
Abstract
Purpose
To investigate whether clinically relevant levels of epigallocatechin gallate (EGCG, a component of green tea) or vitamin C (ascorbic acid) could antagonize bortezomib antitumor activity in CWR22 human prostate xenograft tumors.
Methods
The pharmacokinetics (PK) of EGCG and ascorbic acid were determined in immunocompromised mice and compared with concentrations measured in human PK studies of dietary supplements. Antitumor activity of bortezomib in combination with EGCG or ascorbic acid was determined using several dosing regimens to evaluate different target plasma concentrations of EGCG and ascorbic acid.
Results
Bortezomib dosed twice-weekly at 0.8 mg/kg IV demonstrated tumor growth inhibition (TGI) of 53.9–58.9%. However, when combined with EGCG such that the plasma concentrations of EGCG were >200 μM at the time of bortezomib dosing, all antitumor activity was abrogated (TGI = −17.7%). A lower concentration of EGCG (11–16 μM), which is severalfold higher than measured clinically in humans taking EGCG supplements (0.6–3 μM), was not antagonistic to bortezomib (TGI 63.5%). Pharmacodynamic studies of proteasome inhibition reflected these findings. Ascorbic acid (40 and 500 mg/kg PO daily) was evaluated under a similar study design and did not antagonize bortezomib antitumor activity (TGI 57.2 and 72.2%).
Conclusions
No antagonism of bortezomib is seen in preclinical in vivo experiments, where EGCG or ascorbic acid plasma concentrations are commensurate with dietary or supplemental intake. The data suggest that patients receiving bortezomib treatment do not need to avoid normal dietary consumption of green tea, vitamin C-containing foods, or EGCG or vitamin C dietary supplements.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The first-in-class proteasome inhibitor bortezomib (VELCADE®, PS-341) is approved in the United States for the treatment of patients with multiple myeloma (MM) and for the treatment of patients with mantle cell lymphoma who have received at least one prior therapy [1]. In the European Union, bortezomib is approved in combination with melphalan and prednisone for the treatment of patients with previously untreated MM who are not eligible for high-dose chemotherapy with bone marrow transplant, and as monotherapy for the treatment of progressive MM in patients who have received at least one prior therapy and who have already undergone or are unsuitable for bone marrow transplantation [2]. Bortezomib exerts its antitumor effects by reversibly blocking the activity of the 26S proteasome [3], a critical component of the ubiquitin–proteasome pathway that is responsible for degrading the majority of regulatory intracellular proteins [4]. This inhibition disrupts multiple downstream signaling pathways in cells, inducing apoptosis and inhibiting cell-cycle progression, angiogenesis, cell adhesion, and proliferation [3, 5].
Bortezomib is a modified dipeptidyl boronic acid (Supplemental Fig. 1), which when reconstituted consists of the mannitol ester in equilibrium with its hydrolysis product, the monomeric boronic acid [1]. It has been well documented that boronic acids form tight but reversible bonds with molecules possessing a diol group [6]. This reaction suggests that the activity of bortezomib (and other boronic acid-containing proteasome inhibitors) may be compromised by the presence of 1,2-diol-containing compounds such as epigallocatechin gallate (EGCG; Supplemental Fig. 1), a component of green tea, and vitamin C (ascorbic acid; Supplemental Fig. 1) [7–9], the hypothesis being that the latter compounds form complexes with the boronic acid moiety and thus prevent proteasome binding and inhibition.
Data from in vitro studies have indicated that high concentrations of EGCG and vitamin C have the potential to affect the cytotoxicity of bortezomib. Golden et al. demonstrated that EGCG at concentrations of 2.5 μM and higher antagonized the cytotoxic effect of 10 nM bortezomib on several MM cell lines [8]. In a glioblastoma cell line, 20 μM EGCG blocked proteasome inhibition by 20 nM bortezomib and prevented downstream events including accumulation of ubiquitinated proteins, endoplasmic reticulum stress, and apoptosis [8]. By evaluating six proteasome inhibitors with different chemical structures, Golden et al. demonstrated that EGCG antagonizes proteasome inhibitors containing boronic acid (bortezomib, MG-262, and PS-IX), but has no effect on proteasome inhibitors lacking boronic acid (MG-132, PS-I, and nelfinavir) [8]. This provides evidence that the direct chemical interaction of EGCG with the boronic acid-containing compounds is responsible for the antagonism, rather than placing the antagonism downstream of proteasome inhibition. Similar in vitro experiments identified comparable reductions in bortezomib activity when human cell lines were treated with bortezomib and ascorbic acid [7, 9]. For example, 30 μM ascorbic acid partially antagonized the activity of 5 and 10 nM bortezomib on RPMI8226 cells, while 250 μM ascorbic acid showed nearly complete antagonism of 5–10 nM bortezomib [7]. Thus, as with EGCG, high concentrations of ascorbic acid were required for antagonism.
