
Abstract
Purpose
To evaluate safety of TAS-102 administered twice daily (bid) on days 1–5 and 8–12 of a 4-week cycle, confirm feasibility of the Japanese recommended dose (RD), 35 mg/m2, in Western patients with metastatic colorectal cancer (mCRC) refractory to standard chemotherapies, and describe preliminary antitumor activity.
Methods
This open-label, dose-escalation phase 1 study was conducted at four US centers. Patients were enrolled into two sequential cohorts [30 (cohort 1) or 35 mg/m2/dose bid (cohort 2)]; dose-limiting toxicities (DLT) were evaluated during cycle 1 in dose-escalation cohorts. At RD, 15 additional patients were enrolled in an expansion cohort.
Results
Patients (N = 27) with refractory mCRC received TAS-102; 74 % had received ≥4 prior regimens. DLT was not observed in three patients in cohort 1, and was in one out of nine patients in cohort 2 (grade 3 febrile neutropenia). Therefore, RD was identified as 35 mg/m2 bid. At RD, fatigue (63 %), gastrointestinal disturbances and nausea (46 %), vomiting (46 %), and diarrhea (42 %) were common but rarely grade 3/4. Grade 3/4 nausea, vomiting, and diarrhea occurred at 4 % each. Grade 3/4 toxicity was predominantly hematologic [neutropenia (71 %), anemia (25 %)]; febrile neutropenia was observed in two patients. Stable disease lasting ≥6 weeks was achieved by 16 evaluable patients (70 %); median progression-free survival and overall survival were 5.3 and 7.5 months, respectively.
Conclusions
TAS-102 has an acceptable safety profile and preliminary evidence of disease stabilization in Western patients with refractory mCRC. Results from a randomized phase 3 study have shown survival benefit with disease stabilization evidence in this population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer is the third most common malignancy and the second leading cause of cancer death in men and women in the USA [1, 2]. Fifty to sixty percent of patients diagnosed with colorectal cancer will develop metastatic disease [3]. For patients with unresectable metastatic colorectal cancer (mCRC), initial treatment typically consists of an infusional 5-fluorouracil (5-FU) and leucovorin regimen containing irinotecan (FOLFIRI), oxaliplatin (FOLFOX), or both (FOLFOXIRI) [3–6]. Use of 5-FU, irinotecan, and oxaliplatin, regardless of their sequence in frontline and second-line treatment, is associated with significantly improved survival [7]. The addition of monoclonal antibodies targeting vascular endothelial growth factor (bevacizumab) or epidermal growth factor receptor (cetuximab or panitumumab) to chemotherapy has also led to improved outcomes [8–10]. In the refractory setting, regorafenib has shown modest improvement in overall survival (OS) compared with placebo [11]. More treatment options are needed for patients with disease progression after 5-FU, irinotecan, oxaliplatin, and available monoclonal antibodies.
TAS-102 is a novel, orally active, antineoplastic combination drug consisting of trifluridine (FTD) and tipiracil hydrochloride (TPI) in a molar ratio of 1:0.5. FTD is a thymidine-based nucleoside analogue, which, in its triphosphate form, is incorporated into DNA causing single-strand and double-strand breaks (TS) [12, 13]. TPI is a potent inhibitor of thymidine phosphorylase, which prevents rapid degradation of FTD, thereby allowing sustained FTD concentrations to be maintained after oral administration [14]. The incorporation of FTD into DNA appears to be the primary mechanism of antitumor activity of TAS-102 compared with 5-FU and other fluoropyrimidines that produce cytotoxic effects primarily via TS inhibition [15]. Consistent with this difference, TAS-102 antitumor effects have been observed in colorectal cancer cell lines and xenograft models resistant to 5-FU and other fluoropyrimidines [16, 17]. In preclinical models, TAS-102 induces higher levels of apoptosis compared with 5-FU and, unlike 5-FU, does not elicit an autophagic survival response in colorectal cancer cell lines [18].
