CYP3A4) pathway [35]. In our PK cohort, there was no observed changes in neratinib PKs compared to existing single agent PK data. However, neratinib is a known P-glycoprotein (P-gp) inhibitor and was shown to increase digoxin (a P-gp substrate) plasma concentrations (Cmax) by 54% and the AUC by 32% [36, 37]. Prior human study has shown that trametinib undergoes a non-cytochrome-mediated metabolism, involving deacetylation via hydrolytic enzymes alone or in combination with glucuronidation [38]. Trametinib is also a substrate of drug transporters, P-gp and breast cancer resistant protein (Bcrp) suggesting a P-gp and/or Bcrp substrate or inhibitor can modulate trametinib clearance and exposure [39]. Trametinib may be a low-level CYP3A4 inducer and, therefore, could lead to higher clearance of agents that are mainly metabolized by this same isoenzyme [38]. Fortunately, recent data revealed concomitant administration of trametinib and oral contraceptives (OC), well-known to be metabolized by CYP3A isoenzymes, showed no significant differences in the PK disposition of OC when compared to OC alone, and without any changes in clinical efficacy of OC [40].
We believe the observed drug–drug interaction from our PK analysis is due to combination therapy with neratinib and trametinib, with neratinib inhibiting the clearance of trametinib via the P-gp efflux pathway leading to a very high exposure of trametinib. There may be other factors such as saturation of deacetylation and glucuronidation enzyme pathways which may contribute to additional lowering of trametinib clearance resulting in increased total exposure. With trametinib as a substrate for P-gp, the use of P-gp strong inhibitors (e.g., ketoconazole, itraconazole, clarithromycin, erythromycin, ritonavir, verapamil) or P-gp inducers (e.g.,: phenytoin, rifampin, St. John’s wort, corticosteroids, efavirenz, nevirapine) should be avoided whenever possible when taking trametinib [41]. If clinically necessary, the use of known P-gp inhibitors or inducers when taking trametinib therapy, requires close monitoring and evaluation to avoid potential adverse events or decrease in clinical efficacy.
Limited anti-tumor efficacy was seen across all treated patients. The lack of meaningful response may be due to suboptimal dosing of both neratinib and trametinib. No partial responses were reported, and SD ≥ 4 months was seen in only 2 patients. Both patients were treated at dose level 1. No patients obtained SD ≥ 4 months or PR at dose level minus 1, the declared MTD.
Interestingly, both patients with SD ≥ 4 months harbored EGFR aberrations, including an ovarian cancer patient with KRAS G12D and EGFR G724S mutations and a salivary gland cancer patient with EGFR amplification by IHC. Zhao et al. showed that combination therapy with neratinib and trametinib induced reduction of ERK1/2 phosphorylation but failed to trigger robust anti-apoptotic activity. Zhao et al. also found that trametinib treatment resulted in downregulation of proteins involved in the MAPK and AKT pathways but increased total levels of EGFR [18]. This is in line with a prior study that correlated EGFR aberrations with favorable tyrosine kinase inhibitor treatment outcomes in lung cancer [42]. Taken together, these data may suggest that EGFR aberrations could be a response predictor for neratinib and trametinib treatment but the numbers in our study are too small to make any true assessment.
There are several limitations to this study. First, we were unable to dose escalate to meaningful doses of either agent in combination due to overlapping toxicity resulting in poor patient tolerance. Second, there was heterogeneity in the molecular aberrations allowable for enrollment onto study. Five of 11 (45.5%) patients enrolled on dose level minus 1 had only activating KRAS mutations with no co-occurring pan-ERBB aberrations. It is unclear if the lack of response at this dose level is due to suboptimal dosing versus the type of molecular aberration enrolled. Third, the sample size is small with patients being heavily pre-treated with a median of 3 lines of prior therapy and consisting of multiple solid tumor types making analysis challenging.
In conclusion, neratinib and trametinib combination therapy was not tolerable at dose level 1. MTD was declared as dose level minus 1 (neratinib 160 mg daily with trametinib 1 mg, 5 days on and 2 days off) and had limited clinical activity. This may be due to suboptimal drug dosing of trametinib and neratinib. The increased plasma exposure of trametinib may have contributed to the toxicity observed. Based on the results from our PK cohort, there was significant drug–drug interaction with an increase in trametinib plasma concentrations and exposure due to a decrease in clearance of trametinib. We hypothesize that this could be due to neratinib-induced inhibition of trametinib clearance via P-gp efflux mechanisms. Further work is needed in determining the best dose and/or schedule for treating patients with both agents in combination to improve tolerability. Additionally, further work needs to be done to determine the appropriate tumor types and molecular aberrations for enrollment.