The in vitro experimental conditions, characterized by the constant presence of bortezomib for 48–72 h in cell culture, are quite different from in vivo conditions. Pharmacokinetic studies in human plasma indicate that bortezomib displays a rapid initial distribution phase [10–12], with plasma concentrations dropping more than 10-fold within an hour post-dose [12]. This results in a narrow time window for any potential EGCG:bortezomib or EGCG:ascorbic acid complex formation to occur in the plasma, and cautions against direct application of the in vitro results to the clinical use of bortezomib.
In vivo studies in mouse xenograft models of human MM have also suggested that oral administration of EGCG at 25 or 50 mg/kg and vitamin C at 40 mg/kg/day inhibits the antitumor effects of bortezomib dosed intraperitoneally (i.p.) at 0.5 mg/kg and intravenously (IV) at 0.1 mg/kg, respectively [8, 9]. The findings led the authors to conclude that patients receiving bortezomib treatment should avoid consuming green tea or vitamin C [8, 9], both of which have purported benefits for patients undergoing cancer treatment. However, others have questioned the validity of the recommendation to avoid green tea, given the high concentration of EGCG required for antagonism of bortezomib in cultured cells, and the actual concentrations of EGCG and bortezomib determined in human pharmacokinetic studies [13].
Further evidence is clearly needed in order to provide definitive recommendations on EGCG or vitamin C consumption for patients receiving bortezomib. We therefore set out to better define the concentrations of EGCG and bortezomib showing antagonist effects in cultured cells. We also wanted to determine the doses of EGCG and vitamin C and, for EGCG, a route of administration (oral [PO], subcutaneous [SC], or intravenous [IV]), in mice that resulted in plasma concentrations above the ranges of those measured in human pharmacokinetic studies of EGCG and vitamin C dietary supplements. Oral dosing of EGCG in mice has previously been investigated [14] and results in lower maximal plasma concentrations of EGCG than oral dosing in humans [15, 16]. In humans taking 800 mg of EGCG supplements (equivalent to the amount of EGCG in approximately eight cups of green tea), the maximal plasma concentration was approximately 3 μM (1,522 ng/ml) when EGCG was taken after fasting and approximately 0.6 μM (294 ng/ml) when the supplements were taken with food [15]. In contrast, oral dosing of 50 and 80 mg/kg in mice resulted in maximal plasma concentrations of only 0.06–0.28 μM [14, 17]. Therefore, alternate methods of dosing in mice (SC and IV) were used to investigate the effects of EGCG in a higher concentration range that could be achieved in people taking EGCG supplements. For vitamin C, oral dosing in mice provides a reasonable match to oral dosing in humans in terms of the maximal concentrations, which have been reported to be approximately 150 μM following an oral dose of 1,250 mg [18].
Using this information, we investigated whether clinically relevant plasma concentrations of EGCG or vitamin C could antagonize the antitumor activity of bortezomib in CWR22 human prostate xenograft tumors, a well-validated bortezomib-responsive xenograft model in immunocompromised mice [19, 20]. Although this is not a xenograft model of MM, it is a suitable model for testing the hypothesis that a concentration-dependent interaction of bortezomib and 1,2-diol-containing compounds in the plasma can antagonize bortezomib activity by preventing its binding and inhibition of the proteasome in the target tumor tissue.
Materials and methods
In vitro bortezomib–EGCG combination studies
Serial dilutions of bortezomib (Millennium Pharmaceuticals, Inc., Cambridge, MA) (1:1.5) and EGCG (Sigma–Aldrich, St Louis, MO) (1:3) in RPMI growth media were added to a 384-well plate to create duplicate combination matrices in each plate; 6,000 RPMI-8226 cells were added to each well. After 48 h, cell viability was determined by ATP luminescence (Perkin-Elmer, Waltham, MA) using a Leadseeker imager (GE Life Sciences, Piscataway, NJ). Plotted data represent the average of six points (three plates).
Pharmacokinetic studies
EGCG and vitamin C (Sigma–Aldrich) were prepared fresh for all studies, and the appropriate amount of each compound was formulated with 0.9% saline.
