
Abstract
Purpose
TSU-68 is a low molecular weight inhibitor of the tyrosine kinases for vascular endothelial growth factor receptor 2, platelet-derived growth factor receptor β, and fibroblast growth factors receptor 1. In this study, we assessed the recommended dose with TSU-68 administration of twice-daily (b.i.d.) or thrice-daily (t.i.d.) after meals for 4 weeks in Japanese patients with solid tumors based on the safety and tolerability and investigated the relationship between angiogenesis biomarker and clinical outcomes.
Methods
The study design was a dose-escalation method with alternating enrollment of b.i.d. administration and t.i.d. administration after meal by traditional three-patient cohort.
Results
We enrolled 24 patients at doses of 200, 400, and 500 mg/m2 b.i.d. or 200 and 400 mg/m2 t.i.d. No dose-limiting toxicity (DLT) occurred in the 200 mg/m2 b.i.d. or t.i.d., and 3 patients experienced DLTs at 400 mg/m2 b.i.d. or 400 mg/m2 t.i.d. As main toxicity, blood albumin decreased, malaise, diarrhea, alkaline phosphatase increased, anorexia, abdominal pain, nausea, and vomiting were observed as almost all grade 1–2. There were no apparent differences in pharmacokinetic parameters between days 2 and 28 after the repeated b.i.d. and t.i.d. doses. Although tumor shrinkage was not observed, the disease control rate was 41.7%. As an angiogenesis-related factor of stratified analysis, plasma vascular endothelial growth factor and plasminogen activator inhibitor-1 were detected as a significant increase with progressive disease patients.
Conclusions
A recommended dosage of TSU-68 for this administration schedules was estimated to be 400 mg/m2 or less b.i.d.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Angiogenesis is essential for the proliferation of malignant tumors and development of its metastasis [1]. When a tumor grows to be 2–3 mm or more in the course of proliferation, it may produce angiogenesis-stimulating growth factors by acting on itself and its surrounding normal cells to supply oxygen and nutrition. Such growth factors may induce digestion, migration/proliferation, and formation of lumens of the basement membrane of endothelial cells, leading to formation of a new vascular nest. This may enlarge the lesion, resulting in infiltration and hematogenous metastasis. There are many known growth factors for angiogenesis, including vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and fibroblast growth factor (FGF). Among them, VEGF is regarded as more important since it is reported that the production of VEGF may be increased in many solid tumors and this ability to produce may be correlated with the number of blood vessels and prognosis in breast caner [2], gastric cancer [3], colon cancer [4], lung cancer [5], and others. Angiogenesis by VEGF starts when a VEGF is bound with a VEGF receptor that is a specific receptor appearing in endothelial cells. Subsequently, a VEGF receptor will transmit signals for proliferation and such like following autophosphorylation by tyrosine kinase of an intracellular domain. VEGF receptors comprise Flk-1/KDR, Flt-1, and Flt-4 [6]. In particular, Flk-1/KDR is considered indispensable for the proliferation of endothelial cells as the most important receptor, only appearing in endothelial cells [7]. Neutralization antibodies of VEGF and inhibitors of Flk-1/KDR inhibited proliferation of endothelial cells in vitro, and also suppressed angiogenesis and tumor proliferation in vivo [8, 9]. It is also shown that cellular proliferation was suppressed and tumors had fewer vessels when cell strains derived from ovarian cancer, lung cancer, and glioma were subcutaneously transplanted into SCID mice with a manifestation of Flk-1, the variant lacking tyrosine kinase domain [10, 11].
It has been realized that the susceptibility of established tumor blood vessels to an interference with VEGF/VEGFR2 signaling may be restricted to a fraction of immature vessels that lack co-localization with pericytes. The contact between endothelial cells and periendothelial support cells, such as pericytes or smooth muscle cells, stabilize new blood vessels, promotes endothelial survival, and inhibits endothelial cell proliferation. This is supported by the observation that interference with PDGF-BB/PDGFRb signaling resulted in disruption of already established endothelial/pericytes associations and vessel destabilization [12, 13].