Treatment on days 1–5 and 8–12 of a 4-week cycle was chosen as the recommended phase 2 schedule for TAS-102 administration based on phase 1 clinical studies in patients with advanced solid tumors [19–21]. In Japanese patients, most of whom had mCRC, the maximum tolerated dose (MTD) on this schedule was 35 mg/m2/dose twice daily [22].
However, a previous phase 1 study conducted in the USA in heavily pretreated patients with breast cancer identified 25 mg/m2/dose twice daily as the MTD [23], although higher daily doses were found to be tolerable with other dosing schedules in patients with other advanced tumors, particularly gastrointestinal tumors [21]. Therefore, the present study was designed to evaluate the safety and determine whether the dose and schedule of TAS-102 used in the Japanese mCRC phase 2 trial is feasible in similar patients with refractory mCRC in the USA, and to obtain preliminary information about its antitumor activity.
Methods
Patients
Male or female patients aged ≥18 years of age with histologically or pathologically confirmed refractory mCRC were eligible if they had received at least two prior lines of conventional chemotherapy for metastatic disease, including a fluoropyrimidine, oxaliplatin, and irinotecan, had Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1, were able to take medications orally, had adequate organ function, and had a life expectancy of at least 3 months. Adjuvant therapy with a fluoropyrimidine and oxaliplatin could count as one line of prior therapy if disease recurred within 12 months of completion. Previous treatment with biologics targeting vascular endothelial growth factor, epidermal growth factor, or their receptors was allowed. Patients with known brain metastasis, acute systemic infection, grade ≥3 ascites, or significant cardiovascular disease; those who underwent major surgery or radiotherapy within the previous 4 weeks, had radiation to >25 % of marrow-bearing bone, or had received anticancer therapy within 3 weeks; and women who were pregnant or lactating, were excluded.
Study design
This open-label, non-randomized, sequential-group, dose-escalation phase 1 study was conducted at four centers in the USA from September 2011 to April 2013. The data cutoff date was April 3, 2013, which was 12 months after the last enrolled patient started treatment. The study was conducted in accordance with ethical principles originating in the Declaration of Helsinki (2008) and in compliance with Good Clinical Practice and all local and national regulatory guidelines. Study approval was obtained from the institutional review board at each site before that site enrolled any patients. All patients provided written informed consent before any screening procedures were conducted.
Eligible patients were assigned sequentially to two cohorts corresponding to TAS-102 doses of 30 mg/m2/dose twice daily (cohort 1) or 35 mg/m2/dose twice daily (cohort 2) on days 1–5 and 8–12 of each 4-week cycle. Doses were taken orally within 1 h after completing morning and evening meals.
Three patients were assigned initially to the first dose level using a conventional 3 + 3 dose-escalation design. If a dose-limiting toxicity (DLT) occurred during the first cycle in ≥2 patients, the study was to be stopped. If DLTs were not observed in the first three patients (or in ≤1 of 6 patients), then six patients were to be enrolled at the second dose level, and additional patients were allowed to be enrolled in any cohort at the discretion of the investigator and sponsor if further safety evaluation was warranted. DLTs were defined as grade 4 neutropenia lasting >7 days, febrile neutropenia (absolute neutrophil count <1000/mm3 with a single body temperature of >38.3 °C [101 °F] or sustained temperature of ≥38 °C [100.4 °F] for more than 1 h), grade 4 thrombocytopenia or grade 3 thrombocytopenia with bleeding, grade 3/4 nausea or vomiting lasting >48 h and uncontrolled by aggressive antiemetic therapy, grade 3/4 diarrhea lasting >48 h and unresponsive to antidiarrheal medication, other grade 3/4 non-hematologic toxicity, any drug-related toxicity resulting in >1 week delay in initiation of cycle 2, or any drug-related toxicity preventing administration of ≥80 % of the planned cumulative dose during cycle 1. The recommended dose (RD) for further clinical development was defined as the highest dose level at which <33 % of patients experienced a DLT during the first cycle. After establishing the RD, additional patients were enrolled into the expansion cohort in order to confirm safety and assess preliminary antitumor activity.
