
Abstract
Purpose
Etirinotecan pegol (EP), a long-acting topoisomerase-1 inhibitor, is a polyethylene glycol conjugate of irinotecan, with an intended indication for treatment of breast cancer with brain metastases. The objective of this study was to develop a population pharmacokinetic (popPK) model of EP and four of its metabolites (irinotecan, SN38, SN38-glucuronide, and APC) and determine covariates affecting their pharmacokinetics.
Methods
Data from 83 cancer patients enrolled in phase 1 studies were used. The model was developed in two stages: (1) concentration–time data were analyzed with a 3-analyte model for EP, irinotecan, and SN38; and (2) a 5-analyte model developed based on expansion of 3-analyte model to include concentration–time data for SN38 glucuronide and APC with parameter values from 3-analyte model fixed. Covariate relationships with parameters were selected based on Wald’s test within the Wald’s Approximation Method approach, first for the 3-analyte model then the 5-analyte model.
Results
The final integrated popPK model for the five analytes was a two-compartment per analyte model that followed the metabolic cascade of EP to irinotecan, followed by metabolism of irinotecan to the previously known metabolites, but with altered exposures as compared to administration of irinotecan. With the model developed based on total dose of EP, the population estimates of EP clearance and central volume were 0.237 L/h and 5.5 L, respectively. Patient age, body surface area (BSA), and estimated glomerular filtration rate were found to correlate with EP clearance and BSA with EP central volume. Individuals who were homozygous for UGT1A1*28 genotype had modestly reduced elimination capacity of SN38 compared to heterozygous and wild-type genotypes. Simulations evaluating the clinical importance of significant covariates indicated minimal change in areas under the curve and peak concentrations of EP and SN38.
Conclusions
The pharmacokinetics of EP and four metabolites including the active metabolite SN38 were described by an integrated popPK model. Other than BSA, which was already accounted by a BSA-based dosing scheme, no other covariates were deemed to have clinical implications. No EP starting dose adjustment based on patient demographics and other covariates was deemed necessary.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Irinotecan, a prodrug of 7-ethyl 10-hydroxycamptothecin (SN38), is an antineoplastic agent of the topoisomerase (Top1) inhibitor class that is widely used to treat colorectal and other gastrointestinal cancers. The pharmacokinetics and metabolism of irinotecan and SN38 in humans are well described in the literature [1,2,3,4,5]. Irinotecan is extensively metabolized in the liver to various metabolites. It is cleaved enzymatically by carboxylesterases to form SN38, which has cytotoxic activity that is 100- to 1000-times greater than that of the parent drug [6]. SN38 is subsequently conjugated to an inactive glucuronide (SN38G) by uridine diphosphate glucuronosyltransferases (UGT1A1 and UGT1A9). Other inactive irinotecan metabolites are 7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino]-carbonyloxycampthothecin (APC) and 7-ethyl-10-[4-amino-1-piperidino]-carbonyloxycamptothecin (NPC), resulting from metabolism of irinotecan by CYP3A4 and 3A5 enzymes [3, 7, 8].
Etirinotecan pegol (EP) is a polyethylene glycol (PEG) conjugate of irinotecan, with an intended indication for treatment of breast cancer with brain metastases (BCBM). EP was designed to provide enhanced anti-tumor efficacy and a favorable tolerability profile through a modulated PK profile that facilitates lower peak plasma levels and sustained exposure of tumor tissue to SN38. In animal models, EP provided increased anti-tumor activity and a better safety profile compared with short-acting Top1 inhibitors [9, 10]. EP is a prodrug of irinotecan, consisting of a 20 kDa 4-arm PEG with a single irinotecan molecule conjugated to each arm via a glycine ester; the metabolic pathway of EP is shown in Fig. 1. Chemical hydrolysis of the glycine ester releases irinotecan in vivo; once released, irinotecan is metabolized to the previously described metabolites. The large molecular weight PEG of EP combined with the slow release of irinotecan results in 5–10-fold lower maximal plasma SN38 concentrations and a greatly prolonged half-life compared to that resulting from administration of irinotecan (approximately 40 days compared to about 2 days) [1, 11]. A single intravenous (IV) dose of 145 mg/m2 EP results in approximately the same plasma SN38 AUC as a 350 mg/m2 dose of irinotecan, but exposure is continuous throughout the 21-day cycle, rather than intermittent, and maximal concentrations are less after EP administration than after irinotecan administration [11].