For the EGCG studies, naïve female nude or naïve male CB-17 severe combined immunodeficient (SCID) mice, aged 5–8 weeks, from Charles River Laboratories, Wilmington, MA, received ~0.1 ml doses of EGCG administered PO (50 mg/kg), IV (50 and 100 mg/kg), or SC (50 mg/kg). Animals were dosed on the basis of an average body weight of 25 grams. For the vitamin C studies, male CB-17 SCID mice bearing CWR22 tumors (average body weight of 21 g) received approximately 0.1 ml doses of vitamin C administered PO (40 and 500 mg/kg). In all experiments described in this report, mice had access to food and water ad libitum and were housed and handled in accordance with the Guide for the Care and Use of Laboratory Animals, and Millennium Institutional Animal Care and Use Committee Guidelines.
Animals were anesthetized with CO2, and whole blood samples (~0.5 ml) were collected via cardiac puncture using a 25-gauge needle attached to a 1-ml syringe (Becton–Dickinson [BD], Franklin Lakes, NJ). Samples for EGCG quantification were collected from each animal at 5, 15, and 30 min and at 1, 2, 4, 8, 24, and 48 h post-dose. Samples for vitamin C quantification were collected from each animal at 0.5, 1, 2, 4, and 6 h post-dose. Whole blood samples for the EGCG or vitamin C studies were transferred to BD Microtainer® ethylenediaminetetraacetic acid (EDTA) blood collection tubes or lithium heparin blood collection tubes, respectively, then centrifuged at 10,000 rpm for 10 min at approximately 4°C. Ascorbic acid/EDTA or 10% metaphosphoric acid (MPA; Sigma–Aldrich) buffer solutions were added to the harvested plasma samples (1:1 ratio) to stabilize the EGCG or vitamin C, respectively, before tubes were frozen on dry ice, then stored at −80°C until analysis.
Concentrations of EGCG in prepared plasma samples were quantified using liquid chromatography with tandem mass spectrometry (LC/MS/MS). The LC/MS/MS system consisted of an API 4000 triple quadrupole mass spectrometer (AB Sciex, Foster City, CA) equipped with an Agilent 1100 high-performance liquid chromatography (HPLC) system (Agilent Technologies, Inc, Santa Clara, CA). Analytes were eluted from a Luna C 18 4.6 × 50 mm column (Phenomenex, Torrance, CA) using a linear gradient program consisting of 0.1% (volume-to-volume ratio [v:v]) formic acid (FA) (EMD Chemicals, Inc, Gibbstown, NJ) in water (mobile phase A) and 0.1% (v:v) FA in acetonitrile (mobile phase B) at a flow rate of 1.0 ml/min. EGCG was detected under a negative ion spray mode using the multiple reaction monitoring (MRM) of a mass transition pair at mass-to-charge ratio (m/z) of 457.2/169.0 for EGCG.
The LC/MS/MS system used to determine vitamin C concentrations in prepared plasma consisted of a AB SCIEX QTRAP® 5500 mass spectrometer (AB Sciex, Foster City, CA) equipped with an Shimadzu Prominence high-performance liquid chromatography (HPLC) system (Shimadzu Scientific Instruments, Columbia, MD). Analytes were eluted from a Luna 5 μM HILIC 200 Å, 150 × 4.6 mm column (Phenomenex, Torrance, CA) using an isocratic gradient (mobile phase B 30%) at a flow rate of 2 ml/min. Both mobile phases were prepared according to Phenomenex product guides using three solvent systems: solvent A was acetonitrile (EMD chemicals, Gibbstown, NJ, USA); solvent B was 100 mM ammonium acetate (Sigma–Aldrich), pH 5.8; and solvent C was pure water (Honeywell, Morristown, NJ). Mobile phase A consisted of the solvent ratio mixture A:B:C = 90:5:5. Mobile phase B consisted of the solvent ratio mixture A:B:C = 50:45:5. Ascorbic acid was detected under a negative ion spray mode using the MRM of a mass transition pairs at m/z of 175.0/114.8 for vitamin C.
In vivo antitumor activity studies
Male CB-17 SCID mice aged 5–8 weeks were inoculated SC via trocar in the flank with CWR22 human prostate tumor fragments. Tumor growth was monitored biweekly (BIW) using calipers, and the mean tumor volume was calculated using the formula (0.5 × [length × width2]). When the mean tumor volume reached approximately 150–250 mm3, animals were randomized into treatment groups (n = 9/group). Dosing regimens were selected using the pharmacokinetic profiles established for EGCG and vitamin C to obtain several target plasma concentrations of EGCG and vitamin C to evaluate for antagonism of bortezomib. In the study with IV administration of EGCG, mice were dosed BIW for 7 doses with vehicle (0.9% saline) IV, bortezomib 0.8 mg/kg IV, EGCG 50 mg/kg IV, or a combination of bortezomib 0.8 mg/kg IV and EGCG 50 mg/kg IV. In the combination group, EGCG was dosed first and bortezomib was dosed approximately 2 min after EGCG administration. In the study with SC administration of EGCG, mice received vehicle, bortezomib (0.8 mg/kg IV BIW) alone, EGCG (50 mg/kg SC BIW) alone, or a combination of bortezomib (0.8 mg/kg IV BIW) and EGCG (50 mg/kg SC BIW) for seven doses. There were three different dosing regimens for the combination groups. In the first two combination groups, EGCG was dosed first and bortezomib was dosed approximately 30 or 60 min after EGCG administration. In the third combination group, bortezomib was dosed first and EGCG was dosed approximately 60 min after bortezomib administration.