TSU-68 (chemical name: (Z)-5-[(1, 2-dihydro-2-oxo-3H-indol-3-ylidene) methyl]-2,4-dimethyl-1H-pyrrole-3-propanoic acid) is a new, oral angiogenesis inhibitor. It has a low molecular weight that cuts the supply of oxygen and nutrition by inhibiting tyrosine phosphorylation of Flk-1/KDR, VEGF receptor and suppressing angiogenesis in tumor tissues to suppress tumor proliferation and metastasis [14]. In addition, it was confirmed in pre-clinical studies that the substance inhibited tyrosine phosphorylation of PDGF and FGF receptors that are associated with the transmission of intracellular signals as well as VEGF receptors [15, 16], and also inhibited these related angiogenesis in mice [17].
This study was conducted on Japanese patients with advanced solid tumors to evaluate adverse events and to estimate the recommended dose for twice-daily (b.i.d.) or thrice-daily (t.i.d.) administration after meals for a 4-week treatment of TSU-68. This study was designed to comply with the ethical principals of Good Clinical Practice in accordance with the Declaration of Helsinki.
Patient and methods
Patient selection
Patients with solid tumors whose malignancies were confirmed histopathologically, and patients with malignancies on which the standard therapy had no effect or for which no generally approved standard therapy exists, from a single institution in Japan. They were required to be 20–75 years old, can take the drug orally, have performance statues of 0–2, and expected to survive for a sufficient period of ≥60 days. Patients with physiologically adequate compensatory functions and with parenchymal organs, such as heart, pulmonary, renal, and bone marrow, in particular, functioning sufficiently, were eligible for this study. The following laboratory values, obtained within 15 days before the start of the study, must be satisfied (Leukocyte count: 4,000–12,000/mm3 or neutrophil count: ≥2,000/mm3, platelet count: ≥10 × 104/mm3, hemoglobin level: ≥8.0 g/dl or more, total bilirubin: ≤1.5 mg/dl, glutamic-oxaloacetic transaminase/glutamic-pyruvic transaminase: ≤100 U/l, serum creatinine: ≤1.5 mg/dl, creatinine clearance: ≤50 ml/min, PaO2:≥65 mmHg, no clinical evidence of abnormality on electrocardiogram). These patients needed to have confirmed measurable lesions.
Patients were excluded for the potential influence of previous treatments, such as major invasive surgery, blood transfusion, or administration of G-CSF. Patients with active infections, serious complications, a history of serious thromboembolism, brain metastasis showing clinical symptoms, and colon diseases accompanied by active inflammation were excluded from this study.
It was confirmed that the investigator explained the details of the study to the patients in accordance with the information form before enrollment, and then allowed sufficient time before obtaining written consent. Prior to the conduct of this study, it was reviewed by the institutional review board at the National Cancer Center.
Drug administration
TSU-68 was provided by Taiho Pharmaceutical Co., Ltd (Tokyo, Japan). Twice-daily administration was given within 1 h after meals at about 12 h interval by a dose corresponding to each specified dosage level per body surface area, and thrice-daily administration was given at about 6- or 12-h interval.
TSU-68 was taken for 28 days continuously, and for patients evaluated as being better in evaluation of antitumor effect after completion of 28 days, the administration was to be continued within the range of this study period, unless it became difficult to continue the treatment due to occurrence of any adverse event.
Dose escalation
The dose escalation was a three-patient cohort at each dose level and alternating of b.i.d. and t.i.d. The starting dose and number of TSU-68 was 200 mg/m2 b.i.d.
Dose-limiting toxicity (DLT) was defined as drug-related adverse events (adverse drug reactions) of grade 3 or more severe non-hematological toxicity or grade 4 or more severe hematological toxicity. The doses were increased by 100% for patients showing no adverse drug reaction of grade 2, by 40% for cases showing no DLT and adverse drug reaction of grade 3, or by 33% for cases showing DLT.