TAS-102 treatment was discontinued if patients experienced disease progression, unacceptable toxicity, withdrew consent, or at the investigator’s discretion if felt to be clinically indicated. Study treatment was supplied as 15- and 20-mg immediate-release film-coated tablets; the number of tablets needed to achieve the assigned dose was determined according to the patient’s body surface area. Dose reductions were permitted in 5 mg/m2 decrements to a minimum dose of 20 mg/m2/dose in the event of treatment-related toxicity (grade ≥3 non-hematologic toxicity, grade 4 neutropenia, or grade ≥3 thrombocytopenia). Patients who did not meet resumption criteria within 14 days of the scheduled initiation of the next cycle were discontinued from treatment.
Other anticancer treatments, including palliative radiotherapy, were not allowed. Prophylactic administration of granulocyte colony-stimulating factor (G-CSF), antidiarrheal agents, or antiemetic agents were not permitted during cycle 1.
Assessments
Patients were monitored for adverse events throughout the study and for up to 30 days after the last dose. Adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03) and coded using the Medical Dictionary for Regulatory Activities (version 14.0). Other safety assessments were made before each cycle and at the 30-day safety follow-up visit, including hematology and clinical chemistry (also on days 8, 15, and 22 of cycle 1 and days 1 and 22 of subsequent cycles), physical examination, vital signs, performance status, and electrocardiogram (also on day 12 of cycle 1).
Tumor assessments and imaging studies of the chest, abdomen, and pelvis as clinically indicated were obtained at baseline (within 28 days before first dose) and then every two cycles. KRAS status was determined locally during screening. Tumor response was determined by the investigator according to RECIST criteria (version 1.1) [24]. Patient survival was determined every 2 months after the end of treatment.
Statistical analysis
The sample size was not based on formal hypothesis testing, but was selected to obtain sufficient safety data in order to identify the RD for future clinical studies.
Safety was evaluated in all patients who received at least one dose of TAS-102. DLTs were evaluated in all patients in cohorts 1 and 2 who received at least 80 % of the planned study medication during cycle 1 (or lesser amounts due to occurrence of a DLT). Efficacy was assessed in all patients who completed at least one cycle of treatment and had clinical or radiologic assessments for disease progression.
Statistical analyses were performed using SAS® software (version 9.1; SAS Institute, Inc., Cary, NC). Safety parameters were summarized descriptively. Efficacy parameters were determined for all patients and by KRAS status. The best overall response was based on investigator assessment of radiologic images and classified as complete response (CR), partial response (PR), stable disease (SD), or progressive disease (PD) according to RECIST criteria (version 1.1) [24]. SD required disease maintenance for at least 6 weeks without evidence of progression. Disease control rate (DCR) was defined as the proportion of patients with objective evidence of CR, PR, or SD and was summarized descriptively. Progression-free survival (PFS, defined as the time from the first dose of TAS-102 until radiologic disease progression or death) and OS were evaluated by the Kaplan–Meier method, and 95 % confidence intervals (CI) for median survival values were determined by the methods of Brookmeyer and Crowley [25].
Role of the sponsor
This study was sponsored, designed, and analyzed by Taiho Oncology, Inc. The investigators enrolled and treated the patients and collected the data. All authors had access to the data and vouch for its accuracy. A draft of the manuscript was written by a medical writer who was compensated by Taiho Oncology, Inc. The authors provided guidance for the draft, comments, and feedback on subsequent revised versions. The corresponding author made the final decision to submit the manuscript for publication.
Results
Patient disposition and characteristics
A total of 27 patients received TAS-102, including three patients in cohort 1 (30 mg/m2/dose twice daily), nine patients in cohort 2 (35 mg/m2/dose twice daily), and 15 patients in the expansion cohort (35 mg/m2/dose twice daily) (Fig. 1). All treated patients from cohorts 1 and 2 were included in the DLT evaluable population, and all patients were included in the safety evaluation. One patient without any post-baseline tumor assessments was excluded from the efficacy evaluation. At the time of data cutoff, one patient in the expansion cohort remained on TAS-102 treatment. Most patients discontinued study treatment due to radiologic progression.