For the vitamin C studies, mice were dosed with vehicle (0.9% saline IV BIW), ascorbic acid (40 or 500 mg/kg PO QD) alone, bortezomib (0.8 mg/kg IV BIW) alone, or a combination of ascorbic acid (40 or 500 mg/kg PO QD) and bortezomib (0.8 mg/kg IV BIW) for 22 days. In the combination groups, bortezomib was administered 1 h after ascorbic acid administration.
Antitumor activity was determined by calculating the percent tumor growth inhibition (TGI; [mean tumor volume of the control group—mean tumor volume of a treated group]/mean tumor volume of the control group) on days 22 or 25. Combination treatments were also analyzed at the end of study to evaluate antagonism between EGCG and bortezomib.
Pharmacodynamic studies
Activity of the 20S proteasome in blood and xenograft tumors was measured by an enzymatic assay, and downstream consequences of proteasome inhibition in CWR22 tumors were detected as the accumulation of proteasome substrates and the upregulation of markers of endoplasmic reticulum stress and the unfolded protein response, including GADD34 and ATF3, as previously described [20]. For phospo-β-catenin (Ser33/Ser37/Thr41), primary antibody was Cell Signaling #9561.
Statistical analysis
The differences in the tumor growth trends over time between pairs of treatment groups were assessed using linear mixed effects regression models as previously described [20]. Drug combinations were assessed for synergy or antagonism using observed AUC (area under the curve) values. The change in AUC (ΔAUC) relative to the control was calculated for both single-agent treatment groups and the combination group. The interaction between the two compounds was then assessed by comparing the ΔAUC observed in the combination group to the sum of the changes observed in both single-agent groups. Statistically significant negative (less than zero) synergy scores indicated a synergistic combination. Statistically significant positive (greater than zero) synergy scores indicated a sub-additive or antagonistic combination. Scores of zero and scores that were not statistically significant were considered additive (neither synergistic nor antagonistic). All P values < 0.05 were considered statistically significant.
Results
In vitro combination experiments with EGCG
Experiments in the RPMI8226 MM cell line in a matrix format (Fig. 1) showed that a substantial shift in the potency of bortezomib in cell viability experiments required a high concentration of EGCG (>1 μM). An EGCG concentration of 1.3 μM caused a minor shift in the IC50 of bortezomib from 6.1 to 9.3 nM, whereas 3.8 μM EGCG caused a 2.5-fold increase in the IC50 of bortezomib to 15 nM (Fig. 1). At the highest concentration of bortezomib used in these experiments (40 nM), EGCG concentrations ≥11 μM were required to even partially overcome the cytotoxicity of bortezomib. EGCG 100 μM was cytotoxic on its own, and so antagonism of bortezomib could not be evaluated at this concentration.

In vitro antagonism of bortezomib by high concentrations of EGCG in RPMI-8226 cultured cells. a Matrix format for luminescent ATP-lite cell viability assay. b Data in graph represent an average of six points (three plates with duplicates on each plate)
Pharmacokinetic experiments with EGCG
A single oral dose of EGCG 50 mg/kg in naïve female nude mice resulted in a mean plasma EGCG C max value of 76.3 nM. Mean plasma T max was 30 min (Fig. 2a). These data are consistent with previously published findings in which 50 mg/kg EGCG administered intragastrically resulted in a plasma concentration of approximately 60 nM at 50 min [17].

Pharmacokinetics of EGCG in mice following a oral, b subcutaneous, and c intravenous dosing
Because the EGCG concentrations achieved by oral dosing were so low compared with the levels detected in human pharmacokinetic studies of EGCG supplements, we characterized IV and SC dosing routes of EGCG in mice. In two studies in naïve male CB17 SCID mice, SC EGCG 50 mg/kg resulted in mean C max values of 33.7 and 26.0 μM, with a mean plasma T max of 5 min in both studies. EGCG plasma concentrations at 30 min post-dose were 16.2 and 11.3 μM, respectively, and dropped to 8.4 and 3.9 μM by 1 h, and 2.8 and 2.5 μM by 2 h, respectively (Fig. 2b). IV administration of EGCG at 50 and 100 mg/kg in naïve male CB17 SCID mice resulted in higher mean plasma levels of EGCG than SC dosing, producing mean measured plasma C max values of 224 and 240 μM (Fig. 2c). The earliest IV sampling times in these studies were 2 and 5 min, respectively. Plasma levels of EGCG declined rapidly after IV dosing and were <20 μM within 30 min of dosing.