Each dosage level involved 3 patients, and 3 additional patients were to be administered if one or more of the initial three patients showed DLT. The drug was to be administered to at least 6 patients at the recommended dosage level. The maximum tolerated dose (MTD) was the appearance of DLT in 2/3 patients or 3/6 patients, and provided that those DLT was counted by the same toxicity category. The recommended dosage was determined to be one level lower than the dosage judged as the MTD. If the MTD was not found, the recommended dosage was also determined in consideration of the results of the pharmacokinetic investigation.
Patient evaluation
Patient condition was assessed by hematology/chemistry laboratory data, urinalysis, vital signs, performance status, and clinical findings at least weekly. Symptoms were evaluated in accordance with the Common Toxicity Criteria version 2.0 (NCI-CTC) [18].
Antitumor effects and adverse reactions were evaluated in accordance with the criteria of the Japan Society for Cancer Therapy [19], which is based on criteria established by the WHO. The criteria for the evaluation of antitumor effects were as follows: complete response (CR), eradication of all cancers and maintenance of the condition for 4 weeks or more; partial response (PR), 50% or more reduction in size of lesions and maintenance of the condition for 4 weeks or more; no change (NC), less than 50% reduction in size of lesions or enlargement of lesions within 25% and maintenance of the condition for 4 weeks or more; progressive disease (PD), 25% or more enlargement of lesions or appearance of new lesions.
Pharmacokinetics studies
For twice-daily administration, sequential blood collection was performed after the 1st, 3rd, and 55th doses. Blood samples were collected within 30 min before dosing and at the following times after dosing: 1, 1.5, 2, 3, 4, 6, 8, and 12 h (the 1st, 3rd, and 55th doses), and l, 1.5, 2, 3, 4, and 12 h (the 56th dose).
For three times daily administration, sequential blood collection was performed after the 1st dose and the 2nd, 82nd, and 84th doses (respective about 6, 12 and 6 h after prior dose). Blood samples were collected within 30 and 10 min before dosing for the 1st and 3rd doses, and at the following times after dosing: 1, 1.5, 2, 3, 4, and 5.5 h (the 1st and 82nd doses); 1, 1.5, 2, 3, 4, and 6 h (the 2nd dose); and 1, 1.5, 2, 3, 4 and 14 h (the 84th dose).
Urine samples were collected at the following intervals for measurement of TSU-68: For twice-daily administration, prior to the 1st dose, 0–12 h after the 1st, 2nd, 3rd, 15th, 55th doses, and 0–12 and 12–24 h after the 56th dose. For three times daily administration, prior to the 1st dose, 0–6 h after the 1st, 2nd, 22nd, 82nd, and 83rd doses, 0–12 h after the 3rd dose, and 0–6, 6–14 and 14–24 h after the 84th dose.
TSU-68 concentration was determined using a validated high-performance liquid chromatography method with UV detection, with a lower limit of quantification of 0.1 μg/ml.
In pharmacokinetic analyses, non-compartmental pharmacokinetic parameters including area under the plasma concentration–time curves (AUC) from time 0 to the last measurable time (AUC0–t), maximum concentration (C max), time to maximum concentration (T max), and elimination half-life (T 1/2) were calculated using PhAST (Ver.2.3, MDS Pharma Services, Montreal, Canada).
Biological studies
As for plasma and urine collection before treatment and after day 8 and day 28, plasma VEGF, urinary VEGF, endothelial adhesion molecule-1 (ELAM-1), tissue plasminogen activator (t-PA), plasminogen activator inhibitor-1 (PAI-1), and vascular cell adhesion molecule-1 (VCAM-1) were measured at SRL, Inc. (Tokyo, Japan). Enzyme-linked immunosorbent assay (ELISA) kits for human VEGF, human ELAM-1, and human VCAM-1 were obtained from QuantikineTM from R&D Systems Inc. (Minneapolis, MN). ELISA kit for soluble t-PA was obtained from Calbiochem (La Jolla, CA). Latex photometric immunoassay system for soluble PAI-1 was obtained from Mitsubishi Chemical Medicine Corporation (Tokyo, Japan). The analysis was performed using the SAS®.