Etirinotecan pegol metabolic pathway
This current analysis aimed to establish a semi-mechanistic population pharmacokinetic model that describes the metabolic pathway of EP following intravenous administration of single and multiple EP doses in patients with advanced cancer.
Methods
Clinical studies and patients
Data from two clinical trials of EP, 06-IN-IR001 and 07-PIR-02, in patients with advanced solid tumors were combined for popPK analysis. Both trials were approved by the Institutional Review Board at the study sites and were in compliance with the Declaration of Helsinki; all patients provided written informed consent. Study 06-IN-IR001 was a multicenter, open-label, phase 1 dose-escalation study for finding the maximum tolerated dose, as described previously [11]. Briefly, patients received EP by intravenous infusion over a 90-min period with 3–6 patients per dose cohort in the following regimens: (1) 3 once-weekly doses repeated every 4 weeks (wx3 q4wk); (2) once-biweekly (q14d); and (3) once every 3 weeks (q21d). Patients continued to the next cycle if they did not exhibit unacceptable toxicity or disease progression. Study 06-IN-IR001 enrolled a total of 76 patients; pharmacokinetic data from 67 patients were included for popPK modeling. 07-PIR-02 was the phase 1 portion of a multicenter, open-label, phase 2 safety, and efficacy study of EP in combination with cetuximab in patients with solid tumors refractory to the standard therapies [12]. Eighteen patients received EP (100 or 125 mg/m2) once every 3 weeks. Standard cetuximab schedule was administered as described [13]. Pharmacokinetic data from 16 patients were included in the popPK modeling. Reasons for exclusion of patients from the pharmacokinetic analysis are provided in the supplemental material. PK samples were collected at the beginning of day 1 and up to 4 weeks after the last dose. Rich blood PK sampling was carried out after the first and third doses in 06-IN-IR001 and after the first dose in 07-PIR-02, followed by sparse predose only sampling thereafter for the duration of treatment. All patients were evaluated for UGT1A1*28 genotype.
Bioanalytical assays
Plasma EP and metabolite concentrations were determined using specific and validated LC–MS/MS methods previously described [14]. Prior to popPK analysis, etirinotecan pegol concentrations were converted, such that they reflected the irinotecan content using irinotecan loading factors of 9.4% (06-IN-IR001) and 9.5% (07-PIR-02). All analyte concentrations were converted to molar concentrations based on irinotecan, SN38, SN38G, and APC molecular weights of 586.678, 382.404, 568.53, and 618.687 amu, respectively. Samples below the limit of quantification (BLQ) were excluded and treated as missing in the analysis. The percentage of BLQ samples for EP, irinotecan, SN38, SN38G, and APC were 22.6, 3.8, 4.1, 6.1, and 36.4%, respectively.
Pharmacokinetic model structure
Individual plasma concentration–time data were analyzed by non-linear mixed-effects modeling using stochastic approximation expectation maximization followed by importance sampling methods in Monolix 2016 (Lixoft, Antony, France). Graphical and statistical evaluations used during model development were generated in R 3.0.0 (R Foundation for Statistical Computing, Vienna, Austria) or Monolix. Model evaluation was based on the likelihood objective function value (OFV) result from the importance sampling procedure, Schwartz Bayesian Criterion (SBC), goodness-of-fit plots, and visual predictive check. Corrections for prediction and variability were incorporated in the visual predictive check [15,16,17].
Interindividual variability and residual error models were incorporated in the model, assuming log-normal distribution and a mixture of additive and proportional error models, respectively. A zero-centered mean was also assumed for the variability models.
Covariate screening
The covariates considered in the model are based on the information in irinotecan product labeling, as well as the literature on its pharmacokinetics and metabolism, and are summarized in Table 1. Hepatic and renal functions, age, gender, UGT1A1 status, smoking, race, and concomitant administration of CYP inhibitors and inducers were investigated for their effects on the disposition of EP and metabolites.