Antitumor activity in EGCG/bortezomib combination experiments
Several target plasma concentrations of EGCG were evaluated for antagonism of bortezomib; data from the pharmacokinetic studies provided information on EGCG plasma concentrations at various time points post-SC/IV administration at which time bortezomib was subsequently administered (Table 1). The effect of dosing bortezomib first, followed by SC EGCG dosing 1 h later, was also investigated. The pharmacokinetic profile of bortezomib in mice has a rapid distribution phase, as is the case in humans [20]. Therefore, by 1 h post-dose, very little bortezomib is present in the plasma to potentially interact with EGCG.
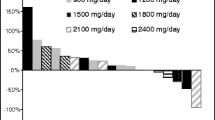
Single-agent bortezomib 0.8 mg/kg IV demonstrated a TGI of 53.9–58.9% versus the control group (Fig. 3; P < 0.001) in CWR22 xenograft-bearing mice, which was consistent with reports of significant antitumor activity seen in previous studies [19, 20]. IV administration of EGCG 50 mg/kg alone did not show any antitumor activity versus control (Fig. 3a; TGI = −15.3%; P > 0.05). EGCG 50 mg/kg IV administered 2 min prior to bortezomib 0.8 mg/kg IV resulted in no antitumor activity (TGI = −17.7%, P > 0.05 versus control), demonstrating antagonism between EGCG and bortezomib when EGCG levels were >200 μM at the time of bortezomib dosing (Fig. 3a). In studies of SC EGCG administration (Fig. 3b), 50 mg/kg EGCG as a single agent did not result in antitumor activity (TGI = 3.6%; P > 0.05 versus control). However, SC EGCG 50 mg/kg followed 30 or 60 min later by bortezomib 0.8 mg/kg IV exhibited significant antitumor activity with a TGI of 63.5 and 46.3%, respectively, compared with controls (P < 0.001). Similarly, bortezomib 0.8 mg/kg IV followed 60 min later by SC EGCG 50 mg/kg also exhibited significant antitumor activity versus controls (TGI = 61.3%; P < 0.001). Thus, there was no antagonism between EGCG and bortezomib when EGCG levels were 16 μM and lower at the time of bortezomib dosing, or when bortezomib was administered 1 h before EGCG (P > 0.05 versus controls for each combination). A repeat of this study confirmed these findings (data not shown).

Evaluation of EGCG antagonism of bortezomib activity in CWR22 xenograft-bearing mice with EGCG administered a intravenously or b subcutaneously
Pharmacodynamic effects of EGCG/bortezomib combination treatment
Pharmacodynamic studies were conducted to determine whether EGCG showed a concentration-dependent ability to antagonize bortezomib-induced proteasome inhibition in blood or tumor tissue. Based on previous PD studies with bortezomib, a time point of 4 h post-dosing was chosen for sample collection. An enzymatic assay for proteasome activity and Western blot-based assays for downstream consequences of proteasome inhibition (accumulation of the proteasome substrate phospho-β-catenin [Ser33/Ser37/Thr41] and upregulation of ATF3 and GADD34, markers of the unfolded protein response) were both employed. No inhibition of proteasome activity compared with vehicle controls was seen with EGCG 50 mg/kg IV alone (Fig. 4a). Four hours after IV administration of bortezomib 0.8 mg/kg, mean 20S proteasome inhibition was 44% in blood and 52% in tumor. This proteasome inhibition was blunted by the combination of EGCG 50 mg/kg IV followed 2 min later by bortezomib 0.8 mg/kg IV, which resulted in only 25 and 33% proteasome inhibition in blood and tumor, respectively, at 4 h post-administration. These results indicate partial antagonism of proteasome inhibition by IV dosing of EGCG, which is further reflected in the analysis of downstream pharmacodynamic markers and ultimately in the antagonism of antitumor activity in this combination regimen. Bortezomib 0.8 mg/kg IV results in elevated levels of phospho-β-catenin, GADD34, and ATF3 compared with vehicle controls (Fig. 4b). EGCG 50 mg/kg IV on its own does not change levels of these pharmacodynamic markers. However, EGCG is able to prevent the elevation of phospho-β-catenin, GADD34, and ATF3 in tumors from mice dosed with the combination schedule of EGCG 50 mg/kg IV followed 2 min later by bortezomib 0.8 mg/kg IV.