Results
Patient characteristics
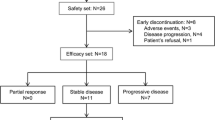
Twenty-four patients were enrolled in this study between June 2001 and March 2002. All patients were evaluable for safety, pharmacokinetics, and biological studies. The patient characteristics are summarized in Table 1. All patients had a good performance status, and their median age was 55 years (range 31–72 years). The site of the primary tumor was colorectal cancer in 9 patients, non-small-cell lung cancer in 7 patients, and others in 8 patients. Twenty-two patients had received prior systemic chemotherapy with standard regimen, and 2 patients had no standard therapy.
Maximum tolerated dose
Patients were enrolled sequentially on the twice-daily/thrice-daily administration cohorts and in parallel within each dosing cohort. No DLT occurred in the 200 mg/m2 b.i.d dose level (3 patients). Three plus three patients were enrolled on the 400 mg/m2 b.i.d. dose level, with 2 patients experiencing DLTs: grade 3 dyspnea, hypoxemia, pleural effusion and anorexia, and unacceptable grade 2 anorexia. The grade 2 anorexia at 400 mg/m2 was an excruciating event with weight loss and blood albumin decrease, and this patient refused drug administration after 6 days. Two patients of DLT in 400 mg/m2 were different toxicity categories. One patient was Pulmonary (grade 3 of dyspnea, hypoxemia, pleural effusion, and anorexia, anorexia were the accompanying events of dyspnea), and the other patient was Gastrointestinal (anorexia of grade 2). Therefore, it did not count 2/3 patients, and enrolled patients with total six at 400 mg/m2. However, the 500 mg/m2 b.i.d. dose level was not found to DLT, and MTD was not reached, because dose escalation was stopped based on pharmacokinetic results. On the other hand, no DLT occurred in the 200 mg/m2 t.i.d. dose level, and one patient experienced a DLT of grade 4 pericardial effusion at 400 mg/m2 t.i.d. dose level. MTD was not reached either because dose escalation was not based on the result of pharmacokinetics, and three patients were enrolled in each dose level.
Toxicity
All 24 patients were evaluated for safety analysis. Major drug-related adverse events for 4-week administration are shown in Table 2. As protocol-defined DLT, there were two patients who had grade 4 pericardial effusion by t.i.d., and grade 3 dyspnea, hypoxemia, pleural effusion, and anorexia by b.i.d. These adverse events were not the defined DLT with revealed characteristics of the TSU-68 safety profile. The main toxicities were almost all grade 1–2, and the toxicities occurring in at least over 30% included urine/stool discoloration, blood albumin decrease, fatigue, diarrhea, blood alkaline phosphatase increase, anorexia, abdominal pain, nausea, and vomiting.
Pharmacokinetics
In the b.i.d. regimen after meal, pharmacokinetic analyses were performed in 12 subjects, at the doses of 200 mg/m2 (n = 3), 400 mg/m2 (n = 6), and 500 mg/m2 (n = 3). In the t.i.d. regimen after meal, pharmacokinetic analyses were performed in 12 subjects, at the doses of 200 mg/m2 (n = 6) and 400 mg/m2 (n = 6). The mean concentration–time profiles in each dose level are shown in the Fig. 1. Pharmacokinetic results are presented in Table 3.

Plasma concentration-versus-time profiles of TSU-68. a Twice-daily administration after meal, b thrice-daily administration after meal
In the b.i.d. and t.i.d. regimens after meal, after the 1st dose, the plasma concentration of TSU-68 increased to reach C max at approximately 3 h, and thereafter disappeared with T 1/2 of approximately 2–3 h. In the b.i.d. regimen, at any dose levels, C max and AUC after the repeated administration of TSU-68 on days 2 and 28 were approximately twofold lower than those after the 1st administration on day 1. The t.i.d. regimen also shows a similar trend. There were no apparent differences in these parameters between days 2 and 28. In addition, no obvious dose-dependent increases were observed with these parameters after the repeated b.i.d. and t.i.d. doses.
Urinary excretion of TSU-68 was below 1% of dose in all dose levels.