Data availability
The dataset generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Baselga J, Swain SM (2009) Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer 9(7):463–475
Drago JZ, Ferraro E, Abuhadra N, Modi S (2022) Beyond HER2: targeting the ErbB receptor family in breast cancer. Cancer Treat Rev 109:102436
Herbst RS, Heymach JV, Lippman SM (2008) Lung cancer. N Engl J Med 359(13):1367–1380
Yarden Y, Pines G (2012) The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer 12(8):553–563
Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC et al (2013) Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov 3(2):224–237
Lee J, Ou SH (2013) Towards the goal of personalized medicine in gastric cancer–time to move beyond HER2 inhibition Part I targeting receptor tyrosine kinase gene amplification. Discov Med 15(85):333–41
Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S et al (2013) Oncogenic ERBB3 mutations in human cancers. Cancer Cell 23(5):603–617
Ueda S, Ogata S, Tsuda H, Kawarabayashi N, Kimura M, Sugiura Y et al (2004) The correlation between cytoplasmic overexpression of epidermal growth factor receptor and tumor aggressiveness: poor prognosis in patients with pancreatic ductal adenocarcinoma. Pancreas 29(1):e1-8
Hatanpaa KJ, Burma S, Zhao D, Habib AA (2010) Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 12(9):675–684
Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI et al (2018) HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 554(7691):189–194
Ciardiello F, Tortora G (2008) EGFR antagonists in cancer treatment. N Engl J Med 358(11):1160–1174
Castellanos E, Feld E, Horn L (2017) Driven by mutations: the predictive value of mutation subtype in EGFR-mutated non-small cell lung cancer. J Thorac Oncol 12(4):612–623
Li BT, Ross DS, Aisner DL, Chaft JE, Hsu M, Kako SL et al (2016) HER2 amplification and HER2 mutation are distinct molecular targets in lung cancers. J Thorac Oncol 11(3):414–419
Deeks ED (2017) Neratinib: First Global Approval. Drugs 77(15):1695–1704
Rabindran SK (2005) Antitumor activity of HER-2 inhibitors. Cancer Lett 227(1):9–23
Roberts PJ, Der CJ (2007) Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26(22):3291–3310
Roskoski R Jr (2014) The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res 79:34–74
Zhao M, Scott S, Evans KW, Yuca E, Saridogan T, Zheng X et al (2021) Combining neratinib with CDK4/6, mTOR, and MEK inhibitors in models of HER2-positive cancer. Clin Cancer Res 27(6):1681–1694
Gayle SS, Castellino RC, Buss MC, Nahta R (2013) MEK inhibition increases lapatinib sensitivity via modulation of FOXM1. Curr Med Chem 20(19):2486–2499
Kruspig B, Monteverde T, Neidler S, Hock A, Kerr E, Nixon C, et al. (2018) The ERBB network facilitates KRAS-driven lung tumorigenesis. Sci Transl Med 10 (446).
Moll HP, Pranz K, Musteanu M, Grabner B, Hruschka N, Mohrherr J, et al. (2018) Afatinib restrains K-RAS-driven lung tumorigenesis. Sci Transl Med. 10 (446).