Baseline values for body weight, gender, age, creatinine, total bilirubin, AST, ALT, and albumin were used. Estimated GFR (eGFR) was calculated using the following equation [18]:
where Scr is serum creatinine in mg/dL [19]. UGT1A1 status was determined based on the presence of the UGT1A1*28 [number of TA(7)] repeats in the promoter region of the UGT1A1 allele in whole blood samples using the FDA-approved Invader® UGT1A1 Molecular Assay kit (Third Wave Technologies Inc., Madison, WC, USA). Results were reported as UGT1A1*28 not detected (wild-type or TA6/TA6), one copy detected (heterozygous or TA6/TA7), two copies detected (homozygous or TA7/TA7), or indeterminate (rare genotype with either TA5 or TA8). Patients were categorized as wild type, heterozygous for UGT1A1*28, or homozygous for UGT1A1*28 (Table 2). Patients reported as indeterminate were placed into the wild-type category.
Visual inspection using plots of covariate vs. standardized random effects of the parameters were utilized during the covariate screening process. Covariates that exhibited trends in the plots of standardized random effects were further screened using the Wald Approximation Method (WAM) developed by Kowalski and Hutmacher [20]. The following covariate relationships were formally evaluated with the WAM algorithm: BSA (vs. CL, V1), gender (vs. \({k_{3{\text{e}}}}\)), age (vs. CL), eGFR (vs. CL), and UGT1A1 (vs. \({k_{3{\text{e}}}}\)). The ten covariate models with the largest decrease in the SBC determined from the WAM algorithm were then incorporated to the popPK model to evaluate for their relationship to specific model parameters. A covariate was considered significant if the p value of the Wald test was ≤ 0.01.
The continuous covariates were incorporated as a power model, such that \({\theta _i}={\theta _{{\text{pop}}}}\prod\nolimits_{j} {{{\left( {\frac{{{\text{co}}{{\text{v}}_{i,j}}}}{{{\text{co}}{{\text{v}}_{{\text{median}},j}}}}} \right)}^{{k_{{\text{cov}},j}}}} \cdot \exp ({\eta _\theta }),}\) whereas categorical covariates were incorporated as an exponential model: \({\theta _i}={\theta _{{\text{pop}}}}\prod\nolimits_{j} {{\text{exp(}}{k_{{\text{cov}},j}}I[{\text{co}}{{\text{v}}_{i,j}}]{\text{)}} \cdot \exp ({\eta _\theta }),}\) with i being an index for individual with a specific covariate characteristic, j being the index for the covariate, pop for the index for the population typical value, θ representing the PK model parameter, cov is either a categorical or continuous covariate, kcov is the coefficient that characterizes the covariate effect on the parameter, and η θ refers to between-subject variability.
The potential clinical impact of statistically significant covariates on analyte exposure was evaluated by comparing the area under the concentration–time curve (AUC) and peak drug concentration (Cmax) values from 500 simulated profiles from virtual patients receiving six cycles of 145 mg/m2 once every 3 weeks. These virtual patients had differing categories of renal function, age (45 and 75 years), and UGT1A1*28 genotype. Simulated parameter values for each candidate covariate were compared to a reference population simulated using median covariate values using Forest plots.
Results
Patient characteristics and pharmacokinetic data set
The data set included data from 83 patients, 67 from study 06-IN-IR001, and 16 from study 07-PIR-02. Baseline characteristics of the patient population in the analysis data set are shown in Table 2. The majority of patients in the PK population were white with a median age of 60 years; 54% were male. Patients had a variety of cancer types, including colorectal (22.9%), lung (18.1%), and pancreatic (10.8%) cancers. Patients were required to have adequate renal function to be eligible to participate in both studies. Nonetheless, several patients exhibited mild and moderate renal impairment based on eGFR, allowing the assessment of the impact of renal function on EP pharmacokinetics. As expected from a mainly Caucasian patient population, 10.8% of patients were homozygous for UGT1A1*28. Fifteen percent and 45% of patients received concomitant weak/moderate CYP3A4 inhibitors and inducers, respectively.
The pharmacokinetic data set consisted of 1414, 1777, 1769, 1731, and 1167 concentration values for model building for EP, irinotecan, SN38, SN38G, and APC, respectively.