Efficacy
Clinical response was estimated by 23 patients with one patient receiving medication of 1 cycle for less than 50%, and the best response of each case is indicated in Table 1. Although a PR patient was not found, the NC patients were observed at 43.5% (10/23 example) in these studies. Notably, two patients with soft tissue sarcoma (Pt.14) and colon cancer (Pt.20) were treated with TSU-68 for 6 and 9 months. In addition, the median time to progression was 28 days (range 27–254+ days), and the median survival period was 218 days (range 79–465+ days).
Biomarkers
As an angiogenesis-related factor, the plasma VEGF, urinary VEGF, ELAM-1, t-PA, PAI-1, and VCAM-1 were investigated in 23 patients at baseline, 23 patients at day 8, and 22 patients at day 28. Median (range) of those factors were as follows: plasma VEGF to 66 pg/ml (<31–183 pg/ml) at baseline, 55 pg/ml (<31–278 pg/ml) at day 8, and 63 pg/ml (<31–270 pg/ml) at day 28; urinary VEGF to 108 pg/ml (<31–491 pg/ml) at baseline, 144 pg/ml (<31–1210 pg/ml) at day 8, and 162.5 pg/ml (68–730 pg/ml) at day 28; ELAM-1 to 51 ng/ml (14–134 ng/ml) at baseline, 50 ng/ml (13–136 ng/ml) at day 8, and 45.5 ng/ml (17–114 ng/ml) at day 28; t-PA to 7.2 mg/ml (2.6–15.4 ng/ml) at baseline, 7.2 ng/ml (3.3–14.3 ng/ml) at day 8, and 6.8 ng/ml (2.9–13.5 ng/ml) at day 28; PAI-1 to 23 ng/ml (10–73 ng/ml) at baseline, 28 ng/ml (11–92 ng/ml) at day 8, and 31 ng/ml (11–120 ng/ml) at day 28; VCAM-1 to 501 ng/ml (373–1,080 ng/ml) at baseline, 562 ng/ml (303–1,140 ng/ml) at day 8, and 545.5 ng/ml (355–988 ng/ml) day 28.
As a result of stratified analysis on efficacy (separated between NC of 10 pts. and PD of 13 pts.) before and after treatment, plasma VEGF and PAI-1 were detected as a significant change by Wilcoxon signed rank-sum test. Figure 2 shows the change in these factors for each patient. Both factors with PD patients were significantly increasing at day 28 compared to baseline. Three progressors in VEGF were the same patients of 3 progressors in PAI-1, and these 3 progressors were Pt.11, Pt.13, and Pt.18 in Table 1. Regarding the other factors, no significant change was found in NC patients or PD patients. Also, the angiogenesis-related factors were compared with NC patients and PD patients on baseline, but a factor in a significant correlation with clinical efficacy could not be detected.

The changing of plasma VEGF and PAI-1 after 8 or 28 days of treatment with TSU-68
Discussion
This study investigated the difference of toxicity and pharmacokinetic data between twice-daily and thrice-daily administering after meal, based on a pre-clinical study, which plasma concentrations of TSU-68 orally administered to fed dogs were higher than that administered to fasted dogs.
In this study, in the time before the advent of molecular-targeted agent, the completed standards systemic chemotherapy population was appropriate for patient evaluation of TSU-68 toxicity and feasibility. The subjects of the protocol-defined DLT were two patients by twice-daily administration and one patient by thrice-daily administration. The plasma concentrations of TSU-68 after day 28 were hardly increased with doses from 200 mg/m2 b.i.d. to 400 mg/m2 t.i.d. For this reason, the dose escalation should stop at 1,200 mg/m2 per total daily dose, and the determination of an unreached MTD was reasonable.
As the main toxicities of TSU-68 in this study, malaise, diarrhea, anorexia, abdominal pain, nausea, and vomiting were observed as subjective symptoms, except the urine/stool discoloration from drug colors. Almost all of the toxicities were grades 1–2, and there were no remarkable differences in the adverse events and grades of side effects by the medication method or the dose level. Moreover, many events of blood albumin decreased and alkaline phosphatase increased as laboratory test values were observed, but there was little evidence of myelosuppression. Consequently, these toxicities were considered acceptable for an oral molecule targeting agent.