Patient disposition
The study population had a median age of 64 years (range, 44–88), most were men (63 %) and white (93 %), and all had ECOG performance status of 0–1 (Table 1). The study population was heavily pretreated; the majority (74 %) had received ≥4 prior regimens, and besides a fluoropyrimidine, irinotecan, and oxaliplatin, most had received bevacizumab (96 %) and either cetuximab or panitumumab (52 %). KRAS status was determined for 25 patients [12 (48 %) wild-type and 13 (52 %) KRAS mutant].
Dose-limiting toxicity
DLT was not observed among the three patients who received TAS-102 at a dose of 30 mg/m2/dose twice daily; therefore, the dose was escalated to 35 mg/m2/dose twice daily. Among the first six patients in cohort 2, one patient had a DLT of grade 3 febrile neutropenia, which was considered related to study treatment and resolved during cycle 1. Three additional patients were enrolled in cohort 2 to confirm tolerability, and none had DLTs. Therefore, the RD was defined as 35 mg/m2/dose twice daily, and the expansion cohort was enrolled to obtain additional safety data at this dose.
Exposure
Patients received TAS-102 for a median of three cycles (range, 1–14). Seven patients (26 %) initiated at least eight cycles of study treatment. Among the 24 patients who received 35 mg/m2 twice daily, the median cumulative dose was 2052 mg/m2 (range, 706–8949), median dose intensity was 154.2 mg/m2/week (range, 108.5–176.6), and relative dose intensity (i.e., ratio of actual to planned dose intensity) was 0.89 (range, 0.62–1.01).
There were no dose reductions in cohort 1, although two of the three patients had the start of a cycle delayed by ≥4 (but <8) days. Ten patients (42 %) in the combined 35 mg/m2 twice daily cohort had dose reductions by their last treatment cycle, including six patients with a single reduction to 30 mg/m2 twice daily and four patients with two dose reductions to 25 mg/m2 twice daily. Fourteen patients (58 %) had at least one cycle delayed by ≥4 days (median, two cycles delayed per patient; range 1–4 cycles), and seven patients had delays of ≥8 days (median, one cycle delayed per patient; range 1–2 cycles). Grade 3/4 neutropenia was the most common reason for dose reductions and delays in cycle initiation.
Safety
Adverse events were observed in all 27 patients. Grade 3/4 toxicity was predominantly hematologic, consisting mostly of neutropenia (70 %) and anemia (22 %) (Table 2). Febrile neutropenia was observed in only two patients (7 %). Prophylactic G-CSF use was not permitted during cycle 1, but was allowed to treat hematologic toxicity according to the American Society of Clinical Oncology guidelines [26]; its use ranged from 20 % of patients (5/25) in cycle 2–67 % of patients (8/12) in cycle 4. Non-hematologic toxicity consisted mainly of fatigue and gastrointestinal disturbances, but rarely was grade 3 or higher (Table 2). Major grade 3/4 toxicities were neutropenia (70 %), anemia (22 %), and leukopenia (19 %). Most of the frequently reported adverse events, including hematologic abnormalities and gastrointestinal disturbances, were considered by the investigator to be related to study treatment.
There were no treatment-related deaths. One patient died of septic shock due to methicillin-resistant Staphylococcus aureus during cycle 12, which was considered unrelated to TAS-102 treatment. Serious adverse events occurred in six patients; two had treatment-related hematologic toxicity, and four had adverse events secondary to metastatic disease or other underlying conditions. Two patients discontinued TAS-102 due to adverse events (grade 3 bile duct stenosis and grade 2 diarrhea, respectively), and only one patient discontinued due to a treatment-related adverse event (grade 2 diarrhea).
