
Abstract
Purpose
Sorafenib, a multikinase inhibitor of Raf and several growth factor receptors, is under investigation in combination with dacarbazine, a commonly used chemotherapeutic agent for the treatment of many cancers. The current phase I study investigates the effects of sorafenib on the pharmacokinetic (PK) profile of dacarbazine and its metabolite 5-amino-imidazole-4-carboxamide (AIC). (AIC is formed in amounts equimolar to the active alkylating moiety, methane diazohydroxide, which is undetectable by known validated assays.)
Methods
Patients with advanced solid tumors received intravenous dacarbazine 1,000 mg/m2 on day 1 of a 21-day cycle to evaluate the PK of dacarbazine alone. Sorafenib 400 mg was administered twice daily continuously starting at day 2 of cycle 1. The PK of dacarbazine in the presence of sorafenib was assessed on day 1 of cycle 2. Sorafenib PK was also assessed at steady state.
Results
PK data were available for 15 of 23 patients. With concomitant administration of sorafenib, the mean AUC and C max values of dacarbazine were reduced by 23 and 16%, respectively. Mean AUC and C max values of AIC were increased by 41 and 45%, respectively, with individual increases of up to 106 and 136%, respectively. The apparent terminal half-lives of the two compounds were not significantly influenced by sorafenib. Based on coefficients of variation, the AUC and C max values for sorafenib and its three metabolites were highly variable with dacarbazine coadministration.
Conclusions
Concomitant administration of sorafenib and dacarbazine as described above may result in decreased dacarbazine exposure but increased AIC exposure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sorafenib is a multikinase inhibitor of Raf, vascular endothelial growth factor (VEGF) receptors, and platelet-derived growth factor receptors [1, 2]. It has been approved as a single agent by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the treatment of advanced renal cell carcinoma and unresectable hepatocellular carcinoma. In addition, sorafenib is being tested in combination with other agents in a variety of advanced solid tumors such as melanoma, breast cancer, renal cell carcinoma, hepatic cancer, and non-small-cell lung cancer [3–16]. Dacarbazine is the most commonly used FDA- and EMA-approved chemotherapeutic agent for the treatment of advanced melanoma. Dacarbazine is metabolized by various cytochrome P450 (CYP) isoenzymes such as CYP1A2, CYP1A1, and CYP2E1 [17]. Sorafenib is primarily metabolized in the liver by CYP3A4 [18]. Therefore, concomitant sorafenib administration is not expected to affect dacarbazine metabolism, and the likelihood of a pharmacokinetic (PK) drug–drug interaction between sorafenib and dacarbazine is low.
An earlier phase I study estimated that the maximum-tolerated dose of sorafenib in combination with dacarbazine 1,000 mg/m2 was 400 mg twice daily (the standard single-agent doses for each agent) [19]. The primary objective of this study was to evaluate the PK profiles of dacarbazine with and without concomitant sorafenib under steady state conditions. A secondary objective was to determine the steady-state PK profiles of sorafenib and its metabolites BAY 67-3472 (M2), BAY 43-9007 (M4), and BAY 68-7769 (M5) in the presence of dacarbazine. This paper reports on PK and safety data. A separate manuscript (in preparation) [20] reports on efficacy and functional analysis using dynamic contrast-enhanced ultrasonography (DCE-US) representing the blood volume and microarray analyses of gene expression obtained in sequential tumor biopsies.
Patients and methods
Patients
Patients with metastatic, histologically confirmed solid tumors were included in this study. Eligible patients had at least one lesion that could be accurately and serially measured per Response Evaluation Criteria in Solid Tumors (RECIST) guidelines [21], were ≥18 years of age with an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, had adequate bone marrow, liver, and renal functions, and had a life expectancy of at least 12 weeks. Patients were excluded if they had previous or concurrent cancer that was distinct from the tumor being evaluated in this study, unless the other cancer was curatively treated more than 3 years prior to study entry; clinically evident congestive heart failure; cardiac arrhythmias; active coronary heart disease or ischemia; uncontrolled hypertension; active clinically serious infections; or active brain metastases. Anticancer chemotherapy, immunotherapy, or vaccine therapy was not permitted during or within 30 days prior to the start of study treatment. Prior treatment with inhibitors of Raf, VEGF, or mTOR signaling pathways or farnesyl transferase inhibitors was not permitted.
