
Abstract
Purpose
To develop a sensitive analytical method to quantify gemcitabine (2′,2′-difluorodeoxycytidine, dFdC) and its metabolites 2′,2′-difluorodeoxyuridine (dFdU) and 2′,2′-difluorodeoxycytidine-5′-triphosphate (dFdCTP) simultaneously from tumour tissue.
Methods
Pancreatic ductal adenocarcinoma tumour tissue from genetically engineered mouse models of pancreatic cancer (KP FL/FL C and KP R172H/+ C) was collected after dosing the mice with gemcitabine. 19F NMR spectroscopy and LC–MS/MS protocols were optimised to detect gemcitabine and its metabolites in homogenates of the tumour tissue.
Results
A 19F NMR protocol was developed, which was capable of distinguishing the three analytes in tumour homogenates. However, it required at least 100 mg of the tissue in question and a long acquisition time per sample, making it impractical for use in large PK/PD studies or clinical trials. The LC–MS/MS protocol was developed using porous graphitic carbon to separate the analytes, enabling simultaneous detection of all three analytes from as little as 10 mg of tissue, with a sensitivity for dFdCTP of 0.2 ng/mg tissue. Multiple pieces of tissue from single tumours were analysed, showing little intra-tumour variation in the concentrations of dFdC or dFdU (both intra- and extra-cellular). Intra-tumoural variation was observed in the concentration of dFdCTP, an intra-cellular metabolite, which may reflect regions of different cellularity within a tumour.
Conclusion
We have developed a sensitive LC–MS/MS method capable of quantifying gemcitabine, dFdU and dFdCTP in pancreatic tumour tissue. The requirement for only 10 mg of tissue enables this protocol to be used to analyse multiple areas from a single tumour and to spare tissue for additional pharmacodynamic assays.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Gemcitabine, 2′,2′-difluorodeoxycytidine, dFdC, is an anticancer drug licensed for the treatment of a number of solid tumour types including pancreatic, non-small cell lung, ovary, bladder and breast cancer.
Gemcitabine is a prodrug that is phosphorylated intra-cellularly by deoxycytidine kinase (dCK), thymidine kinase (TK2), dCMP kinase and nucleoside diphosphate kinase into the active metabolites 2′,2′-difluorodeoxycytidine-5′-diphosphate (dFdCDP) and triphosphate (dFdCTP). The cytotoxic effects of gemcitabine have been attributed to a combination of two mechanisms of action: (1) inhibition of ribonucleotide reductase, which reduces the pool of dNTPs available for DNA synthesis, and (2) competition with dCTP for utilisation by DNA polymerase, leading to incorporation into DNA and chain termination [14, 26].
Gemcitabine is metabolically inactivated by cytidine deaminase (CDA) into the metabolite 2′,2′-difluorodeoxyuridine (dFdU) [1]. CDA enzyme is present in normal and tumour cells [24, 30] as well as in blood [3]. The cytotoxic effects of gemcitabine are also modulated by the cell membrane nucleoside transporters ENT1, ENT2, CNT1 and CNT3, without which gemcitabine cannot enter cells and be activated by phosphorylation [6, 20, 21]. Survival after gemcitabine treatment has been linked to expression of hENT1 and hCNT3 [21, 33].
Despite being licenced for use in pancreatic adenocarcinoma, gemcitabine exhibits only a modest improvement in patient survival [7], and it has been suggested that its activity may be limited by poor drug delivery, particularly in the case of pancreatic adenocarcinoma (PDA) tumours, which tend to be hypovascular with extensive desmoplastic stroma [23, 32]. Previous studies have demonstrated that the combination treatment of gemcitabine with an agent that depletes the tumour stroma by inhibition of the Hedgehog signalling pathway enhances drug delivery and efficacy in a mouse model of pancreatic cancer [23].
Fluorine-19 nuclear magnetic resonance spectroscopy (19F NMR) is used commonly to detect fluorine-containing compounds and their metabolites that are non-naturally occurring in biological tissues and fluids. Among NMR methods, 19F NMR has advantages due to the characteristics of the natural fluorine nucleus, which includes a nuclear spin (I) of ½, 100% natural abundance, high sensitivity (83% of 1H NMR), a large chemical shift range (500 ppm) and short longitudinal relaxation times (T1) [22]. It offers a unique method of observing fluorinated drugs, their pharmacokinetics and metabolism either in vivo or in vitro (including ex vivo tissue samples [4, 9, 39].
We have now improved a previously published 19F NMR method for gemcitabine [9, 23] and used it to quantify gemcitabine in mouse pancreatic tumour tissue. However, large tissue samples were required for processing and the sample run-time required to achieve acceptable signal to noise for metabolite quantification became limiting factors. To perform a detailed analysis of gemcitabine tumour pharmacokinetics, further improved analytical methods are required.
Several LC–MS/MS methods to measure gemcitabine and dFdU in plasma have been described [5, 34, 41]. Honeywell et al. [13] described a method for analysis of gemcitabine and dFdU in plasma and tissue. However, intra-tumoural levels of the active metabolite dFdCTP would clearly be the preferred correlant with pharmacodynamics of the drug. Levels of dFdCTP measured in peripheral blood mononuclear cells have been used as a substitute for tissue concentrations [36]. Recently, Jansen et al. [16] described a method for quantifying gemcitabine, dFdU and their phosphorylated metabolites including dFdCTP in PBMCs. To the best of our knowledge, five studies have reported tissue concentrations of dFdCTP but these either measured only dFdCTP and not tissue dFdC and dFdU [10, 26, 29], or used radioisotope-labelled dFdC [37], or used enzymatic dephosphorylation of dFdCTP and then measured the resulting dFdC [31]. An analytical method capable of simultaneous quantification of dFdC, dFdU and dFdCTP in tumour tissue would be very useful.