Efficacy
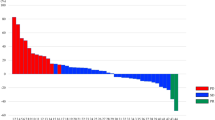
The efficacy evaluation was conducted in 26 patients with post-baseline tumor assessments. Although no patients had a CR or PR, 17 patients achieved an SD lasting for >6 weeks, resulting in a DCR of 65.4 %. Seven of 11 patients with wild-type KRAS (64 %) and eight of 13 patients with mutant KRAS (62 %) achieved SD. Among the patients receiving 35 mg/m2 twice daily (RD level), 16 out of 23 patients achieved an SD, resulting in a DCR of 69.6 % (Fig. 2).

Waterfall plot of target lesion change and best overall response. The asterisks denote patients who received TAS-102 at a dose of 30 mg/m2 twice daily. Four patients in the efficacy population did not have all target lesions evaluated post-baseline and are excluded from this graph. The second bar from the right denotes a patient who had progressive disease due to development of a new lesion despite shrinkage of the target lesion
In the entire study population, median PFS and OS were 4.1 months (95 % CI, 2.0–7.9) and 8.9 months (95 % CI, 4.9–14.4), respectively (Fig. 3). Seven patients receiving TAS-102 at a dose of 35 mg/m2 twice daily did not have survival events by the data cutoff date and were censored at the date they were last known to be alive. Among the 24 patients receiving 35 mg/m2, median PFS and OS were 5.3 months (95 % CI, 2.4–8.0) and 7.5 months (95 % CI, 4.6–14.4), respectively.

a Kaplan–Meier plot of progression-free survival. b Kaplan–Meier plot of overall survival
Among patients with KRAS wild-type and mutant tumors, median PFS was 3.5 months (95 % CI, 2.0–7.9) and 4.1 months (95 % CI, 2.0–8.0), respectively, and median OS was 7.3 months (95 % CI, 4.9–11.5) and 6.0 months (95 % CI, 4.0–14.4), respectively.
Discussion
In the initial phase 1 trial of TAS-102 in Japanese patients, the RD of 35 mg/m2 twice daily on days 1–5 and 8–12 of a 28-day cycle produced a well-tolerated safety profile [22]. Common grade 3/4 adverse events were hematologic, including neutropenia (38 %) and leukopenia (33 %). This was similar to the safety profile observed in previous phase 1 trials in Western patients, though one of these trials conducted in patients with heavily pretreated breast cancer determined an MTD of 25 mg/m2 twice daily [19, 20, 23].
Based on preliminary antitumor activity observed in the cohort of patients with colorectal cancer treated in the Japanese phase 1 study (DCR of 50 % [22]), a randomized, double-blind, placebo-controlled, phase 2 trial in Japan was conducted. Patients with metastatic colorectal adenocarcinoma who had received ≥2 prior standard chemotherapy regimens were randomized 2:1 to receive either TAS-102 35 mg/m2 or placebo twice daily (days 1–5 and 8–12 of 28-day cycles). Compared with placebo, TAS-102 significantly improved OS (9.0 vs. 6.6 months, HR 0.56, P = 0.0011) and PFS with a manageable safety profile [27]. The present trial was designed to confirm the safety of the 35 mg/m2 twice daily dose used in the Japanese phase 2 trial in the Western population of patients with colorectal cancer prior to embarking on a global phase 3 trial of TAS-102 in patients with refractory mCRC. In this study, we treated a total of 27 patients, 74 % of whom had received ≥4 prior treatment regimens, in two cohorts: 30 mg/m2/dose twice daily and 35 mg/m2/dose twice daily. There were no DLTs observed in the first cohort. One patient in the second cohort had dose-limiting grade 3 febrile neutropenia.
Similar to previous studies of TAS-102 conducted in both Japanese and Western populations, grade 3/4 toxicity observed in this trial was predominantly hematologic. Compared with the Japanese randomized phase 2 trial in patients with colorectal cancer, rates of grade 3/4 neutropenia (71 vs. 50 %), febrile neutropenia (8 vs. 4 %), and anemia (25 vs. 17 %) were modestly increased among Western patients, though these findings may be due to the sample size differences between the two trials. In general, toxicities were manageable by appropriate dose delay and/or reduction. Only one patient discontinued study treatment due to a drug-related adverse event. Scheduling prophylactic G-CSF (except during cycle 1) may have avoided dose reductions in some patients who experienced a dose delay. The preliminary efficacy endpoints observed in this phase 1 trial were a DCR of 65 % and a median OS of 8.9 months, which were similar to the results of the Japanese phase 2 trial.