Study design
This phase I, single-center, open-label, uncontrolled study was conducted in France between September 2005 (date of first patient first visit) and August 2006 (data cutoff date). On day 1 of a 21-day cycle, dacarbazine 1,000 mg/m2 was administered as a 1-h infusion. Sorafenib 400 mg was administered twice daily continuously starting on day 2 of cycle 1. Toxicity-related dose modifications of sorafenib and dacarbazine were performed in accordance with protocol-specified guidelines. Treatment continued until the occurrence of unacceptable toxicity, tumor progression, or death. Sorafenib tablets were supplied by Bayer HealthCare AG (Leverkusen, Germany); dacarbazine was supplied by Faulding Pharmaceuticals SA (Asnieres, France). The study was conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonization Good Clinical Practice guidelines, the EU-Directive 2001/20/EC, and local applicable laws. All patients provided signed informed consent before starting study treatment.
Study outcomes
The primary endpoint was the determination of the PK profile of dacarbazine with and without sorafenib. Secondary endpoints included evaluation of the PK profile of sorafenib in the presence of dacarbazine, safety and efficacy of the combination treatment, novel biomarker analyses using DCE-US, and gene profile analyses. This paper reports on PK and safety data; the other results are reported in a separate manuscript (in preparation) [20].
Pharmacokinetic variables and sampling schedules
Dacarbazine is a prodrug from which the active alkylating moiety methane diazohydroxide is formed by metabolization. In this metabolic process, the inactive metabolite 5-amino-imidazole-4-carboxamide (AIC) is formed in equimolar quantities as methane diazohydroxide, which cannot be analytically measured [17]. Therefore, in addition to the PK of dacarbazine, we also studied the PK of AIC to understand the changes in the exposure of the active alkylating moiety in the presence of sorafenib.
On day 1 of cycle 1, plasma samples were obtained prior to dacarbazine administration and at 0.5, 1.0, 1.25, 1.5, 2.0, 4.0, 8.0, 12.0, and 24.0 h following dacarbazine administration to assess the PK of dacarbazine and AIC in the absence of sorafenib. On day 1 of cycle 2, samples were collected from the same patients to assess the PK of dacarbazine in the presence of sorafenib at the same time points as above. Additional samples were collected on day 1 of cycle 2 prior to sorafenib dosing and at 0.5, 1.0, 2.0, 4.0, 8.0, 10.0, and 12.0 h thereafter to evaluate the PK profile of sorafenib and its main metabolites, M2, M4, and M5, in the presence of dacarbazine. Samples for dacarbazine measurements were stored at or below −70°C; samples for sorafenib measurements were stored below −15°C. Stability data indicated that all analytes were stable during analysis.
The following PK variables were determined for dacarbazine and AIC on day 1 of cycle 1 and day 1 of cycle 2: area under the plasma concentration–time curve (AUC) from zero to infinity after a single dose [AUC(0–inf)], AUC from time zero to the last data point [AUC(0–tn)], maximum concentration of drug in plasma (C max), time to reach maximum drug concentration in plasma (t max), and apparent terminal half-life (t 1/2). The following variables were determined for sorafenib, M2, M4, and M5 on day 1 of cycle 2: AUC from time 0–12 h after dose at steady state [AUC(0–12)ss], AUC(0–12)ss normalized with respect to dose (in mg) per kg body weight [AUC(0–12)ss,norm], C max at steady state (C max,ss), C max,ss normalized with respect to dose (in mg) per kg body weight (C max,ss,norm), and t max at steady state (t max,ss).
Pharmacokinetic assay methods and analyses
All analytes in plasma samples were quantified using a fully validated liquid chromatography–tandem mass spectrometry assay method with a lower limit of quantification of 40.6 μg/l for dacarbazine, 40.7 μg/l for AIC, and 0.01 mg/l for sorafenib and its metabolites. The assay for each sample set was performed once. Mean inter-assay precision ranges as determined by analysis of quality control samples were 7.0–8.7% for dacarbazine, 2.8–9.3% for AIC, 1.5–12.4% for sorafenib, 2.6–4.5% for M2, 3.9–5.2% for M4, and 3.2–6.7% for M5. Corresponding mean inter-assay accuracy ranges were 97.1–104.7% for dacarbazine, 99.3–105.3% for AIC, 102.3–107.0% for sorafenib, 97.9–103.2% for M2, 98.1–100.3% for M4, and 95.5–101.0% for M5. The parameters AUC, AUC(0–tn), and C max of dacarbazine and AIC were analyzed after logarithmic transformations applying an analysis of variance assuming a log-normal distribution.