Etirinotecan pegol pharmacokinetics and covariate selection
3-Analyte pharmacokinetic model
Prior to the development of an integrated pharmacokinetic model, two- and three-compartment pharmacokinetic models were evaluated for each analyte using data from study 06-IN-IR001 (see supplemental material). Fitting a three-compartment model to EP or any of its metabolites was either not successful or resulted in a non-parsimonious fit with greater variability in parameter estimates as compared to fitting with a two-compartment model. Hence, we decided to use a two-compartment model for each analyte in the integrated model. A graphical representation of the 3-analyte model is shown in Fig. 2a. As EP is administered by intravenous infusion, dosing starts in the first compartment, the central compartment for EP. Irinotecan is subsequently released from the four-arm polymer backbone. The transformation rate \(\frac{{{F_{12}}{\text{CL}}}}{{{V_1}}}\) for the release of irinotecan represents the formation of irinotecan regardless of whether the original precursor has one, two, three, or four irinotecan moieties still attached; this rate constant, hence, is the aggregate release rate describing irinotecan formation. Irinotecan is subsequently metabolized to either SN38 or APC. In the 3-analyte model, this rate is represented by \({F_{23}}{k_{2{\text{e}}}}\). \(V_{3}^{*}\) is the aggregated single term for \({V_3}/({F_{12}} \cdot {F_{23}})\). Hence, estimated volumes of distribution are not the true volumes of the analytes in humans, with the exception of EP volume of distribution. The scaling factor for the central and peripheral compartments for each analyte is represented by \({V_i}\) and \({V_{i{\text{p}}}}\), respectively, where i assumes numerical values starting with 1, representing each analyte. The elimination rate constants are \({k_{i0}}\), with increasing numerical values for i representing each analyte. The rate constants \({k_{i{\text{p}}}}\) and \({k_{{\text{p}}i}}\) represent the transfer from the central to the peripheral compartments and vice versa, respectively. A linear elimination process for all analytes was assumed, resulting in the following equations:

Model structure to describe etirinotecan pegol metabolism using a 3-analyte model (a) and 5-analyte model (b)
where \(V_{2}^{*}={V_2}/{F_{12}}\)
where \(V_{3}^{*}={V_3}/({F_{12}} \cdot {F_{23}})\) for the 3-analyte model.
\({\text{CL}}/{V_1}\) represents both the EP elimination rate and its conversion to irinotecan; \({k_{{\text{2e}}}}\) is the rate constant for irinotecan elimination and its conversion to SN38; \({k_{3{\text{e}}}}\) for SN38 elimination and its conversion to SN38G. Metabolite conversion fractions and volumes of distribution are non-identifiable, and thus, only ratios of metabolic conversion fraction to volume of distribution were estimated. The F and V parameters were indistinguishable by the model, and thus, aggregate parameters represented by \(V_{i}^{*}\) for \(i=2,3\) were determined instead. For example, \({F_{12}}\) and \({V_{2~}}\) cannot be separately distinguished by the model, and thus, \(V_{2}^{*}={V_2}/{F_{12}}\) was determined. The same approach was applied to the other volume terms.
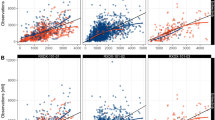
Goodness-of-fit plots for all analytes showed good agreement between individual-predicted and observed concentrations without noticeable bias in individual weighted residuals across predicted drug concentration values (first 3 rows of Fig. 3). The majority of individual weighted residuals were within the ± 2 units from the zero ordinate.

Goodness-of-fit plots for all analytes of etirinotecan pegol from the intital 3-analyte (a) and final 5-analyte (b) population pharmacokinetic models. Left, observed vs. individual-predicted concentrations; center, observed vs. population-predicted concentrations; right, individual weighted residuals vs. population-predicted concentrations. Solid lines represent the line of unity in left and center and zero residuals in right. Dashed lines represent loess smoothing
Graphical outputs for covariate screening are shown in Figures S2 to S5 in supplemental material. The full covariate model for the 3-analyte model was the first ranked model in Table 3, which included BSA on CL and V1, age on CL, and UGT1A1 on k3e; eGFR was a significant covariate of CL in subsequent stepwise addition. The parameters identified as impacted by covariates are described below:
Table 4 provides a summary of the parameter estimates for the 3-analyte model. The aggregate effect of incorporating covariates to EP CL and V1 parameters reduced interindividual variability expressed in CV% from 34 to 27% and from 26 to 22%, respectively. UGT1A1 polymorphism as a covariate of k3e reduced the interindividual variability of k3e from 51 to 47%. The magnitude of the covariate effects was small.