Storer BE (1989) Design and analysis of phase I clinical trials. Biometrics 45(3):925–937
Johnson A, Zeng J, Bailey AM, Holla V, Litzenburger B, Lara-Guerra H et al (2015) The right drugs at the right time for the right patient: the MD Anderson precision oncology decision support platform. Drug Discov Today 20(12):1433–1438
Ito Y, Suenaga M, Hatake K, Takahashi S, Yokoyama M, Onozawa Y et al (2012) Safety, efficacy and pharmacokinetics of neratinib (HKI-272) in Japanese patients with advanced solid tumors: a Phase 1 dose-escalation study. Jpn J Clin Oncol 42(4):278–286
Ouellet D, Kassir N, Chiu J, Mouksassi MS, Leonowens C, Cox D et al (2016) Population pharmacokinetics and exposure-response of trametinib, a MEK inhibitor, in patients with BRAF V600 mutation-positive melanoma. Cancer Chemother Pharmacol 77(4):807–817
Alshammari K, Aung KL, Zhang T, Razak ARA, Serra S, Stockley T et al (2021) Phase II trial of trametinib and panitumumab in RAS/RAF wild type metastatic colorectal cancer. Clin Colorectal Cancer 20(4):334–341
Wong KK, Fracasso PM, Bukowski RM, Lynch TJ, Munster PN, Shapiro GI et al (2009) A phase i study with neratinib (HKI-272), an irreversible pan ErbB receptor tyrosine kinase inhibitor, in patients with solid tumors. Clin Cancer Res 15(7):2552–2558
Planchard D, Besse B, Groen HJM, Souquet PJ, Quoix E, Baik CS et al (2016) Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 17(7):984–993
Gandara DR, Leighl N, Delord JP, Barlesi F, Bennouna J, Zalcman G et al (2017) A Phase 1/1b study evaluating trametinib plus docetaxel or pemetrexed in patients with advanced non-small cell lung cancer. J Thorac Oncol 12(3):556–566
Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J et al (2012) Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 367(18):1694–1703
Hurvitz SA, Saura C, Oliveira M, Trudeau ME, Moy B, Delaloge S et al (2021) Efficacy of Neratinib plus capecitabine in the subgroup of patients with central nervous system involvement from the NALA Trial. Oncologist 26(8):e1327–e1338
Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C et al (2012) Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol 13(8):773–781
White PS, Pudusseri A, Lee SL, Eton O (2017) Intermittent dosing of dabrafenib and trametinib in metastatic BRAF(V600E) mutated papillary thyroid cancer: two case reports. Thyroid 27(9):1201–1205
Leonowens C, Pendry C, Bauman J, Young GC, Ho M, Henriquez F et al (2014) Concomitant oral and intravenous pharmacokinetics of trametinib, a MEK inhibitor, in subjects with solid tumours. Br J Clin Pharmacol 78(3):524–532
Abbas R, Hug BA, Leister C, Burns J, Sonnichsen D (2011) Pharmacokinetics of oral neratinib during co-administration of ketoconazole in healthy subjects. Br J Clin Pharmacol 71(4):522–527
Zhang H, Ou YC, Su D, Wang F, Wang L, Sahasranaman S et al (2021) In vitro investigations into the roles of CYP450 enzymes and drug transporters in the drug interactions of zanubrutinib, a covalent Bruton’s tyrosine kinase inhibitor. Pharmacol Res Perspect 9(6):e00870
Nerlynx Prescribing Information (2020).
Lugowska I, Kosela-Paterczyk H, Kozak K, Rutkowski P (2015) Trametinib: a MEK inhibitor for management of metastatic melanoma. Onco Targets Ther 8:2251–2259
Vaidhyanathan S, Mittapalli RK, Sarkaria JN, Elmquist WF (2014) Factors influencing the CNS distribution of a novel MEK-1/2 inhibitor: implications for combination therapy for melanoma brain metastases. Drug Metab Dispos 42(8):1292–1300
Arkenau HT, Taylor D, Xu X, Chitnis S, Llacer-Perez C, Moore K et al (2022) Pharmacokinetic interaction between the MEK1/MEK2 inhibitor trametinib and oral contraceptives containing norethindrone and Ethinyl estradiol in female patients with solid tumors. Clin Pharmacol Drug Dev 11(5):585–596
Andrew Finch pp (2014) P-glycoprotein and its role in drug-drug interactions. Aust Prescr 37:137–139
Bose P, Ozer H (2009) Neratinib: an oral, irreversible dual EGFR/HER2 inhibitor for breast and non-small cell lung cancer. Expert Opin Investig Drugs 18(11):1735–1751
Acknowledgements
The authors are thankful to the patients who took part in this study and Qianqian Wang for her support in patient recruitment and collection of data.
Funding
This study was sponsored by Puma Biotechnology. Puma Biotechnology reviewed the manuscript before journal submission. The authors are fully responsible for the content of this manuscript, and the views and opinions described in the publication reflect only those of the authors.