Antagonism of the pharmacodynamic effects of bortezomib with intravenous administration (a, b) but not subcutaneous administration (c, d) of EGCG demonstrates concentration-dependence of antagonism. a, c Blood and tumor proteasome inhibition 4 h post-dose measured by enzymatic assay of β5 specific activity. b, d Western blot of xenograft tumors to measure proteasome substrate [phospho-β-catenin (Ser33/Ser37/Thr41)] and pathway markers of ER stress response (ATF3 and GADD34) in the same tumor samples used in a and c
The antagonistic effect of EGCG on pharmacodynamic markers of proteasome inhibition appears to be concentration dependent, because EGCG 50 mg/kg SC followed 30 or 60 min later by bortezomib 0.8 mg/kg IV had little or no antagonistic effect on proteasome inhibition in blood or tumor (Fig. 4c). In this experiment, bortezomib 0.8 mg/kg IV alone resulted in mean proteasome inhibition of 59% in blood, compared with 54 and 56% using the combination schedules, respectively. Proteasome inhibition in tumor was >50% in all groups, consistent with the findings from the antitumor activity studies. Reflecting these results, phospho-β-catenin, GADD34, and ATF3 elevations were detected in all groups, with no substantial difference between the single-agent bortezomib and EGCG/bortezomib combination groups (Fig. 4d).
Pharmacokinetic experiments with ascorbic acid
Levels of endogenous ascorbic acid measured in naïve or CWR22 tumor-bearing male CB-17 SCID mice were in the range 48.3–59.2 μM, consistent with previously reported mouse [21] and human [9, 18, 22] endogenous plasma levels. Oral dosing of ascorbic acid at 40 and 500 mg/kg results in the elevation of plasma concentrations above the endogenous level by 30 min post-dose, and the elevated level is maintained for at least 6 h. Following a single oral dose of ascorbic acid 40 mg/kg, mean C max was 72.8 μM in the tumor-bearing mice at T max of 4 h (Fig. 5a). Following a single oral dose of ascorbic acid 500 mg/kg, mean C max was 193 μM at T max of 2 h (Fig. 5a). The pharmacokinetic profile in nontumor-bearing mice was similar (data not shown).

a Pharmacokinetics of orally administered ascorbic acid in male CB-17 SCID mice and b the effect of concomitant administration of ascorbic acid on the antitumor activity of bortezomib in CWR22 xenograft-bearing mice
Antitumor activity in bortezomib/ascorbic acid combination treatment
Based on pharmacokinetic findings, a 1 h interval between ascorbic acid and bortezomib administration was selected for antitumor activity experiments. The plasma levels at this time point following the 500 mg/kg dose are in line with human ascorbic acid exposures in subjects who had taken 1–1.25 g of dietary supplements [9, 18].
Ascorbic acid 40 or 500 mg/kg alone did not exhibit any antitumor activity (TGI = −24.4% and −15.5%, respectively; P > 0.05 versus controls), while bortezomib 0.8 mg/kg IV alone had significant antitumor activity compared with controls (TGI = 62.8%; P < 0.001) (Fig. 5b). Ascorbic acid 40 or 500 mg/kg in combination with bortezomib also exhibited significant antitumor activity compared with controls (TGI = 57.2 and 72.2%, respectively; P < 0.001). No antagonism was seen between ascorbic acid and bortezomib in any of the combination groups (P > 0.05).
Discussion
The findings from our studies reported herein provide important new evidence regarding the potential effect of EGCG or vitamin C consumption on bortezomib activity in patients receiving therapy. Our results demonstrate that no antagonistic interactions between EGCG and bortezomib, or ascorbic acid and bortezomib, are seen in preclinical models at plasma concentrations of EGCG and ascorbic acid commensurate with human dietary or supplemental intake.
Our in vitro EGCG experiments were intended to characterize the minimum concentration at which antagonism was detected. Golden et al. demonstrated that the activity of bortezomib was affected by EGCG in vitro, but the lowest EGCG concentration used was 2.5 μM [8]. Our studies showed that EGCG concentrations of up to 0.4 μM had no impact on the cytotoxicity of bortezomib in RPMI8226 cells. The IC50 of bortezomib did double (from 7 to 15 nM) with an increase in EGCG concentration from 0.4 to 3.8 μM, indicating partial antagonism. Importantly, even at an EGCG concentration of 3.8 μM, there was no antagonism of 40 nM bortezomib. Antagonism of 40 nM bortezomib only became evident once EGCG concentrations reached 11 μM and above. Thus, substantial changes in the in vitro potency of bortezomib only occurred at micromolar concentrations of EGCG. The maximal plasma concentration of EGCG measured in people taking 800 mg supplements (equivalent to the amount of EGCG in eight cups of green tea) is approximately 3 μM, but this is reached only when the supplements are taken after fasting [15, 16].