5-Analyte pharmacokinetic model
Upon completion of the final 3-analyte model, individual parameter estimates for EP, irinotecan, and SN38 were fixed [with the exception for the Corr (\({\text{CL}},{V_1}\)) and Corr (\({k_{2{\text{e}}}},V_{2}^{*}\)) which could not be fixed in the software], and SN38G and APC concentration–time data were added to the model, thus generating the 5-analyte model, graphically represented in the last two rows in Fig. 3. Assuming linear elimination processes for SN38G and APC, the following additional equations were incorporated:
where \(V_{4}^{*}={V_4}/({F_{12}} \cdot ({F_{23}}+{F_{25}}) \cdot {F_{34}})\)
where \(V_{5}^{*}={V_5}/{F_{25}}=V_{3}^{*}\).
\(V_{3}^{*}\) in this 5-analyte model represents the aggregated term for \({V_3}/({F_{12}} \cdot ({F_{23}}+{F_{25}}))\). Glucuronidation of SN38 to SN38G is governed by the rate constant \({F_{34}}{k_{3{\text{e}}}}\). The volume of the central compartment for APC was assumed to be the same as that for SN38. The fraction for the formation of SN38 (Firinotecan→SN38) and subsequently one minus that fraction (1 − Firinotecan→SN38) to describe the formation of APC accounted for the difference in the concentrations of the two metabolites. Hence, estimated volumes of distribution are not the true volumes for APC and SN38G in humans. The scaling factor for the central and peripheral compartments, elimination rate constants, and rate constants from the central to the peripheral compartments and vice versa for SN38G and APC are represented equivalent to those described for the 3-analyte model. As described for the 3-analyte model, only ratios of metabolic conversion fraction to volume of distribution were estimated.
Final parameters are shown in Table 4. In addition to the covariates identified in the 3-analyte model, the impact of UGT1A1 status on the ratio of central volume of distribution of SN38G to its conversion fractions (\(V_{4}^{*}\)) was evaluated; however, the covariate was not found significant. CYP3A4/5 inducer effect was significant on k25, which is the conversion of irinotecan to APC. However, the direction of the effect indicates that CYP3A4 inducers reduced the conversion of irinotecan to APC, contrary to the expected effect of CYP3A4/5 inducers. Thus, this covariate was not included in the final model. The 5-analyte model maintained all of the covariates in the 3-analyte model.
Goodness-of-fit plots for all analytes showed good agreement between individual-predicted and observed concentrations without noticeable bias in individual weighted residuals across predicted drug concentration values (Fig. 3). The majority of individual weighted residuals were within the ± 2 units from the zero ordinate.
Prediction- and variability-corrected visual predictive checks, stratified by dosing schedules, in supplemental material Figures S6 and S7, captured the majority of the observed data within the 95% prediction interval.
Clinical impact of significant covariates
To assess the clinical impact of significant covariates, we simulated exposure for specified patient populations. A reference population was generated by setting each covariate to the median values of those in the study population for age (60 years) and renal function (84.1 mL/min), and UGT1A1 to non-homozygous UGT1A1*28. To assess the clinical relevance of age, renal function, and UGT1A1*28, their impacts on EP and SN38 exposure were evaluated by simulating 500 virtual patients in the following scenarios: (1) 75 years old; (2) 45 years old; (3) 45 mL/min eGFR for moderate renal impairment; (4) 75 mL/min eGFR for mild renal impairment; (5) 105 mL/min eGFR for normal renal function. Figure 4 shows forest plots of the impact of age and eGFR on EP AUC and Cmax. In the top panel, the EP exposures in these scenarios were close to that of the reference. The 95% prediction interval of each scenario overlapped considerably, suggesting limited clinical impact. As illustrated in the bottom panel of Fig. 4, EP Cmax was not impacted by any of the covariates.