We now describe a novel LC–MS/MS protocol for simultaneous quantification of gemcitabine, the active metabolite dFdCTP and the inactive metabolite dFdU in tumour tissue from a mouse model of pancreatic cancer. This LC–MS/MS protocol was also used to quantify gemcitabine and dFdU in plasma. We have assessed the performance of the method and believe that it is faster and at least as sensitive as previously published methods [10, 37].
This study provides insights into the intra-tumoural distribution of the three analytes. We compare the 19F NMR and LC–MS/MS protocols and discuss the relative merits of each method for detection of gemcitabine and its metabolites.
Methods
Chemicals
dFdCTP was generously provided by Infinity Pharmaceutical Co. (MA, USA) or was purchased from Sierra Biotech. Gemcitabine hydrochloride was obtained from Tocris Bioscience (UK), and dFdU was purchased from Carbosynth (UK). GemzarTM (Eli Lilly), a ~48% preparation of dFdC, was obtained from Hannas (Delaware) or Addenbrooke’s Hospital Pharmacy (Cambridge, UK) and was used at 10.2 mg/ml in sterile normal saline to dose the mice. Stable isotope-labelled 13C9, 15N3-cytidine 5′-triphosphate, dFUR, 2-fluoro-2′-deoxyadenosine (2F2dA) and tetrahydrofuran (THF) were obtained from Sigma–Aldrich (UK). Tetrahydrouridine (THU) was purchased from Calbiochem (Merck Chemicals, UK). Methanol, acetonitrile trifluoroacetic acid (TFA), sodium hydroxide and ammonia solution were obtained from Fisher Scientific (UK).
Tissue extraction for 19F NMR
The snap-frozen tissues collected at necropsy were weighed. Four volumes of ice-cold acetonitrile were added to homogenise the sample using the Qiagen TissueLyser with a 5-mm ball bearing for two rounds of 6 min at 25 kHz. An equal volume of ice-cold water (to acetonitrile) was added, and samples were incubated on ice for 10 min. Samples were centrifuged at 14,000 rpm for 10 min at 4°C. Supernatants were transferred to cold-resistant vials and snap-frozen in liquid nitrogen. Holes were punctured in the tops once samples were frozen. Vials were transferred into a Heto PowerDry LL1500 Freeze-Dryer (Thermo) and allowed to freeze-dry for at least 24 h.
19F NMR spectroscopy on tumour/tissue extracts
Freeze-dried tumour tissue extracts (from at least 100 mg tissue wet weight) were re-suspended in 600 μl of D2O, spiked with 40 nmoles of internal standard 2F2dA, adjusted to pH5 using 20 μl of Universal pH Indicator solution (Sigma) and diluted HCl and then transferred to a 5-mm standard NMR tube (Wilmad) for 19F NMR analysis. All 19F NMR measurements were carried out on a Bruker 600 MHZ (14.1T) Avance NMR spectrometer, and a QNP probe was used for acquisition of 19F NMR spectra. Acquisition parameters included a 1-D pulse sequence of 19F observation and inverse-gated 1H decoupling, spectral sweep width of 177 ppm (100,000 Hz), 4,096 scans and 1.65 s of repetition time; total acquisition time was about 1 h 55 min. Chemical shift assignments of reference 2F2dA at −52.06 ppm, gemcitabine (dFdC) at −116.33 ppm, dFdU distributed over 4 peaks at −115.70, −116.12, −116.95, −117.08 ppm and dFdCTP at −116.92 ppm in 19F NMR spectra were determined by spiking the samples with appropriate standards. A broad hump observed in the baseline of 19F NMR spectra was removed by application of linear prediction (LP) back-projection to the time domain data by using 2,000 (number of LP) coefficients and 128 back-prediction points prior to Fourier transformation and phase correction. dFdC, dFdU and dFdCTP peak areas were integrated using the Bruker Topspin software processing package. The drug (dFdC) and its metabolite absolute concentrations were estimated in terms of nmoles by using the internal standard reference 2F2dA, and normalised to the tissue wet weights, and 19F metabolite concentrations are shown in ng/mg tissue.
Sample preparation for LC–MS/MS
In order to minimise enzyme-mediated degradation of the analytes ex vivo, all manipulations were done on ice. A minimum of 10 mg of tumour tissue was required, and extraction of gemcitabine, dFdU and dFdCTP was achieved by homogenising the tissue in ice-cold acetonitrile (50% v/v) containing tetrahydrouridine (25 μg/ml) in a Precellys 24 tissue homogeniser to give a final concentration of 0.05 mg/μl of tissue homogenate. An aliquot (50 μl) of the homogenate was added to a microfuge tube with 200 μl of ice-cold acetonitrile (50% v/v) containing 50 ng/ml each of 5′-deoxy-5-fluorouridine (dFUR) and 13C9, 15N3-cytidine triphosphate as internal standards (previously used as internal standards [36, 41]). Vortex mixing was followed by centrifugation at 20,000×g for 25 min, and the resultant supernatant was evaporated to dryness in a Speedvac. The residue was reconstituted in water (100 μl), and 20 μl was injected into the mass spectrometer. With respect to plasma, 25 μl was processed in the same way as the 50 μl of tumour homogenate, by adding 200 μl of ice-cold acetonitrile (85% v/v) containing the internal standards.