The toxicity of TSU-68 did not demonstrate gastrointestinal perforation of characteristic of monoclonal VEGF antibody [20], or a high incidence of a hypertension or skin rash in VEGFR tyrosine kinase inhibitors [21, 22]. On the other hand, as a toxicity characteristic of TSU-68, fluid retention such as face edema, pleural effusion, and pericardia effusions were seen with a relatively high frequency. As similar characteristics, tyrosine kinases inhibitor of a high incidence of fluid retention was Imatinib, targeted for Bsc-Abl, KIT and PDGFR, and was reported at 54% [23].
As a biomarker, although the factor relevant to angiogenesis was measured before and after TSU-68 administration, no factor changed significantly. When this biomarker was stratified by NC patients and PD patients, VEGF and PAI-1 increased significantly after TSU-68 administration compared with before administration in PD patients. These stratification factors suggest the possibility that a significant increase in both VEGF and PAI-1 become response markers of early tumor progression for TSU-68. It is known that VEGF participates in the blood vessel rebirth of tumor multiplication [5], and PAI-1 was increased by the effect of PDGF in a vascular smooth muscle cell [24, 25]. Since the changes of PAI-1 levels show the difference between NC patients and PD patients, there is some potential for inhibitory activity in PDGFR tyrosine kinases by TSU-68. However, as the values of the biomarkers prior to the administration were not related by NC patients and PD patients stratification, a marker could not be used to predict the clinical effect for TSU-68.
The inhibition of PDGFR tyrosine kinase by TSU-68 enforced tumor vessel regression by interfering with pericyte-mediated endothelial cell survival [15, 26] and inhibitory effect on PDGFR strengthened inhibitory effect of VEGFR to induce antitumor effects. The phenomena showing PDGF-related toxicity and efficacy may be a characteristic of TSU-68, which is dissimilar from the other multi-kinase inhibitors for anti-angiogenesis.
In the pharmacokinetics of TSU-68, the C max and AUC after the repeated doses on days 2 and 28 were lower than those after the 1st dose on day 1. These parameters on day 28 were comparable with those on days 2. This suggests that the decreased plasma exposure to TSU-68 rapidly reaches a steady state and is maintained over therapeutic cycles. This trend is consistent with a published clinical result showing that AUC of TSU-68 on day 56 was similar to that on day 28 [27]. Furthermore, PK parameters of TSU-68 were not apparently different between the 55th and 56th doses (b.i.d.), and between the 82nd and 84th (t.i.d.), suggesting that a circadian rhythm had no influence on PK of TSU-68. The observed decrease in the exposure is probably due to autoinduction of TSU-68 metabolism. Since urinary excretion accounted for a very low percentage of the dose, predominant elimination of TSU-68 can be regarded as hepatic metabolism. In the non-clinical studies [28, 29], TSU-68 was found to cause induction of liver cytochrome P450, CYP1A1/2 involved in its own metabolism, leading to the decrease in the TSU-68 plasma concentrations.
No obvious dose-dependent increases were observed with C max and AUC after the repeated doses. The most likely reason for this observation is a saturation of absorption based on its lower solubility. A previous clinical study reported that absolute bioavailability of TSU-68 administered at 100 mg/m2 after meals was 42% [30]. In this study, AUC of TSU-68 administered after meals tended to be slightly higher, compared to a previous study [31], in which TSU-68 was administered between meals. Another Japanese clinical study of TSU-68 has also reported that the AUC under fed conditions was higher than that under fasted conditions [30]. These findings may suggest that absorption of TSU-68 tends to increase when food is taken just before TSU-68 dosing. When total daily AUC was compared between the b.i.d. and t.i.d. regimens, the AUC was estimated to be similar between the two regimens. Therefore, the b.i.d. regimen would be preferable in therapeutic use from the viewpoint of compliance.
As an efficacy of TSU-68, there was no tumor reduction and a few patients were treated for more than 6 months. However, the tumor control rate was 43.5% (10/23 patients). These patients had no effects with standard therapy, or did not have standard therapy previously. In light of this population, it was decided that antitumor effect not be pursued.