Author information
Authors and Affiliations
Contributions
All authors conceived and/or designed the work that led to the submission, acquired data and/or played an important role in interpreting the results; drafted or revised the manuscript; approved the final version and agreed to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Conflict of interest
SPP reports clinical trial research funding/grant support through the institution from AbbVie, ABM Therapeutics, Acepodia, Alkermes, Aminex Therapeutics, Amphivena Therapeutics, BioMarin Pharmaceutical, Boehringer Ingelheim, Bristol Myers Squib, Cerulean Pharma, Chugai Pharmaceutical, Curis, Cyclacel Pharmaceuticals, Daiichi Sankyo, Eli Lilly, ENB Therapeutics, Epigenetix, Five Prime Therapeutics, F-Star Beta Limited, F-Star Therapeutics, Gene Quantum, Genmab A/S, Gilead Sciences, GlaxoSmithKline, Helix BioPharma, Hengrui Pharmaceuticals, HiberCell, Immorna Biotherapeutics, Immunomedics, Incyte, Jacobio Pharmaceuticals, Jiangsu Simcere Pharmaceutical, Lytix Biopharma AS; Medimmune, Medivation, Merck Sharp and Dohme Corp., Nectin Therapeutics, Novartis Pharmaceuticals, Pieris Pharmaceuticals, Pfizer, Phanes Therapeutics, Principia Biopharma, Puma Biotechnology, Purinomia Biotech, Rapt Therapeutics, Replimune; Seattle Genetics; Silverback Therapeutics; Synlogic Therapeutics; Taiho Oncology; Tesaro, Inc., TransThera Bio, ZielBio, NCI/NIH; P30CA016672 – Core Grant (CCSG Shared Resources) and also worked as a consultant for CRC Oncology. DDK reports research funding/grant support for clinical trials from Pfizer, Phoplatin Therapeutic, Symphogen, NIH clinical translation science award, clinical translation NCI grant and consultancy/advisory board participation for Black Beret Life Science, Affigen and Phosplatin.VS reports research funding/grant support for clinical trials from AbbVie, Agensys, Inc., Alfasigma, Altum, Amgen, Bayer, BERG Health, Blueprint Medicines Corporation, Boston Biomedical, Inc., Boston Pharmaceuticals, Celgene Corporation, D3 Bio, Inc., Dragonfly Therapeutics, Inc., Exelixis, Fujifilm, GlaxoSmithKline, Idera Pharmaceuticals, Inc., Incyte Corporation, Inhibrx, Loxo Oncology, MedImmune, MultiVir, Inc., NanoCarrier, Co., National Comprehensive Cancer Network, NCI-CTEP, Northwest Biotherapeutics, Novartis, PharmaMar, Pfizer, Relay Therapeutics, Roche/Genentech, Takeda, Turning Point Therapeutics, UT MD Anderson Cancer Center, and Vegenics Pty Ltd.; travel support from ASCO, ESMO, Helsinn Healthcare, Incyte Corporation, Novartis, and PharmaMar; consultancy/advisory board participation for Helsinn Healthcare, Jazz Pharmaceuticals, Incyte Corporation, Loxo Oncology/Eli Lilly, MedImmune, Novartis, QED Therapeutics, Relay Therapeutics, Daiichi-Sankyo, and R-Pharm US; and other relationship with Medscape. AT reports research funding/grant support for clinical trials from OBI Pharma USA Inc., Baxalta, Bayer, Boston Biomedical, EMD Serono, Parker Institute for Cancer Immunotherapy, Placon Therapeutics, Agenus, Tempus Labs, Tvardi Therapeutics, Karus Therapeutics, Novocure, NIH/NCI P30CA016672 – Core Grant (CCSG Shared Resources). AT served as consulting or advisory role for Vincerx, Diaccurate. BrYet, NEX-II, Macrogenics. SQF reports research funding/grant support for clinical trials from NIH/NCI P30CA016672 – Core Grant (CCSG Shared Resources), Abbisko, BeiGene, BioAtla, Boehringer Ingelheim, CUE Biopharma, Eli Lilly & Co., Exelisis, Greenfire Bio, Hookipa Biotech, IMV, Innovent Biologics, K-Group Beta, Lyvgen Biopharm, MacroGenics, MediLink Therapeutics, Millennium Pharmaceuticals, Nerviano Medical Sciences, NeuPharma, NextCure, Ningbo NewBay Technology Development, Novartis, NovoCure, Nykode Therapeutics AS., Parexel International, Pionyr Immunotherapeutics, PureTech Health, Sellas Life Sciences Group, Soricimed Biopharma, SQZ Biotechnologies, Sumitomo Dainippon, Taiho Oncology and NCCN; Treadwell Therapeutics, Turnstone Biologics, Tyligand Bioscience, Virogin Biotech. SP reports clinical trial research funding/grant support through the