Preparation of calibration standards for LC–MS/MS
One milligram per millilitre stock solutions of gemcitabine and dFdCTP were made in water, while dFdU was dissolved in methanol. The appropriate working solutions were used to spike tumour homogenate prepared from untreated mice as described above to give calibration standards with the following ranges: 20–5,000 ng/ml (0.4–100 ng/mg) for dFdU, 10–2,500 ng/ml (0.2–50 ng/mg) for gemcitabine and dFdCTP. Quality control samples were prepared in the same way to give the appropriate concentrations.
Chromatography
The analysis of adenosine phosphates using porous graphitic carbon (PGC) by Wang et al. [38] served as an important reference for the assay development work in this study. Chromatography was performed on an Accela pump and Accela autosampler (Thermo Fisher Scientific, USA). The analytes were separated on a PGC Hypercarb column (100 × 2.1 ID, 5 μm; Thermo Fisher Scientific) fitted with a guard column (Hypercarb 10 × 2.1, 5 μm; Thermo Fisher Scientific) with (A) 10 mM ammonium acetate, pH 10 and (B) acetonitrile as mobile phase. The autosampler and column temperatures were maintained at 4 and 30°C, respectively. The gradient program at a flow rate of 300 μl/min started with 95% A for 2 min, a decrease to 80% in 0.2 min and held for 5.6 min, back to 95% over 0.2 min and held at 95% for 7 min to give a total run-time of 15 min. In order to minimise carry-over between injections, the needle and injection path were flushed using the external wash procedure (Thermo Scientific, surveyor autosampler plus hardware manual: Post injection events) first with 6 ml of water, 150 μl of 100% acetonitrile followed by 3 × 150 μl of 50% acetonitrile and finally with 400 μl water.
Column regeneration
Regeneration of the column was sometimes necessary to avoid loss of retention capacity, as has been reported by other groups [12, 15, 28]. The manufacturer’s instructions were used to restore the column to its original state. Briefly, the column was inverted and flushed at 0.2 ml/min with 25 ml of THF/water (1:1) containing 0.1% TFA, followed by 25 ml of THF/water containing 0.1% sodium hydroxide and a further flush with 25 ml of THF/water. The column was then re-equilibrated with methanol/water (95:5) before use.
Mass spectrometry
A TSQ Vantage triple stage quadrupole mass spectrometer (Thermo Scientific, USA) fitted with a heated electrospray ionisation (HESI-II) probe operated in positive and negative mode at a spray voltage of 2.5 kV, capillary temperature of 150°C and vaporizer temperature of 250°C. Sheath and auxiliary gas pressures were set at 50 and 20 units, respectively. Compound optimisation was done manually using Thermo TSQ Tune Master 2.1.0.1028 (Thermo Fisher Scientific, USA) by infusion into the mass spectrometer using a T-connector, and the scan parameters are shown in Table 1. Quantitative data acquisition was done using LC Quan2.5.6 (Thermo Fisher Scientific, USA).
Assessment of LC–MS/MS assay performance
Assessment of assay performance was performed using the FDA Guidance for Industry 2001 document “Bioanalytical Method Validation” as a guide, determining the linearity, precision, accuracy, recovery, matrix effects and stability of the analytes. The linearity of the assay was tested using eight non-zero standards with the back-calculated concentration of each standard values not exceeding ±15% of the spiked value (±20% at the lower limit of quantification). The precision and accuracy were assessed by the replicate analysis (n = 5) of QC samples at four different concentrations. A blank sample injected after the highest calibration standard was used to determine carry-over. The short-term stability of the analytes in homogenate and plasma containing THU on ice for 4 h and three freeze–thaw cycles at −80°C were assessed using three aliquots of QC samples at two concentrations. The matrix effects of tumour homogenate were determined by comparing peak areas of analytes spiked after extraction with neat standards in triplicate at 10 ng/mg (dFdU) and 5 ng/mg (dFdC and dFdCTP). The recovery was assessed by comparing the peak response ratios of analytes spiked before extraction with those spiked after extraction at the same concentrations used for matrix effect determination.
Use of the assays to determine analyte levels in mouse tumour tissue and plasma
The assays were used to study the levels of gemcitabine, dFdU and dFdCTP in mouse tumour tissue and plasma. Mouse studies were performed in accordance with the UK Animals (Scientific Procedures) Act 1986 and the NCRI 2010 Guidelines for the welfare and use of animals in cancer research [40] with approval from the local Animal Ethics Committee. Mice were of either Kras G12D ; p53 flox/flox ; Pdx1-Cre (KP FL/FL C) or Kras G12D ; p53 R172H; Pdx1-Cre (KP R172H/+ C) strains as previously described [2, 11]. Pancreatic ductal adenocarcinoma development was monitored by ultrasound and/or abdominal palpation for KP R172H/+ C and KP FL/FL C mice. Three of the mice (MH1011, MH1015 and MH1019) were found to have haemorrhagic ascites as a result of their tumour.