Subsequently, the global phase 3 RECOURSE trial of TAS-102 in patients with refractory mCRC who had at least two prior lines of treatment was initiated based on the RD established in this phase 1 trial. Results from this multicenter trial showed improvement in the primary endpoint of OS compared with placebo [28]. In conclusion, the dose of 35 mg/m2 twice daily is generally well tolerated in the treatment of patients with refractory mCRC in a Western population.
References
Siegel R, Ma J, Zou Z, Jemal A (2014) Cancer statistics, 2014. CA Cancer J Clin 64:9–29
Siegel R, Desantis C, Jemal A (2014) Colorectal cancer statistics, 2014. CA Cancer J Clin 64:104–117
National Comprehensive Cancer Network (2014). NCCN clinical practice guidelines in oncology (NCCN Guidelines®). Colon cancer, version 3. http://www.nccn.org. Accessed 26 March 2014
Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, Jandik P, Iveson T, Carmichael J, Alakl M, Gruia G, Awad L, Rougier P (2000) Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet 355:1041–1047
Tournigand C, André T, Achille E, Lledo G, Flesh M, Mery-Mignard D, Quinaux E, Couteau C, Buyse M, Ganem G, Landi B, Colin P, Louvet C, de Gramont A (2004) FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol 22:229–237
Falcone A, Ricci S, Brunetti I et al (2007) Phase III trial of infusional fluorouracil, leucovorin, oxaliplatin, and irinotecan (FOLFOXIRI) compared with infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) as first-line treatment for metastatic colorectal cancer: the Gruppo Oncologico Nord Ovest. J Clin Oncol 25:1670–1676
Grothey A, Sargent D, Goldberg RM, Schmoll HJ (2004) Survival of patients with advanced colorectal cancer improves with the availability of fluorouracil-leucovorin, irinotecan, and oxaliplatin in the course of treatment. J Clin Oncol 22:1209–1214
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350:2335–2342
Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D’Haens G, Pintér T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, Nippgen J, Rougier P (2009) Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 360:1408–1417
Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, André T, Chan E, Lordick F, Punt CJ, Strickland AH, Wilson G, Ciuleanu TE, Roman L, Van Cutsem E, Tzekova V, Collins S, Oliner KS, Rong A, Gansert J (2010) Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol 28:4706–4713
Grothey A, Van Cutsem E, Sobrero A et al (2013) Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicenter, randomised, placebo-controlled phase 3 trial. Lancet 381:303–312
Emura T, Suzuki N, Yamaguchi M, Ohshimo H, Fukushima M (2004) A novel combination antimetabolite, TAS-102, exhibits antitumor activity in FU-resistant human cancer cells through a mechanism involving FTD incorporation in DNA. Int J Oncol 25:571–578
Suzuki N, Nakagawa F, Nukatsuka M, Fukushima M (2011) Trifluorothymidine exhibits potent antitumor activity via the induction of DNA double-strand breaks. Exp Ther Med 2:393–397
Fukushima M, Suzuki N, Emura T, Yano S, Kazuno H, Tada Y, Yamada Y, Asao T (2000) Structure and activity of specific inhibitors of thymidine phosphorylase to potentiate the function of antitumor 2′deoxyribonucleosides. Biochem Pharmacol 59:1227–1236
Longley DB, Harkin DP, Johnston PG (2003) 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 3:330–338
Murakami Y, Kazuno H, Emura T, Tsujimoto H, Suzuki N, Fukushima M (2000) Different mechanisms of acquired resistance to fluorinated pyrimidines in human colorectal cancer cells. Int J Oncol 17:277–283