TSU-68 phase I studies in Japan have three different dosing regimens, including the twice-daily between meals [31] and the twice-daily or thrice-daily after meals. The recommended dosage of TSU-68 was concluded to be ≤800 mg/m2 b.i.d. under the between-meal conditions, and ≤400 mg/m2 b.i.d. and ≤400 mg/m2 t.i.d. under the after-meal conditions. As mentioned above, AUC of TSU-68 tend to be somewhat increased by administering the drug after meals, and the total daily AUC was similar between the b.i.d. and t.i.d. regimens. In addition, marked differences in the types or severity of drug-related adverse events were not observed between the three regimens. Therefore, for further studies, the recommended dose schedule of TSU-68 administration was finally considered “b.i.d. after meals”, in which higher AUC and more convenient therapeutic use would be achieved.
An Independent Data Monitoring Committee discussed data obtained from the three studies to evaluate the recommended dose of TSU-68 for further studies. The number of TSU-68 tablets administered to patients should not be adjusted by patients according to body surface area, because of no obvious dose-dependency in the steady-state AUC. Regarding safety, the incidences of drug-related adverse events showed no marked dose dependency, although the number of drug-related adverse events by patient tended to increase when patients were treated at doses of 400 mg/m2 or more. Additionally, out of 6 patients enrolled in dose level 400 mg/m2, which was the maximum recommended dose of TSU-68, one patient (Pt.4 in Table 1) experienced DLT (grade 3 dyspnoea, hypoxia, pleural effusion and anorexia) and one patient (Pt.5 in Table 1) experienced grade 2 anorexia which caused discontinuation of TSU-68 administration (DLT), and 2 patients were treated at a dose of 600 mg/body b.i.d. after meals. Since TSU-68 was a tablet of 200 mg, the next dose level down was 400 mg/body; both of the 2 patients (Pt.1 and Pt.2 of Table 1) in 400 mg/body b.i.d. had no DLT. Therefore, the Independent Data Monitoring Committee considered that 400 mg/body b.i.d. was more tolerable than 600 mg/body b.i.d. for safety and suggested that “400 mg/body b.i.d. after meals” was proper as the recommended dose of TSU-68.
TSU-68 is a medicine comparably safe in receptor tyrosine kinase inhibitors and is particularly useful at prolonging the survival of patients with cancer in combinations of standard chemotherapies; further combinations studies of TSU-68 with standard chemotherapy are planned in several solid tumors.
References
Folkman J, Klagsbrun M (1987) Angiogenic factors. Science 235:442–447
Toi M, Hoshina S, Takayanagi T, Tominaga T (1994) Association of vascular endothelial growth factor expression with tumor angiogenesis and with early relapse in primary beast cancer. Jpn J Cancer Res 85:1045–1049
Maeda K, Chung YS, Ogawa Y, Takatsuka S, Kang SM, Ogawa M, Sawada T, Sowa M (1996) Prognostic value of vascular endothelial growth factor expression in gastric carcinoma. Cancer 77:858–863
Takahashi Y, Kitadai Y, Bucana CD, Cleary KR, Ellis LM (1995) Expression of vascular endothelial growth factor and its receptor, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer. Cancer Res 55:3964–3968
Giatromanolaki A, Koukourakis MI, Kakolyris S, Turley H, O’Byrne K, Scott PA, Pezzella F, Georgoulias V, Harris AL, Gatter KC (1998) Vascular endothelial growth factor, wild-type p53, and angiogenesis in early operable non-small cell lung cancer. Clin Cancer Res 4:3017–3024
Hicklin DJ, Ellis LM (2005) Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol 23:1011–1027
Kowanetz M, Ferrara N (2006) Vascular endothelial growth factor signaling pathways: therapeutic perspective. Clin Cancer Res 12:5018–5022