Mice were dosed with 50 or 100 mg/kg gemcitabine by IP injection in saline approximately 1 h before collecting blood and tissues. These doses of gemcitabine are the same as was used previously for efficacy studies in the KP R172H/+ C mouse model [23] and are estimated to be equivalent to 150 and 300 mg/m2 (using a mass-to-body surface area factor of 3 for mouse [27]). Tumours were rapidly excised at necropsy, soft surrounding tissue was trimmed off, and the tissue was snap-frozen in liquid nitrogen then stored at −80°C until required. Blood was taken into tubes containing EDTA (1.75 mg/ml) and THU (25 μg/ml) on ice and centrifuged, and plasma was stored at −80°C.
The amount of compound was measured by LC–MS/MS in ng/ml of tissue homogenate or plasma. The tumour homogenate contained 50 mg of tissue per ml, and ng/ml values were converted to ng/mg tissue (e.g. 50 ng/ml measured concentration is equivalent to 1 ng/mg of tumour tissue).
Results
19F NMR
Previous laboratory experience suggested that in order to quantify minute amounts of fluorinated species in a poorly perfused tissue sample, the 19F NMR method required further optimisation to increase the sensitivity. The chemical environment of the sample is responsible for the coupling and de-coupling of the spins as well as the chemical shift of peaks. To identify the typical spectra of gemcitabine (dFdC), dFdU and dFdCTP, standards were spiked into an untreated homogenised mouse tumour sample.
Under the conditions in our recently published paper [23], dFdCTP produced one broad and shallow peak, making it difficult to quantify. The most optimal condition identified was an adjustment of the sample to pH 5, where the standards appeared the sharpest in their resulting spectra. Figure 1 shows a cumulative representation of peaks of the three fluorinated standards when run individually on fractions of the same tumour tissue homogenate. Here, the fluorine signals for dFdC and dFdCTP each show as one distinct peak, whereas the signal for dFdU appears spread over 4 peaks. The relative ratios between the 4 peaks of dFdU always remain the same, as the two fluorine atoms remain decoupled (i.e. peak 1:2 is 0.32:1; peak 2:3 is 1:0.67; peak 3:4 is 1:0.29). Using these ratios, the overlap between the smallest peak of dFdU and dFdCTP can be deduced from the integral of the combined peak and the respective values derived.

Overlaid spectra of 19F NMR peaks for 30 nmoles of dFdC (green), dFdU (blue), dFdCTP (red), spiked and quantified separately into an untreated KP FL/FL C tumour sample
These optimised conditions were then used to measure gemcitabine and its metabolite concentrations in tumours from mice dosed with gemcitabine. Representative calculated values are shown in Table 2. There was considerable inter-mouse variation both in the absolute analyte concentrations and in the relative ratios of the analytes (e.g. the dFdCTP/dFdC ratio ranged from 0.33 for TB7798 to 18 for MH733).
Despite efforts to optimise this method for analysing tumour samples, some restricting factors still remained. At least 100 mg of tissue was necessary for processing, which limits the amount of tissue available from any one tumour for other assays (such as histology and pharmacodynamic assays). Secondly, the time required for each sample run to gain an acceptable signal to noise ratio (1 h 55 min) hindered the possibility of high-throughput analysis.
LC–MS/MS
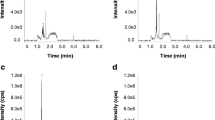
In light of the limitations of the NMR method (100 mg of tissue required and 1 h 55 min acquisition time), the goal was set of developing a sensitive LC–MS/MS assay, suitable for high throughput, to measure dFdC, dFdU and dFdCTP, using a minimum of 10 mg of mouse tumour tissue. Typical chromatograms from this new method, at the limit of quantification, are shown in Fig. 2. There were no significant interfering peaks for all the analytes in blank tumour homogenate and plasma. With respect to plasma, only 25 μl of plasma was required for quantification of gemcitabine and dFdU. dFdCTP was not assayed for in plasma because negligible amounts were expected in the plasma and initial studies showed poor recovery of spiked dFdCTP after acetonitrile precipitation from plasma. The recoveries of all analytes from tumour tissue were greater than 90%. A study of matrix effects in tumour homogenate showed signal reduction for dFdU (21%) and enhancement for gemcitabine (7.3%) and dFdCTP (1.4%).The calibration ranges for gemcitabine, dFdCTP and dFdU were from 0.2 to 50, 0.2–50 and 0.4–100 ng/mg tissue, respectively. The calibration ranges in plasma were from 10 to 2,500 and 20–5,000 ng/ml for gemcitabine and dFdU, respectively. Linear regression and 1/x weighting were used for gemcitabine and dFdCTP while a quadratic fit was used for dFdU. The correlation coefficients (r 2) of the curves were greater than 0.99. The intra-assay precision and accuracy values for tissue samples were well within recommended levels with accuracy values of 91.7–103.9% and precision (% CV) less than 6% for all analytes in tumour tissue homogenate (Table 3). For plasma, the intra-assay precision and accuracy for gemcitabine and dFdU were also within recommended levels with accuracy values of 80.4–112.7% (80.4% at the LLOQ where ±20% is allowed, all other concentrations were within ±15%), and the % CV was less than 6% (Table 3). The freeze–thaw stability was assessed using five replicates at each of the concentrations shown in Table 4. After each freeze cycle, the samples were allowed to thaw on ice. All analytes were stable in tumour tissue homogenate and plasma containing THU after three freeze–thaw cycles at −80°C. Long-term stability during storage at −80°C is ongoing. In order to assess short-term stability, three replicates at each of the concentrations were kept on ice for 4 h. All analytes were stable on ice for the 4 h. The quality control samples were always within ±15% of nominal concentrations for gemcitabine. The carry-over was assessed by injecting a blank sample after the highest standard with values in both tumour tissue homogenate and plasma less than 15%.