Forest plots showing the effect of age and eGFR on the cumulative AUC and Cmax of etirinotecan pegol over six cycles relative to a reference population. The eGFR used in each category was the midpoint of the normal renal function and mild, moderate and severe renal impairment groups. The reference population reflects the typical patient’s characteristics, i.e., demographics, baseline lab values and UGT1A1 status, in phase I and II studies
In addition to assessing the impact of age and renal function on EP exposure, their impact on SN38 exposure was also investigated. Depicted in Fig. 5, and consistent with the limited impact of age and eGFR on EP exposure, the downstream effects on SN38 AUC and Cmax were negligible. In comparison to a patient who is wild-type or heterozygous for the UGT1A1 promoter region polymorphism, the medians of the estimated SN38 AUC and Cmax increased by about 1.7- and 1.3-fold, respectively, for a patient homozygous for UGT1A1*28. The evaluation of the impact of UGT1A1*28 on SN38 exposure was based on a limited sample size, as only nine patients in the data set were homozygous for UGT1A1*28 and additional data are required to adequately assess the true magnitude of the impact of UGT1A1*28 on SN38 pharmacokinetics and the potential clinical implication for EP safety.

Forest plots showing the effect of UGT1A1 polymorphism, age, and eGFR on cumulative SN38 AUC and Cmax values of over six treatment cycles relative to a reference population. The eGFR used in each category was the midpoint of the normal renal function and mild, moderate, and severe renal impairment groups. The reference population reflects the typical patient’s characteristics, i.e., demographics, baseline lab values, and UGT1A1 status, in phase I and II studies
Discussion
The pharmacokinetics of EP and its metabolites irinotecan, SN38, SN38G, and APC were well described by a semi-mechanistic model that included all five analytes. The integrated model of all analytes describes known mechanisms of the EP metabolic pathway. Nonetheless, limitations to the model do exist. Due to the complexity of the model, the fractions that characterize a specific metabolic direction and the volume of distribution of the central compartment of the metabolite were aggregated into a single volume parameter. Even with these limitations, the integrated model was sufficient for characterization of the individual concentration–time profile for all analytes and enabled subsequent simulations of the effects of various characteristics of the population.
The clinical relevance of the observed covariate effects was evaluated through a series of simulations with varying covariate values. Baseline BSA had an effect on EP clearance and volume of distribution, supporting the BSA-based dosing of EP. An additional covariate for EP clearance was eGFR, consistent with its elimination pathways, hydrolysis to irinotecan, and renal clearance. EP clearance decreases with decreasing kidney function. However, the effect of mild and moderate renal impairment was minimal. The small increase in EP exposure with increasing renal impairment had no impact on the exposure of the active metabolite SN38. Consequently, dose adjustments are not required in patients with mildly or moderately impaired renal function.
Age was a significant covariate of EP CL. However, varying the age from 45 to 75 years had minimal impact on EP or SN38 exposures, suggesting that no dose adjustment of EP is required in the elderly population. In addition to the covariates impacting EP clearance, the UGT1A1*28 polymorphism was identified as a statistically significant covariate for SN38 elimination. The increase in the number of TA repeats in its promoter region is associated with decreased enzymatic activity [21, 22]. Patients homozygous for UGT1A1*28 genotype were projected to exhibit a 1.8-fold higher SN38 exposure, which could potentially impact the safety and tolerability of EP. However, the variability explained by UGT1A1 polymorphism was relatively small. The maximum SN38 concentrations resulting from EP administration are lower than those for a 350 mg/m2 as well as a 60 mg/m2 irinotecan single dose administration [8]; the standard dosing regimen for irinotecan is 350 mg/m2 once every 3 weeks in colon cancer patients. A toxicokinetic study in dogs comparing irinotecan and EP showed that SN38 Cmax but not AUC was associated with drug-induced neutropenia [10]. A meta-analysis consisting of 878 patients reported that the association between UGT1A1*28 genotype and irinotecan-induced hematologic toxicity was significant only at higher irinotecan doses that resulted in higher maximum SN38 concentrations [23]. Another meta-analysis consisting of 1998 patients suggested that a dose-dependent association existed between homozygous UGT1A1*28 patients and the risk of irinotecan-induced neutropenia with the high dose (≥ 250 mg/m2) compared to the low (80–145 mg/m2) and medium (150–200 mg/m2) doses (relative risk: 7.22 vs. 2.04) [24]. A separate meta-analysis by the latter group also found a dose-dependent increased risk of diarrhea and UGT1A1*28 homozygotes at medium and high doses but not at low doses of irinotecan [25]. We recognize that the small number of homozygous UGT1A1*28 patients in the current analysis population limits broad generalization of the true impact of UGT1A1*28 polymorphism in the general population.