Emura T, Murakami Y, Nakagawa F, Fukushima M, Kitazato K (2004) A novel antimetabolite, TAS-102 retains its effects on FU-related resistant cancer cells. Int J Mol Med 13:545–549
Bijnsdorp IV, Peters GJ, Temmink OH, Fukushima M, Kruyt FA (2010) Differential activation of cell death and autophagy results in an increased cytotoxic potential for trifluorothymidine compared with 5-fluorouracil in colon cancer cells. Int J Cancer 126:2457–2468
Hong DS, Abbruzzese JL, Bogaard K, Lassere Y, Fukushima M, Mita A, Kuwata K, Hoff PM (2006) Phase I study to determine the safety and pharmacokinetics of oral administration of TAS-102 in patients with solid tumors. Cancer 107:1383–1390
Overman MJ, Varadhachary G, Kopetz S, Thomas MB, Fukushima M, Kuwata K, Mita A, Wolff RA, Hoff P, Xiong H, Abbruzzese JL (2008) Phase 1 study of TAS-102 administered once daily on a 5-day-per-week schedule in patients with solid tumors. Invest New Drugs 26:445–454
Overman MJ, Kopetz S, Varadhachary G, Fukushima M, Kuwata K, Mita A, Wolff RA, Hoff P, Xiong H, Abbruzzese JL (2008) Phase I clinical study of three times a day oral administration of TAS-102 in patients with solid tumors. Cancer Invest 26:794–799
Doi T, Ohtsu A, Yoshino T, Boku N, Onozawa Y, Fukutomi A, Hironaka S, Koizumi W, Sasaki T (2012) Phase I study of TAS-102 treatment in Japanese patients with advanced solid tumors. Br J Cancer 107:429–434
Green MC, Pusztai L, Theriault RL, Adinin RB, Hofweber M, Fukushima M, Mita A, Bindra N, Hortobagyi GN (2006) Phase I study to determine the safety of oral administration of TAS-102 on a twice daily (BID) schedule for five days a week (wk) followed by two days rest for two wks, every (Q) four wks in patients (pts) with metastatic breast cancer (MBC). J Clin Oncol 24(18S):10576
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247
Brookmeyer R, Crowley J (1982) A confidence interval for the median survival time. Biometrics 38:29–41
Smith TJ, Khatcheressian J, Lyman GH et al (2006) 2006 update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline. J Clin Oncol 24:3187–3205
Yoshino T, Mizunuma N, Yamazaki K, Nishina T, Komatsu Y, Baba H, Tsuji A, Yamaguchi K, Muro K, Sugimoto N, Tsuji Y, Moriwaki T, Esaki T, Hamada C, Tanase T, Ohtsu A (2012) TAS-102 monotherapy for pretreated metastatic colorectal cancer: a double-blind, randomised, placebo-controlled phase 2 study. Lancet Oncol 13:993–1001
Mayer RJ, Van Cutsem E, Falcone A et al (2015) Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med 372:1909–1919
Acknowledgments
The authors thank the patients and their families and study site personnel who participated in this study. This study was sponsored by Taiho Oncology Inc. Medical writing assistance was provided by Taiho Oncology Inc. Many thanks to Paul Bebeau, Gihan Atalla, Susan Darrah, and Hiroshi Ambe of Taiho, and John Ilgenfritz of United Biosources whose help was instrumental in the conduct of this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
J. C. Bendall, R. J. Mayer, J. W. Goldman, J. R. Infante, and M. R. Patel declare that they have no conflict of interest. L. S. Rosen’s institution (University of California, Los Angeles) has received funding to support this trial from Taiho Oncology. F. Benedetti, H. Mizuguchi, and C. Zergebel are employees of Taiho Oncology Inc. D. Lin is an employee of Taiho Pharmaceutical of Beijing Co., Ltd.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bendell, J.C., Rosen, L.S., Mayer, R.J. et al. Phase 1 study of oral TAS-102 in patients with refractory metastatic colorectal cancer. Cancer Chemother Pharmacol 76, 925–932 (2015). https://doi.org/10.1007/s00280-015-2850-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-015-2850-4