Typical LC–MS/MS chromatogram for tumour tissue homogenate spiked with concentrations of analytes at the lower limit of quantification, a gemcitabine (0.2 ng/mg), b dFdU (0.4 ng/mg) and d dFdCTP (0.2 ng/mg). Internal standards: c dFUR and e 13C, N15CTP
The LC–MS/MS assay was then used to measure the analytes in tumours and plasma from mice dosed with gemcitabine at 50 or 100 mg/kg (Table 5). Samples were collected 75 min after dosing mice MH1015, MH1019 and MH1014, 70 min after dosing mouse MH1011, 65 min after dosing mouse MH963 and 60 min after dosing mouse MH959. Quantification of gemcitabine and dFdU in the plasma samples required dilution by a factor of hundred and a dilution QC of the same factor was run together with the samples.
As with 19F-NMR, there was inter-mouse variation in absolute concentrations and ratios of the analytes. The amounts of dFdU observed in tumour tissue in these mice were similar to the plasma concentration (assuming tissue density is 1 g/ml, 1 ng/mg tissue = 1 μg/ml). The plasma concentrations of gemcitabine and dFdU are comparable to those previously reported, using an HPLC method [23].
In order to investigate the distribution of analytes within a tumour, multiple pieces from different regions of each of one KP R172H/+ C and one KP FL/FL C tumour were assayed for the 3 analytes (Table 6). These samples were taken 60 min after a single dose of 100 mg/kg gemcitabine. There was variation in the total amount of analytes and the ratios of analytes in different regions of the same tumour. The KP R172H/+ C tumour appeared to have generally higher levels of dFdC and dFdU than KP FL/FL C tumour, but lower dFdCTP concentrations.
Discussion
Comparing 19F NMR with LC–MS/MS
The advantage of 19F NMR is the detection of all fluorinated species and metabolites of a compound in any tissue type simultaneously. Once optimised, the 19F NMR protocol can be used directly whereas the LC–MS/MS method requires optimising for each type of tissue sample and compound metabolite analysed. However, the sensitivity of 19F NMR is generally quite low. In principle, sensitivity can be improved by increasing the sample size (processing enough tissue with very low concentrations of drug to have quantifiable signal), the magnetic field (defined by the instrument used) and the number of transients accumulated (run-time). Even with a prolonged acquisition time (1 h 55 min in this study), 19F NMR required more tissue (100 mg) than the LC–MS/MS protocol which can quantify with greater resolution down to 0.2 ng/mg (0.4 pmol/mg) of dFdCTP from as little as 10 mg of tissue in only 15 min per sample.
If the signal from a fluorinated compound is high enough, it is possible to do non-invasive in vivo 19F MRS spectroscopy, as demonstrated with 5-FU and capecitabine in cancer patients [17, 18, 35]. Unfortunately, the sensitivity of MRS in vivo is usually lower than for ex vivo NMR due to the lower power of whole body magnets and the transient accumulation time that is limited by patient tolerance. Another major problem of MRS in vivo is poor peak resolution, making it extremely difficult to resolve the gemcitabine metabolites detected in the present ex vivo studies. Three in vivo 19F NMR studies of gemcitabine in mouse xenograft tumours have been reported; two studies used very high-dose gemcitabine (500–800 mg/kg), and none were able to distinguish between the parent compound and its metabolites [4, 8, 19], which is key for gemcitabine because of significant amounts of the inactive metabolite dFdU. Another attractive ex vivo method to detect fluorinated drugs and their metabolites in intact tissue (samples are not destroyed during analysis) is high-resolution magic angle spinning (HRMAS) 19F NMR, but in general this has lower peak resolution than the extraction method employed in the present study, since spinning the samples may not eliminate all the line-broadening dipolar coupling mechanisms. To our knowledge, HRMAS NMR has not been reported for gemcitabine.
A shortcoming of the LC–MS/MS method is that it requires excised tissue samples for analysis. However, the LC–MS/MS protocol is higher throughput than the stated NMR method, because analysis is faster and it can be automated for sample preparation and autoloading of multiple samples sequentially. The major advantage of the LC–MS/MS method over NMR is the requirement for a small sample, of as little as 10 mg (discussed further below).
Chromatography
We have overcome the well-documented analytical challenges (such as simultaneous chromatographic resolution) involving nucleotides and nucleosides to develop an attractive LC–MS/MS method. The generally poor separation of nucleotides and nucleosides on many stationary phases including C-18 has resulted in the use of mass spectrometry-incompatible methods such as ion-exchange chromatography. Porous graphitic carbon (PGC) has, however, been shown by others to obviate the need for ion-exchange with excellent peak shapes. The analysis of adenosine phosphates using PGC by Wang et al. [38] was an important reference for the method development in this study. We, however, observed shifts in retention times with continued column usage, as reported by others [12, 15, 16]. In our view, reproducibility of retention on PGC represents the most important challenge. Regeneration of the column was sufficient to restore the column to its original conditions and reproducibility of retentions times within each run appeared to be dependent on the cleanness of the samples. The samples prepared using procedures described in this study were sufficiently clean to give good quantitative data for the analytes investigated.