Replicating these high concentrations in mice required IV or SC dosing of EGCG. Maximal EGCG concentrations after IV and SC dosing at 50 mg/kg were much higher than human plasma C max values, at >200 and >25 μM, respectively, and plasma concentration remained higher than 3 μM for up to almost 2 h. In contrast, oral dosing at 50 mg/kg, the dose used in a previous study evaluating EGCG antagonism of bortezomib [8], resulted in maximal plasma concentrations of <0.1 μM in our studies and in other mouse pharmacokinetic studies [17]. These pharmacokinetic findings informed the selection of several dosing regimens providing a range of EGCG plasma concentrations for investigation of antagonistic effects on bortezomib antitumor activity. Data from our experiments in CWR22 xenograft-bearing mice showed significant antitumor activity with single-agent bortezomib that was consistent with previous studies [19, 20], with this activity being antagonized by the presence of EGCG at very high concentrations (224 μM, achieved via IV administration 2 min before bortezomib dosing). This concentration is approximately 80-fold higher than the 3 μM maximal concentration observed in human pharmacokinetic studies of EGCG dietary supplements taken while fasting. In contrast, results from studies using SC administration of EGCG, in which EGCG was present at concentrations up to 16 μM at the time of bortezomib dosing, demonstrated that EGCG was not antagonistic to bortezomib even at concentrations up to 4- to 5-fold higher than maximal concentrations of EGCG observed in human pharmacokinetic studies [15, 16].
Our pharmacodynamic analyses of proteasome inhibition by bortezomib in the presence and absence of EGCG showed findings consistent with the results of the antitumor activity studies, with antagonism of pharmacodynamic effects observed only with the very high concentrations of EGCG (>200 μM) achieved with the combination of EGCG 50 mg/kg IV followed 2 min later by bortezomib 0.8 mg/kg IV. We hypothesize that ester formation in blood at high EGCG concentrations prevents distribution of free bortezomib to the tumor and therefore prevents proteasome inhibition. The markers reflecting the downstream consequences of proteasome inhibition (accumulation of phospho-β-catenin, GADD34, and ATF3) may be the most reliable pharmacodynamic markers in this context, because they cannot be influenced by ex vivo changes in the EGCG–bortezomib complex. For example, if EGCG–bortezomib ester were present in a blood sample but hydrolyzed during sample processing, then newly liberated bortezomib could inhibit the proteasome ex vivo, thus resulting in an overestimate of proteasome inhibition. However, the consistent results of our multiple assays tracking different stages in the proteasome inhibition pathway, from enzyme inhibition to endoplasmic reticulum stress response, provide confidence in the robustness of our findings.
Our studies with ascorbic acid and bortezomib found no antagonistic effects on bortezomib antitumor activity at plasma concentrations of ascorbic acid recorded in pharmacokinetic studies in humans taking oral vitamin C supplements [9, 18]. While studies in cell culture have indicated that high concentrations of ascorbic acid (250 μM) can almost completely antagonize the ability of 5–10 nM bortezomib to inhibit the proteasome and kill cells [7, 9], in vivo plasma concentrations of up to 200 μM did not affect the antitumor activity of bortezomib in our xenograft experiment. This highlights the difficultly of extrapolating from in vitro results and the importance of evaluating drug interactions in a physiologically relevant system.
In conclusion, while the findings of our studies are consistent with a concentration-dependent antagonism of the antitumor activity of bortezomib by EGCG in CWR22 xenograft-bearing mice, antagonism is only seen when very high levels of EGCG (80-fold greater than those reported in human subjects taking EGCG supplements while fasting) are present at the time of bortezomib dosing. Plasma concentrations of EGCG and ascorbic acid reported in human subjects taking EGCG or vitamin C supplements show no antagonism to the antitumor activity of bortezomib in CWR22 xenograft-bearing mice, and therefore there appears no need for patients receiving bortezomib therapy to avoid normal dietary consumption of green tea, vitamin C-containing foods, or EGCG or vitamin C dietary supplements.