Safety
Safety was evaluated in all patients who had received at least one dose of either study treatment. Safety was assessed through observed adverse events (AEs) and results of physical examination, laboratory tests, and vital signs measurement. Safety assessment took place at baseline and weekly starting from day 1 of cycle 1. AEs were coded and graded using version 3.0 of the National Cancer Institute Common Toxicity Criteria.
Determination of sample size
As this was a descriptive PK and safety phase I study, no formal sample size estimation was performed. The planned enrollment of approximately 25 patients was based on the requirements of relevant PK data sampling in a phase I trial.
Results
The study enrolled 24 patients, one of whom developed progressive disease before study treatment. The remaining 23 patients underwent treatment and were evaluable for safety analysis. Twenty-one patients (91%) discontinued treatment owing to progressive disease and two patients (9%) discontinued owing to AEs. Complete PK data were available for 15 patients. The other 8 patients had incomplete or no PK data on sorafenib and/or dacarbazine and were not included in the PK evaluation. The baseline characteristics of patients are reported in Table 1. Detailed dosing and drug exposure data are reported in the supplementary table.
Pharmacokinetics
The 15 patients included in the PK analysis did not undergo any dose modifications during the PK evaluation period. For 13 of the 15 patients, the PK profile of dacarbazine was determined on day 1 of cycle 1 in the absence of sorafenib and repeated on day 1 of cycle 2 following a 20-day treatment period of sorafenib, as planned. For two patients, the second dacarbazine PK sampling was done on day 1 of cycle 3 and day 1 of cycle 6. For all the PK analyses, these were combined with data obtained from other patients on day 1 of cycle 2. Sorafenib PK sampling was performed in all patients during the second dacarbazine PK sampling.
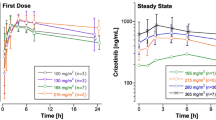
Geometric mean plasma concentration–time data for dacarbazine, AIC, and sorafenib are shown in Fig. 1a–c, respectively. While plasma concentrations of dacarbazine were slightly lower in cycle 2 at 4 h after start of infusion and following times when compared with those in cycle 1, the corresponding mean plasma concentrations of AIC were distinctly higher in cycle 2 compared with cycle 1. The apparent t 1/2 of either dacarbazine or AIC was not altered on concomitant administration of sorafenib. Mean plasma concentrations of sorafenib ranged between 1.7 and 3.0 mg/l.

a Plasma concentrations (geometric means/geometric standard deviation) of dacarbazine after a 1-h intravenous infusion of 1,000 mg/m2 dacarbazine without (cycle 1) or with (cycle 2) concomitant multiple oral doses of 400 mg bid sorafenib (n = 15) b Plasma concentrations (geometric means/geometric standard deviation) of AIC after a 1-h intravenous infusion of 1,000 mg/m2 dacarbazine without (cycle 1) or with (cycle 2) concomitant multiple oral doses of 400 mg bid sorafenib (n = 15) c Plasma concentrations (geometric means/geometric standard deviation) of sorafenib after multiple oral doses of 400 mg bid sorafenib and following a concomitant 1-h intravenous infusion of 1,000 mg/m2 dacarbazine on day 1 of cycle 2 (n = 15)
Table 2 summarizes the PK results for dacarbazine and AIC. While the mean AUC and C max of dacarbazine were reduced by 23 and 16%, respectively, mean AUC and C max of AIC were increased by 41 and 45%, respectively, with individual increases of up to 106 and 136%, respectively. The apparent t 1/2 of either compound was not significantly influenced by concomitant administration of sorafenib. Table 3 reports the steady state PK data for sorafenib and its metabolites. Sorafenib contributes approximately 83% to the sum of AUC(0–12)ss values, while the metabolites contribute approximately 9% (M2), 4% (M4), and 3% (M5). From the values of the coefficients of variation, it is evident that the PK parameters of sorafenib and its metabolites showed a high degree of variability.