Analyte concentrations in tumour tissue and plasma measured by LC–MS/MS
The method was used to measure concentrations of the analytes in pancreatic tumour tissue and plasma from mice treated with gemcitabine. There were inter-mouse differences in absolute concentrations and ratios of analytes, which were not obviously due to the range in sampling times from 60 to 75 min after dosing. There was also no obvious difference between those mice with haemorrhagic ascites and those without. Similar variation was observed in the tumour samples measured by 19F NMR. Concentrations of the 3 analytes were generally higher in those dosed with 100 mg/kg gemcitabine than with 50 mg/kg. It is clear that dFdCTP concentrations in tumour cannot be predicted from either tumour or plasma gemcitabine or dFdU concentrations, underlining the value of this analytical assay for investigation of tumour PK. The concentrations of dFdCTP detected in the tumours (ranging from <0.2 to 1.54 ng/mg in KP R172H/+ C tumours, and 0.5–13.4 ng/mg in KP FL/FL C tumours) are similar to those reported in a human glioblastoma biopsy 1 h after a gemcitabine dose (3,000 pmol/g, which is equivalent to 1.5 ng/mg)[31], and to head and neck tumour concentrations 2 h after a 300 mg/m2 dose of gemcitabine (2.13 pmol dFdCTP/mg, equivalent to 1.07 ng/mg) [10, 31].
Analysis of multiple areas of a tumour is made feasible by the small sample size requirements of this analytical assay. We analysed multiple samples from different regions of one KP R172H/+ C and one KP FL/FL C tumour. These showed little intra-tumour variation in gemcitabine and dFdU, but significant variation in dFdCTP, which may reflect different cellularity in different parts of the tumour, because dFdCTP is formed intra-cellularly, whereas gemcitabine and dFdU are expected to be present both intra-cellularly and extra-cellularly in the interstitial fluid. The KP R172H/+ C tumour showed higher concentrations of gemcitabine and dFdU than the KP FL/FL C tumours, but the dFdCTP concentrations were lower in the KP R172H/+ C than the KP FL/FL C. The vascular function and density of the KP FL/FL C tumours in comparison to the hypovascular KP R172H/+ C tumours is currently under investigation (M. Jacobetz, personal communication).
The requirement for only 10 mg of tumour tissue raises the possibility of measuring tumour PK in biopsy samples from human tumours, such as those obtained from a Tru-cut biopsy (as used for liver tumours, for example) or an endoscopic microcore biopsy (being developed for pancreatic adenocarcinoma [R. Brais et al. manuscript in preparation]). It may be possible to use the LC–MS/MS method for multiple biopsies or repeat biopsies. Also, using only a small piece for PK analysis allows tissue to be spared for additional pharmacodynamic assays.
Conclusions
We have improved a 19F NMR protocol and described a sensitive HPLC–MS/MS method to quantify gemcitabine, dFdU and dFdCTP in mouse pancreatic tissue. As little as 10 mg of tissue is required for LC–MS/MS, which makes it ideal when tissue specimens are also required for other assessments. The method can be applied to preclinical and clinical studies. The protocol will now be used to investigate tumour pharmacokinetics of gemcitabine in more detail and to correlate results with tumour histology and pharmacodynamic endpoints. We are also developing combination therapy strategies, using agents targeting the tumour stroma, which may increase the delivery of gemcitabine to tumour tissue. This has already been demonstrated in studies with the Hedgehog pathway inhibitor IPI-926 [23], and this assay will be instrumental in the development of this combination approach.