References
Millennium Pharmaceuticals Inc (2009) VELCADE® (bortezomib) for Injection. Prescribing information. Cambridge, MA, USA Issued December 2009, version 10
Janssen-Cilag International NV (2010) VELCADE® (bortezomib). Summary of product characteristics. Beerse, Belgium Issued July 2010
Adams J (2004) The development of proteasome inhibitors as anticancer drugs. Cancer Cell 5:417–421
Ciechanover A (1998) The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J 17:7151–7160
Boccadoro M, Morgan G, Cavenagh J (2005) Preclinical evaluation of the proteasome inhibitor bortezomib in cancer therapy. Cancer Cell Int 5:18
Springsteen G, Wang BA (2002) Detailed examination of boronic acid-diol complexation. Tetrahedron 58:5291–5300
Zou W, Yue P, Lin N et al (2006) Vitamin C inactivates the proteasome inhibitor PS-341 in human cancer cells. Clin Cancer Res 12:273–280
Golden EB, Lam PY, Kardosh A et al (2009) Green tea polyphenols block the anticancer effects of bortezomib and other boronic acid-based proteasome inhibitors. Blood 113:5927–5937
Perrone G, Hideshima T, Ikeda H et al (2009) Ascorbic acid inhibits antitumor activity of bortezomib in vivo. Leukemia 23:1679–1686
Papandreou CN, Daliani DD, Nix D et al (2004) Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. J Clin Oncol 22:2108–2121
Moreau P, Coiteux V, Hulin C et al (2008) Prospective comparison of subcutaneous versus intravenous administration of bortezomib in patients with multiple myeloma. Haematologica 93:1908–1911
Reece DE, Sullivan D, Lonial S et al (2010) Pharmacokinetic and pharmacodynamic study of two doses of bortezomib in patients with relapsed multiple myeloma. Cancer Chemother Pharmacol e-pub ahead of print. doi:10.1007/s00280-010-1283-3
Shah JJ, Kuhn DJ, Orlowski RZ (2009) Bortezomib and EGCG: no green tea for you? Blood 113:5695–5696
Lambert JD, Lee MJ, Lu H et al (2003) Epigallocatechin-3-gallate is absorbed but extensively glucuronidated following oral administration to mice. J Nutr 133:4172–4177
Chow HH, Hakim IA, Vining DR et al (2005) Effects of dosing condition on the oral bioavailability of green tea catechins after single-dose administration of Polyphenon E in healthy individuals. Clin Cancer Res 11:4627–4633
Chow HH, Cai Y, Hakim IA et al (2003) Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals. Clin Cancer Res 9:3312–3319
Lambert JD, Lee MJ, Diamond L et al (2006) Dose-dependent levels of epigallocatechin-3-gallate in human colon cancer cells and mouse plasma and tissues. Drug Metab Dispos 34:8–11
Padayatty SJ, Sun H, Wang Y et al (2004) Vitamin C pharmacokinetics: implications for oral and intravenous use. Ann Intern Med 140:533–537
Williamson MJ, Silva MD, Terkelsen J et al (2009) The relationship among tumor architecture, pharmacokinetics, pharmacodynamics, and efficacy of bortezomib in mouse xenograft models. Mol Cancer Ther 8:3234–3243
Kupperman E, Lee EC, Cao Y et al (2010) Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res 70:1970–1980
Tsao CS, Leung PY, Young M (1987) Effect of dietary ascorbic acid intake on tissue vitamin C in mice. J Nutr 117:291–297
Lykkesfeldt J, Loft S, Nielsen JB, Poulsen HE (1997) Ascorbic acid and dehydroascorbic acid as biomarkers of oxidative stress caused by smoking. Am J Clin Nutr 65:959–963
Acknowledgments
The authors would like to thank John Donovan, Khristofer Garcia, Michael Pickard, Ben Knight, Ruju Srivastava, Karthik Venkatakrishnan, and Dixie-Lee Esseltine from Millennium Pharmaceuticals, Inc. for their contributions. The authors also acknowledge the editorial assistance of Steve Hill, a medical writer with FireKite, during the development of this manuscript, which was funded by Millennium Pharmaceuticals, Inc. These studies were funded by Millennium Pharmaceuticals, Inc.
Conflict of interest
B. B., L. X., M. J., C. T., J. Y., P. H., P. F., L. D., M. M., C. C., J. B., E. K., and A. B. are employees of Millennium Pharmaceuticals, Inc., and J. M. is an employee of Janssen Pharmaceutica and holds stocks in Johnson & Johnson.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
280_2011_1591_MOESM1_ESM.tif
Supplemental Fig. 1 Chemical structures of (A) bortezomib, (B) EGCG, (C) vitamin C, and (D) representative structure of a bortezomib–EGCG ester. Supplementary material 1 (TIFF 66 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bannerman, B., Xu, L., Jones, M. et al. Preclinical evaluation of the antitumor activity of bortezomib in combination with vitamin C or with epigallocatechin gallate, a component of green tea. Cancer Chemother Pharmacol 68, 1145–1154 (2011). https://doi.org/10.1007/s00280-011-1591-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-011-1591-2