In conclusion, EP pharmacokinetics are well described by the proposed model. Other than the effect of BSA, which is already taken into account by a BSA-based dosing scheme, no other covariates were deemed to have clinical implications. No EP dose adjustment based on the covariates investigated appears necessary.
References
Chabot GG (1997) Clinical pharmacokinetics of irinotecan. Clin Pharmacokinet 33(4):245–259. https://doi.org/10.2165/00003088-199733040-00001
Mathijssen RH, van Alphen RJ, Verweij J, Loos WJ, Nooter K, Stoter G, Sparreboom A (2001) Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin Cancer Res 7(8):2182–2194
Sparreboom A, de Jonge MJ, de Bruijn P, Brouwer E, Nooter K, Loos WJ, van Alphen RJ, Mathijssen RH, Stoter G, Verweij J (1998) Irinotecan (CPT-11) metabolism and disposition in cancer patients. Clin Cancer Res 4(11):2747–2754
Slatter JG, Schaaf LJ, Sams JP, Feenstra KL, Johnson MG, Bombardt PA, Cathcart KS, Verburg MT, Pearson LK, Compton LD, Miller LL, Baker DS, Pesheck CV, Lord RS 3rd (2000) Pharmacokinetics, metabolism, and excretion of irinotecan (CPT-11) following I.V. infusion of [(14)C]CPT-11 in cancer patients. Drug Metab Dispos 28(4):423–433
Xie R, Mathijssen RH, Sparreboom A, Verweij J, Karlsson MO (2002) Clinical pharmacokinetics of irinotecan and its metabolites: a population analysis. J Clin Oncol 20(15):3293–3301
Kawato Y, Aonuma M, Hirota Y, Kuga H, Sato K (1991) Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res 51(16):4187–4191
Santos A, Zanetta S, Cresteil T, Deroussent A, Pein F, Raymond E, Vernillet L, Risse ML, Boige V, Gouyette A, Vassal G (2000) Metabolism of irinotecan (CPT-11) by CYP3A4 and CYP3A5 in humans. Clin Cancer Res 6(5):2012–2020
Shingyoji M, Takiguchi Y, Watanabe-Uruma R, Asaka-Amano Y, Matsubara H, Kurosu K, Kasahara Y, Tanabe N, Tatsumi K, Kuriyama T (2004) In vitro conversion of irinotecan to SN-38 in human plasma. Cancer Sci 95(6):537–540
Hoch U, Staschen CM, Johnson RK, Eldon MA (2014) Nonclinical pharmacokinetics and activity of etirinotecan pegol (NKTR-102), a long-acting topoisomerase 1 inhibitor, in multiple cancer models. Cancer Chemother Pharmacol 74(6):1125–1137. https://doi.org/10.1007/s00280-014-2577-7
Sy SK, Sweeney TD, Ji C, Hoch U, Eldon MA (2017) Etirinotecan pegol administration is associated with lower incidences of neutropenia compared to irinotecan administration. Cancer Chemother Pharmacol 79(1):57–67. https://doi.org/10.1007/s00280-016-3192-6
Jameson GS, Hamm JT, Weiss GJ, Alemany C, Anthony S, Basche M, Ramanathan RK, Borad MJ, Tibes R, Cohn A, Hinshaw I, Jotte R, Rosen LS, Hoch U, Eldon MA, Medve R, Schroeder K, White E, Von Hoff DD (2013) A multicenter, phase I, dose-escalation study to assess the safety, tolerability, and pharmacokinetics of etirinotecan pegol in patients with refractory solid tumors. Clin Cancer Res 19(1):268–278. https://doi.org/10.1158/1078-0432.CCR-12-1201
ClinicalTrials.gov (2008) A phase 2a/2b multicenter, open-label study to evaluate NKTR 102 in combination with cetuximab versus irinotecan in combination with cetuximab in second line colorectal cancer patients. Identifier NCT00598975, National Library of Medicine (US), Bethesda, MD. https://clinicaltrials.gov/ct2/show/NCT00598975?term=NCT00598975&recrs=e&rank=1
Lilly Bristol-Myers-Squibb (2004) Cetuximab (Erbitux package insert). http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/125084s225lbl.pdf. Accessed 23 Jan 2017
Eldon M, Hoch U (2011) Population pharmacokinetics of NKTR-102, a topoisomerase I inhibitor-polymer conjugate, in patients with advanced solid tumors. J Clin Oncol 29 (15):Abstr 2598