Safety
Overall, toxicities were manageable, with the vast majority of grade 3/4 AEs improving or resolving upon transient study drug dose reduction or discontinuation. No patient died of treatment-related causes; 10 patients (43.5%) died of progressive disease, 5 within 30 days after the last dose of a study drug and 5 thereafter.
Table 4 summarizes the incidence of treatment-emergent AEs related to one or both of the study drugs and affecting at least two patients. The most common grade 3/4 toxicities included amylase or lipase elevation, which was attributed to sorafenib and asymptomatic in all cases. The hematologic toxicities were attributed to both study drugs. The most common drug-related toxicities of any grade were fatigue, nausea, diarrhea, hand–foot skin reaction, and rash/desquamation, each affecting a minimum of just under half of the patients. The most common categories of toxicities of any grade were gastrointestinal (19 patients [83%]), constitutional (18 patients [78%]), and dermatologic (15 patients [65%]).
In Table 5, we report selected PK parameters of AIC in each of the 15 patients included in the PK analysis and the associated percent changes in hematologic parameters (i.e. platelet, leukocyte, and neutrophil levels). It can be seen that four patients (reference numbers 01, 06, 11, and 14) with grade 4 platelets and grade 3/4 neutrophils showed an increase in C max and AUC(0–inf) of AIC on concomitant sorafenib administration. However, we also see an increased incidence of hematologic toxicities without an associated increase in the C max and AUC(0–inf) on concomitant administration of sorafenib (patients with reference numbers 12 and 13) as well as an increase in C max and AUC(0–inf) without an associated increase in hematologic toxicities (patient with reference number 09).
Discussion
In this paper, we report PK and safety data from the combination of sorafenib and dacarbazine in patients with advanced solid tumors. Our results indicate that while concomitant administration of sorafenib and dacarbazine decreased dacarbazine exposure, it resulted in increased AIC exposure. We also found that increased AIC exposure might be associated with an increased incidence of hematologic toxicities, likely because of the interference of methane diazohydroxide with erythropoiesis [22–24]. However, because of the small sample size, no statistically significant correlations between increased AIC exposure and hematologic toxicities could be established. Because of the study design, we could obtain PK profiles of sorafenib and its metabolites only in the presence of dacarbazine. Similar to other studies, we found that the PK parameters of sorafenib and its metabolites showed a high degree of variability [25]. Our data also show that sorafenib contributes approximately 83% to the sum of AUC(0–12)ss values, while the metabolites contribute approximately 9% (M2), 4% (M4), and 3% (M5). This is comparable with data obtained from previous single-agent studies (data on file, Bayer HealthCare AG).
The combination of sorafenib and dacarbazine was associated with a clinically acceptable toxicity profile, with the vast majority of the grades 3/4 AEs improving or resolving upon transient discontinuation and/or dose reduction of the study drugs. No unexpected serious adverse reactions were reported. The sorafenib–dacarbazine combination has also been investigated in randomized [14] and open-label [26] phase II studies, and in another phase I study [19] with similar safety results. Currently, the combination is being investigated in a phase II trial for sarcoma (Clinicaltrials.gov identifier: NCT00837148).
In conclusion, the combined treatment with sorafenib and dacarbazine may result in an increased exposure to AIC, which may be considered an indicator for the exposure to the active alkylating agent methane diazohydroxide. Due to the small number of patients in the present study, a statistically significant correlation between AIC exposure and observed hematologic toxicities, even if present, could not be established. Further studies may be necessary to more clearly characterize this potential drug–drug interaction.