References
Abbruzzese JL, Grunewald R, Weeks EA, Gravel D, Adams T, Nowak B, Mineishi S, Tarassoff P, Satterlee W, Raber MN et al (1991) A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J Clin Oncol 9:491–498
Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, Hanahan D, Redston MS, Chin L, Depinho RA (2006) Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci USA 103:5947–5952
Besnard T, Renee N, Etienne-Grimaldi MC, Francois E, Milano G (2008) Optimized blood sampling with cytidine deaminase inhibitor for improved analysis of capecitabine metabolites. J Chromatogr B Analyt Technol Biomed Life Sci 870:117–120
Blackstock AW, Lightfoot H, Case LD, Tepper JE, Mukherji SK, Mitchell BS, Swarts SG, Hess SM (2001) Tumor uptake and elimination of 2′,2′-difluoro-2′-deoxycytidine (gemcitabine) after deoxycytidine kinase gene transfer: correlation with in vivo tumor response. Clin Cancer Res 7:3263–3268
Bowen C, Wang S, Licea-Perez H (2009) Development of a sensitive and selective LC-MS/MS method for simultaneous determination of gemcitabine and 2,2-difluoro-2-deoxyuridine in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 877:2123–2129
Burke T, Lee S, Ferguson PJ, Hammond JR (1998) Interaction of 2′,2′-difluorodeoxycytidine (gemcitabine) and formycin B with the Na+-dependent and -independent nucleoside transporters of Ehrlich ascites tumor cells. J Pharmacol Exp Ther 286:1333–1340
Burris H, Storniolo AM (1997) Assessing clinical benefit in the treatment of pancreas cancer: gemcitabine compared to 5-fluorouracil. Eur J Cancer 33(Suppl 1):S18–S22
Cron GO, Beghein N, Ansiaux R, Martinive P, Feron O, Gallez B (2008) 19F NMR in vivo spectroscopy reflects the effectiveness of perfusion-enhancing vascular modifiers for improving gemcitabine chemotherapy. Magn Reson Med 59:19–27
Edzes HT, Peters GJ, Noordhuis P, Vermorken JB (1993) Determination of the antimetabolite Gemcitabine (2′,2′-difluoro-2′-deoxycytidine) and of 2′,2′-difluoro-2′-deoxyuridine by 19F nuclear magnetic resonance spectroscopy. Anal Biochem 214:25–30
Eisbruch A, Shewach DS, Bradford CR, Littles JF, Teknos TN, Chepeha DB, Marentette LJ, Terrell JE, Hogikyan ND, Dawson LA, Urba S, Wolf GT, Lawrence TS (2001) Radiation concurrent with gemcitabine for locally advanced head and neck cancer: a phase I trial and intracellular drug incorporation study. J Clin Oncol 19:792–799
Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA (2005) Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7:469–483
Holmgren E, Carlsson H, Goede P, Crescenzi C (2005) Determination and characterization of organic explosives using porous graphitic carbon and liquid chromatography-atmospheric pressure chemical ionization mass spectrometry. J Chromatogr A 1099:127–135
Honeywell R, Laan AC, van Groeningen CJ, Strocchi E, Ruiter R, Giaccone G, Peters GJ (2007) The determination of gemcitabine and 2′-deoxycytidine in human plasma and tissue by APCI tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 847:142–152
Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W (1991) Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res 51:6110–6117
Jansen RS, Rosing H, Schellens JH, Beijnen JH (2009) Retention studies of 2′-2′-difluorodeoxycytidine and 2′-2′-difluorodeoxyuridine nucleosides and nucleotides on porous graphitic carbon: development of a liquid chromatography-tandem mass spectrometry method. J Chromatogr A 1216:3168–3174
Jansen RS, Rosing H, Schellens JH, Beijnen JH (2009) Simultaneous quantification of 2′,2′-difluorodeoxycytidine and 2′,2′-difluorodeoxyuridine nucleosides and nucleotides in white blood cells using porous graphitic carbon chromatography coupled with tandem mass spectrometry. Rapid Commun Mass Spectrom 23:3040–3050
Kamm YJ, Heerschap A, van den Bergh EJ, Wagener DJ (2004) 19F-magnetic resonance spectroscopy in patients with liver metastases of colorectal cancer treated with 5-fluorouracil. Anticancer Drugs 15:229–233
Klomp D, van Laarhoven H, Scheenen T, Kamm Y, Heerschap A (2007) Quantitative 19F MR spectroscopy at 3 T to detect heterogeneous capecitabine metabolism in human liver. NMR Biomed 20:485–492
Kristjansen PE, Quistorff B, Spang-Thomsen M, Hansen HH (1993) Intratumoral pharmacokinetic analysis by 19F-magnetic resonance spectroscopy and cytostatic in vivo activity of gemcitabine (dFdC) in two small cell lung cancer xenografts. Ann Oncol 4:157–160
Mackey JR, Yao SY, Smith KM, Karpinski E, Baldwin SA, Cass CE, Young JD (1999) Gemcitabine transport in xenopus oocytes expressing recombinant plasma membrane mammalian nucleoside transporters. J Natl Cancer Inst 91:1876–1881
Marechal R, Mackey JR, Lai R, Demetter P, Peeters M, Polus M, Cass CE, Young J, Salmon I, Deviere J, Van Laethem JL (2009) Human equilibrative nucleoside transporter 1 and human concentrative nucleoside transporter 3 predict survival after adjuvant gemcitabine therapy in resected pancreatic adenocarcinoma. Clin Cancer Res 15:2913–2919
Martino R, Malet-Martino M, Gilard V (2000) Fluorine nuclear magnetic resonance, a privileged tool for metabolic studies of fluoropyrimidine drugs. Curr Drug Metab 1:271–303
Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Ruckert F, Grutzmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA (2009) Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324:1457–1461