institution from Mirati Therapeutics, Merck, Pfizer, Lilly, Xencor, Novartis, Bristol-Myers Squibb, Ipsen, Astellas, Purple Biotech, 4D Pharma, Boehringer Ingelheim, NGM Biopharmaceuticals, Janssen, Arcus Biosciences, Elicio Therapeutics, Zymeworks, BioNtech and receives personal fees for advisory/consulting from Zymeworks, Ipsen,Novartis, Janssen, Boehringer Ingelheim. TAY reports research funding/grant support for clinical trials from Acrivon, Artios, AstraZeneca, Bayer, Beigene, BioNTech, Blueprint, BMS, Clovis, Constellation, Cyteir, Eli Lilly, EMD Serono, Forbius, F-Star, GlaxoSmithKline, Genentech, Haihe, ImmuneSensor, Ionis, Ipsen, Jounce, Karyopharm, KSQ, Kyowa, Merck, Mirati, Novartis, Pfizer, Ribon Therapeutics, Regeneron, Repare, Rubius, Sanofi, Scholar Rock, Seattle Genetics, Tesaro, Vivace and Zenith, served as a consultant for AbbVie, AstraZeneca, Acrivon, Adagene, Almac, Aduro, Amphista, Artios, Athena, Atrin, Avoro, Axiom, Baptist Health Systems, Bayer, Beigene, Boxer, Bristol Myers Squibb, C4 Therapeutics, Calithera, Cancer Research UK, Clovis, Cybrexa, Diffusion, EMD Serono, F-Star, Genmab, Glenmark, GLG, Globe Life Sciences, GSK, Guidepoint, Idience, Ignyta, I-Mab, ImmuneSensor, Institut Gustave Roussy, Intellisphere, Jansen, Kyn, MEI pharma, Mereo, Merck, Natera, Nexys, Novocure, OHSU, OncoSec, Ono Pharma, Pegascy, PER, Pfizer, Piper-Sandler, Prolynx, Repare, resTORbio, Roche, Schrodinger, Theragnostics, Varian, Versant, Vibliome, Xinthera, Zai Labs and ZielBio and a stockholder in Seagen. VKM discloses research funding (to institution) from EMD Serono, Pfizer, Novartis, Bicara Pharmaceuticals, Bristol Myers Squibb, RedX, and BioNTech. BK declares ownership medtronic corporation stock that is less than $10,000. FM-B reports research funding/grant support for clinical trials from Aileron Therapeutics, AstraZeneca, Bayer Healthcare Pharmaceutical, Calithera Biosciences, Curis, CytomX Therapeutics, Daiichi Sankyo, Debiopharm International, eFFECTOR Therapeutics, Genentech, Guardant Health, Klus Pharma, Takeda Pharmaceutical, Novartis, Puma Biotechnology, Taiho Pharmaceutical. FM-B served as advisory committee for Black Diamond, Biovica, Eisai, FogPharma, Immunomedics, Inflection Biosciences, Karyopharm Therapeutics, Loxo Oncology, Mersana Therapeutics, OnCusp Therapeutics, Puma Biotechnology Inc., Seattle Genetics, Sanofi, Silverback Therapeutics, Spectrum Pharmaceuticals, Zentalis, personal fees for consulting/travel related from AbbVie, Aduro BioTech, Alkermes, AstraZeneca, Daiichi Sankyo, DebioPharm, Ecor1 Capital, eFFECTOR Therapeutics, F. Hoffman-La Roche, GT Apeiron, Genentech, Harbinger Health, IBM Watson, Infinity Pharmaceuticals, Jackson Laboratory, Kolon Life Science, Lengo Therapeutics, Menarini Group, OrigiMed, PACT Pharma, Parexel International, Pfizer, Protai Bio, Samsung Bioepis, Seattle Genetics, Tallac Therapeutics, Tyra Biosciences, Xencor, Zymeworks, European Organisation for Research and Treatment of Cancer (EORTC), European Society for Medical Oncology (ESMO), personal fees for honoraria from Chugai Biopharmaceuticals. No potential conflicts of interest were disclosed by the other authors.
Ethical approval and consent to participate
All participants signed the consent form prior to study enrollment. The study was approved by MD Anderson Institutional Review Board (IRB) in accordance with the Declaration of Helsinki, Good Clinical Practice, and all federal, state and local regulatory guidelines.
Consent to publish
A signed informed consent was obtained from each participant prior to any study procedure.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Piha-Paul, S.A., Tseng, C., Tran, H.T. et al. A phase I trial of the pan-ERBB inhibitor neratinib combined with the MEK inhibitor trametinib in patients with advanced cancer with EGFR mutation/amplification, HER2 mutation/amplification, HER3/4 mutation or KRAS mutation. Cancer Chemother Pharmacol 92, 107–118 (2023). https://doi.org/10.1007/s00280-023-04545-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-023-04545-4