References
Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M (2008) Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther 7:3129–3140
Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA (2004) BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 64:7099–7109
Duran I, Hotte SJ, Hirte H, Chen EX, MacLean M, Turner S, Duan L, Pond GR, Lathia C, Walsh S, Wright JJ, Dancey J, Siu LL (2007) Phase I targeted combination trial of sorafenib and erlotinib in patients with advanced solid tumors. Clin Cancer Res 13:4849–4857
Kupsch P, Henning BF, Passarge K, Richly H, Wiesemann K, Hilger RA, Scheulen ME, Christensen O, Brendel E, Schwartz B, Hofstra E, Voigtmann R, Seeber S, Strumberg D (2005) Results of a phase I trial of sorafenib (BAY 43-9006) in combination with oxaliplatin in patients with refractory solid tumors, including colorectal cancer. Clin Colorectal Cancer 5:188–196
Mross K, Steinbild S, Baas F, Gmehling D, Radtke M, Voliotis D, Brendel E, Christensen O, Unger C (2007) Results from an in vitro and a clinical/pharmacological phase I study with the combination irinotecan and sorafenib. Eur J Cancer 43:55–63
Richly H, Henning BF, Kupsch P, Passarge K, Grubert M, Hilger RA, Christensen O, Brendel E, Schwartz B, Ludwig M, Flashar C, Voigtmann R, Scheulen ME, Seeber S, Strumberg D (2006) Results of a phase I trial of sorafenib (BAY 43-9006) in combination with doxorubicin in patients with refractory solid tumors. Ann Oncol 17:866–873
Siu LL, Awada A, Takimoto CH, Piccart M, Schwartz B, Giannaris T, Lathia C, Petrenciuc O, Moore MJ (2006) Phase I trial of sorafenib and gemcitabine in advanced solid tumors with an expanded cohort in advanced pancreatic cancer. Clin Cancer Res 12:144–151
Ryan CW, Goldman BH, Lara PN Jr, Mack PC, Beer TM, Tangen CM, Lemmon D, Pan CX, Drabkin HA, Crawford ED (2007) Sorafenib with interferon alfa-2b as first-line treatment of advanced renal carcinoma: a phase II study of the Southwest Oncology Group. J Clin Oncol 25:3296–3301
Gollob JA, Rathmell WK, Richmond TM, Marino CB, Miller EK, Grigson G, Watkins C, Gu L, Peterson BL, Wright JJ (2007) Phase II trial of sorafenib plus interferon alfa-2b as first- or second-line therapy in patients with metastatic renal cell cancer. J Clin Oncol 25:3288–3295
Escudier B, Lassau N, Angevin E, Soria JC, Chami L, Lamuraglia M, Zafarana E, Landreau V, Schwartz B, Brendel E, Armand JP, Robert C (2007) Phase I trial of sorafenib in combination with IFN alpha-2a in patients with unresectable and/or metastatic renal cell carcinoma or malignant melanoma. Clin Cancer Res 13:1801–1809
Adjei AA, Molina JR, Mandrekar SJ, Marks R, Reid JR, Croghan G, Hanson LJ, Jett JR, Xia C, Lathia C, Simantov R (2007) Phase I trial of sorafenib in combination with gefitinib in patients with refractory or recurrent non-small cell lung cancer. Clin Cancer Res 13:2684–2691
Gridelli C, Rossi A, Mongillo F, Bareschino M, Maione P, Ciardiello F (2007) A randomized phase II study of sorafenib/gemcitabine or sorafenib/erlotinib for advanced non-small-cell lung cancer in elderly patients or patients with a performance status of 2: treatment rationale and protocol dynamics. Clin Lung Cancer 8:396–398
Agarwala SS, Keilholz U, Hogg D, Robert C, Hersey P, Eggermont A, Grabbe S, Gonzalez R, Patel K, Hauschild A (2007) Randomized phase III study of paclitaxel plus carboplatin with or without sorafenib as second-line treatment in patients with advanced melanoma [abstract]. J Clin Oncol ASCO Annual Meeting Proceedings Part I. (June 20 Supplement) 25:8510
McDermott DF, Sosman JA, Gonzalez R, Hodi FS, Linette GP, Richards J, Jakub JW, Beeram M, Tarantolo S, Agarwala S, Frenette G, Puzanov I, Cranmer L, Lewis K, Kirkwood J, White JM, Xia C, Patel K, Hersh E (2008) Double-blind randomized phase II study of the combination of sorafenib and dacarbazine in patients with advanced melanoma: a report from the 11715 Study Group. J Clin Oncol 26:2178–2185