Onodera H, Kuruma I, Ishitsuka H, Horii I (2000) Pharmacokintic study of capecitabine in monkeys and mice: species differences in distribution of the enzymes responsible for its activation to 5-FU. Xenobio Metabol and Dispos 15:439–451
Peters GJ, Clavel M, Noordhuis P, Geyssen GJ, Laan AC, Guastalla J, Edzes HT, Vermorken JB (2007) Clinical phase I and pharmacology study of gemcitabine (2′,2′-difluorodeoxycytidine) administered in a two-weekly schedule. J Chemother 19:212–221
Plunkett W, Huang P, Gandhi V (1995) Preclinical characteristics of gemcitabine. Anticancer Drugs 6(Suppl 6):7–13
Reagan-Shaw S, Nihal M, Ahmad N (2008) Dose translation from animal to human studies revisited. FASEB J 22:659–661
Reepmeyer JC, Brower JF, Ye H (2005) Separation and detection of the isomeric equine conjugated estrogens, equilin sulfate and delta 8,9-dehydroestrone sulfate, by liquid chromatography–electrospray-mass spectrometry using carbon-coated zirconia and porous graphitic carbon stationary phases. J Chromatogr A 1083:42–51
Ruiz van Haperen VW, Veerman G, Boven E, Noordhuis P, Vermorken JB, Peters GJ (1994) Schedule dependence of sensitivity to 2′,2′-difluorodeoxycytidine (Gemcitabine) in relation to accumulation and retention of its triphosphate in solid tumour cell lines and solid tumours. Biochem Pharmacol 48:1327–1339
Ruiz van Haperen VW, Veerman G, Braakhuis BJ, Vermorken JB, Boven E, Leyva A, Peters GJ (1993) Deoxycytidine kinase and deoxycytidine deaminase activities in human tumour xenografts. Eur J Cancer 29A:2132–2137
Sigmond J, Honeywell RJ, Postma TJ, Dirven CM, de Lange SM, van der Born K, Laan AC, Baayen JC, Van Groeningen CJ, Bergman AM, Giaccone G, Peters GJ (2009) Gemcitabine uptake in glioblastoma multiforme: potential as a radiosensitizer. Ann Oncol 20:182–187
Sofuni A, Iijima H, Moriyasu F, Nakayama D, Shimizu M, Nakamura K, Itokawa F, Itoi T (2005) Differential diagnosis of pancreatic tumors using ultrasound contrast imaging. J Gastroenterol 40:518–525
Tanaka M, Javle M, Dong X, Eng C, Abbruzzese JL, Li D (2010) Gemcitabine metabolic and transporter gene polymorphisms are associated with drug toxicity and efficacy in patients with locally advanced pancreatic cancer. Cancer 116:5325–5335
Vainchtein LD, Rosing H, Thijssen B, Schellens JH, Beijnen JH (2007) Validated assay for the simultaneous determination of the anti-cancer agent gemcitabine and its metabolite 2′,2′-difluorodeoxyuridine in human plasma by high-performance liquid chromatography with tandem mass spectrometry. Rapid Commun Mass Spectrom 21:2312–2322
van Laarhoven HW, Klomp DW, Kamm YJ, Punt CJ, Heerschap A (2003) In vivo monitoring of capecitabine metabolism in human liver by 19fluorine magnetic resonance spectroscopy at 1.5 and 3 Tesla field strength. Cancer Res 63:7609–7612
Veltkamp SA, Hillebrand MJ, Rosing H, Jansen RS, Wickremsinhe ER, Perkins EJ, Schellens JH, Beijnen JH (2006) Quantitative analysis of gemcitabine triphosphate in human peripheral blood mononuclear cells using weak anion-exchange liquid chromatography coupled with tandem mass spectrometry. J Mass Spectrom 41:1633–1642
Veltkamp SA, Pluim D, van Tellingen O, Beijnen JH, Schellens JH (2008) Extensive metabolism and hepatic accumulation of gemcitabine after multiple oral and intravenous administration in mice. Drug Metab Dispos 36:1606–1615
Wang J, Lin T, Lai J, Cai Z, Yang MS (2009) Analysis of adenosine phosphates in HepG-2 cell by a HPLC-ESI-MS system with porous graphitic carbon as stationary phase. J Chromatogr B Analyt Technol Biomed Life Sci 877:2019–2024
Wolf W, Presant CA, Servis KL, el-Tahtawy A, Albright MJ, Barker PB, Ring R 3rd, Atkinson D, Ong R, King M et al (1990) Tumor trapping of 5-fluorouracil: in vivo 19F NMR spectroscopic pharmacokinetics in tumor-bearing humans and rabbits. Proc Natl Acad Sci USA 87:492–496
Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, Double JA, Everitt J, Farningham DA, Glennie MJ, Kelland LR, Robinson V, Stratford IJ, Tozer GM, Watson S, Wedge SR, Eccles SA, Committee of the National Cancer Research I (2010) Guidelines for the welfare and use of animals in cancer research. Br J Cancer 102:1555–1577
Xu Y, Keith B, Grem JL (2004) Measurement of the anticancer agent gemcitabine and its deaminated metabolite at low concentrations in human plasma by liquid chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 802:263–270
Acknowledgments
We thank Michael Williams (CRUK CRI) for advice on LC–MS/MS protocol, and Paul Mackin and staff of the CRI BRU, especially Steve Kupzac, for animal colony maintenance and assistance with animal studies. We thank Infinity Pharmaceuticals for providing dFdCTP. Funding for this work was provided by Cancer Research UK. The authors would like to particularly acknowledge the advice, support and encouragement given by Dr Merrill Egorin, who passed away earlier this year. Without his outstanding mentorship of a young investigator, this Pharmacology and Drug Development Group would not exist. He is sadly missed.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bapiro, T.E., Richards, F.M., Goldgraben, M.A. et al. A novel method for quantification of gemcitabine and its metabolites 2′,2′-difluorodeoxyuridine and gemcitabine triphosphate in tumour tissue by LC–MS/MS: comparison with 19F NMR spectroscopy. Cancer Chemother Pharmacol 68, 1243–1253 (2011). https://doi.org/10.1007/s00280-011-1613-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-011-1613-0