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO (2011) Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J 13(2):143–151. https://doi.org/10.1208/s12248-011-9255-z
de Kock L, Sy SK, Rosenkranz B, Diacon AH, Prescott K, Hernandez KR, Yu M, Derendorf H, Donald PR (2014) Pharmacokinetics of para-aminosalicylic acid in HIV-uninfected and HIV-coinfected tuberculosis patients receiving antiretroviral therapy, managed for multidrug-resistant and extensively drug-resistant tuberculosis. Antimicrob Agents Chemother 58(10):6242–6250. https://doi.org/10.1128/AAC.03073-14
Sy SK, de Kock L, Diacon AH, Werely CJ, Xia H, Rosenkranz B, van der Merwe L, Donald PR (2015) N-acetyltransferase genotypes and the pharmacokinetics and tolerability of para-aminosalicylic acid in patients with drug-resistant pulmonary tuberculosis. Antimicrob Agents Chemother 59(7):4129–4138. https://doi.org/10.1128/AAC.04049-14
Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J, Ckd EPI (2009) A new equation to estimate glomerular filtration rate. Ann Intern Med 150(9):604–612
Albiero J, Sy SK, Mazucheli J, Caparroz-Assef SM, Costa BB, Alves JL, Gales AC, Tognim MC (2016) Pharmacodynamic evaluation of the potential clinical utility of fosfomycin and meropenem in combination therapy against KPC-2-producing Klebsiella pneumoniae. Antimicrob Agents Chemother 60(7):4128–4139. https://doi.org/10.1128/AAC.03099-15
Kowalski KG, Hutmacher MM (2001) Efficient screening of covariates in population models using Wald’s approximation to the likelihood ratio test. J Pharmacokinet Pharmacodyn 28(3):253–275
Cote JF, Kirzin S, Kramar A, Mosnier JF, Diebold MD, Soubeyran I, Thirouard AS, Selves J, Laurent-Puig P, Ychou M (2007) UGT1A1 polymorphism can predict hematologic toxicity in patients treated with irinotecan. Clin Cancer Res 13(11):3269–3275. https://doi.org/10.1158/1078-0432.CCR-06-2290
Iyer L, Das S, Janisch L, Wen M, Ramirez J, Karrison T, Fleming GF, Vokes EE, Schilsky RL, Ratain MJ (2002) UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenom J 2(1):43–47
Hoskins JM, Goldberg RM, Qu P, Ibrahim JG, McLeod HL (2007) UGT1A1*28 genotype and irinotecan-induced neutropenia: dose matters. J Natl Cancer Inst 99(17):1290–1295. https://doi.org/10.1093/jnci/djm115
Hu ZY, Yu Q, Pei Q, Guo C (2010) Dose-dependent association between UGT1A1*28 genotype and irinotecan-induced neutropenia: low doses also increase risk. Clin Cancer Res 16(15):3832–3842. https://doi.org/10.1158/1078-0432.CCR-10-1122
Hu ZY, Yu Q, Zhao YS (2010) Dose-dependent association between UGT1A1*28 polymorphism and irinotecan-induced diarrhoea: a meta-analysis. Eur J Cancer 46(10):1856–1865. https://doi.org/10.1016/j.ejca.2010.02.049
Funding
This study was funded by Nektar Therapeutics.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
SKBS and YC are former employees of Nektar Therapeutics; TG, UH, and MAE are employees of Nektar Therapeutics and own stocks and stock options of Nektar Therapeutics.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of institutional and/or national research committees and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sy, S.K.B., Chia, Y.L., Gordi, T. et al. Integrated population pharmacokinetics of etirinotecan pegol and its four metabolites in cancer patients with solid tumors. Cancer Chemother Pharmacol 81, 897–909 (2018). https://doi.org/10.1007/s00280-018-3562-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-018-3562-3