Azad NS, Posadas EM, Kwitkowski VE, Steinberg SM, Jain L, Annunziata CM, Minasian L, Sarosy G, Kotz HL, Premkumar A, Cao L, McNally D, Chow C, Chen HX, Wright JJ, Figg WD, Kohn EC (2008) Combination targeted therapy with sorafenib and bevacizumab results in enhanced toxicity and antitumor activity. J Clin Oncol 26:3709–3714
Richly H, Kupsch P, Passage K, Grubert M, Hilger RA, Voigtmann R, Schwartz B, Brendel E, Christensen O, Haase CG, Strumberg D (2004) Results of a phase I trial of BAY 43-9006 in combination with doxorubicin in patients with primary hepatic cancer. Int J Clin Pharmacol Ther 42:650–651
Reid JM, Kuffel MJ, Miller JK, Rios R, Ames MM (1999) Metabolic activation of dacarbazine by human cytochromes P450: the role of CYP1A1, CYP1A2, and CYP2E1. Clin Cancer Res 5:2192–2197
Lathia C, Lettieri J, Cihon F, Gallentine M, Radtke M, Sundaresan P (2006) Lack of effect of ketoconazole-mediated CYP3A inhibition on sorafenib clinical pharmacokinetics. Cancer Chemother Pharmacol 57:685–692
Eisen T, Ahmad T, Gore ME, Marais R, Gibbens I, James MG, Schwartz B, Bergamini L (2005) Phase I trial of BAY 43-9006 (sorafenib) combined with dacarbazine (DTIC) in metastatic melanoma patients [abstract]. J Clin Oncol ASCO Annual Meeting Proceedings Part I. (June 1 Supplement) 23:7508
Soria J, Lazar V, Lassau N, Pena C, Massard C, Robert C, Koscielny S, Deutsch E, Zafarana E, Armand JP (2007) Sorafenib (S) and dacarbazine (D) combination in patients (pts) with advanced malignant solid tumors: phase I study with tumor biopsy genomic analysis and dynamic contrast enhanced ultrasonography (DCE-US) [abstract]. J Clin Oncol ASCO Annual Meeting Proceedings Part I. (June 20 Supplement) 25:3556
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216
Ahmann DL, Bisel HF, Edmonson JH, Hahn RG, Eagan RT, O’Connell MJ, Frytak S (1976) Clinical comparison of adriamycin and a combination of methyl-CCNU and imidazole carboxamide in disseminated malignant melanoma. Clin Pharmacol Ther 19:821–824
Gutterman JU, Mavligit G, Gottlieb JA, Burgess MA, McBride CE, Einhorn L, Freireich EJ, Hersh EM (1974) Chemoimmunotherapy of disseminated malignant melanoma with dimethyl triazeno imidazole carboxamide and bacillus calmette–guerin. N Engl J Med 291:592–597
Costanza ME, Nathanson L, Schoenfeld D, Wolter J, Colsky J, Regelson W, Cunningham T, Sedransk N (1977) Results with methyl-CCNU and DTIC in metastatic melanoma. Cancer 40:1010–1015
Strumberg D, Clark JW, Awada A, Moore MJ, Richly H, Hendlisz A, Hirte HW, Eder JP, Lenz HJ, Schwartz B (2007) Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumors. Oncologist 12:426–437
Eisen T, Marais R, Affolter A, Lorigan P, Ottensmeier C, Robert C, Corrie P, Chevreau C, Erlandsson F, Gore M, investigators Fts (2007) An open-label phase II study of sorafenib and dacarbazine as first-line therapy in patients with advanced melanoma [abstract]. J Clin Oncol ASCO Annual Meeting Proceedings Part I. (June 20 Supplement) 25:8529
Acknowledgments
This study was sponsored by Bayer HealthCare AG, Leverkusen, Germany. We acknowledge the medical writing assistance provided by Meenakshi Subramanian, PhD, Evidence Scientific Solutions, which was supported by Bayer HealthCare AG and Onyx Pharmaceuticals, Inc.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Brendel, E., Ludwig, M., Lathia, C. et al. Pharmacokinetic results of a phase I trial of sorafenib in combination with dacarbazine in patients with advanced solid tumors. Cancer Chemother Pharmacol 68, 53–61 (2011). https://doi.org/10.1007/s00280-010-1423-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-010-1423-9