Asano M, Yukita A, Matsumoto T, Matsumoto T, Kondo S, Suzuki H (1995) Inhibition of tumor growth and metastasis by an immunoneutralizing monoclonal antibody to human vascular endothelial growth factor/vascular permeability factor 121. Cancer Res 55:5296–5301
Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N (1993) Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumor growth in vivo. Nature 362:841–844
Millauer B, Shawver LK, Plate KH, Risau W, Ullrich A (1994) Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature 367:576–579
Millauer B, Longhi MP, Plate KH, Shawver LK, Risau W, Ullrich A, Strawn LM (1996) Dominant-negative inhibition of Flk-1 suppresses the growth of many tumor types in vivo. Cancer Res 56:1615–1620
Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E (1999) Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest 103:159–165
Abramsson A, Lindblom P, Betsholtz C (2003) Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest 112:1142–1151
Laird AD, Vajkoczy P, Shawver LK, Thurnher A, Liang C, Mohammadi M, Schlessinger J, Ullrich A, Hubbard SR, Blake RA, Fong TA, Strawn LM, Sun L, Tang C, Hawtin R, Tang F, Shenoy N, Hirth KP, McMahon G, Cherrington JM (2000) SU6668 is a potent antiangiogenic and antitumor agent that induces regression of established tumors. Cancer Res 60:4152–4160
Laird AD, Christensen JG, Li G, Carver J, Smith K, Xin X, Moss KG, Louie SG, Mendel DB, Cherrington JM (2002) SU6668 inhibits Flk-1/KDR and PDGFRB in vivo, resulting in rapid apoptosis of tumor vasculature and tumor regression in mice. FASEB J 16:681–690
Zhang H, Issekutz AC (2002) Down-modulation of monocyte transendothelial migration and endothelial adhesion molecule expression by fibroblast growth factor: reversal by the anti-angiogenic agent SU6668. Am J Pathol 160:2219–2230
Machida S, Saga Y, Takei Y, Mizuno I, Takayama T, Kohno T, Konno R, Ohwada M, Suzuki M (2005) Inhibition of peritoneal dissemination of ovarian cancer by tyrosine kinase receptor inhibitor SU6668 (TSU-68). Int J Cancer 114:224–229
Japan Clinical Oncology Group (1999) National Cancer Institute—common toxicity criteria version 2.0, Japanese translations first version. Jpn J Cancer Chemother 26:1084–1144
Japan Society Cancer Therapy (1993) Criteria for the evaluation of the clinical effects of solid cancer chemotherapy. J Jpn Soc Cancer Ther 28:101–130
Gordon MS, Margolin K, Talpaz M, Sledge GW Jr, Holmgren E, Benjamin R, Stalter S, Shak S, Adelman D (2001) Phase I safety and pharmacokinetic study of recombinant human anti-vascular endothelial growth factor in patients with advanced cancer. J Clin Oncol 19:843–850
Faivre S, Delbaldo C, Vera K, Robert C, Lozahic S, Lassau N, Bello C, Deprimo S, Brega N, Massimini G, Armand JP, Scigalla P, Raymond E (2006) Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol 24:25–35
Clark JW, Eder JP, Ryan D, Lathia C, Lenz HJ (2005) Safety and pharmacokinetics of the dual action Raf kinase and vascular endothelial growth factor receptor inhibitor, BAY 43-9006, in patients with advanced, refractory solid tumors. Clin Cancer Res 11:5472–5480
Deininger MWN, O’Brien SG, Ford JM, Druker BJ (2003) Practical management of patients with chronic myeloid leukemia receiving imatinib. J Clin Oncol 21:1637–1647
Reilly CF, McFall RC (1991) Platelet-derived growth factor and transforming growth factor-beta regulate plasminogen activator inhibitor-1 synthesis in vascular smooth muscle cells. J Biol Chem 266:9419–9427
van Leeuwen RT, Kol A, Andreotti F, Kluft C, Maseri A, Sperti G (1994) Angiotensin II increases plasminogen activator inhibitor type 1 and tissue-type plasminogen activator messenger RNA in cultured rat aortic smooth muscle cells. Circulation 90:362–368
Erber R, Thurnher A, Katsen AD, Groth G, Kerger H, Hammes HP, Menger MD, Ullrich A, Vajkoczy P (2004) Combined inhibition of VEGF and PDGF signaling enforces tumor vessel regression by interfering with pericyte-mediated endothelial cell survival mechanisms. FASEB J 18:338–340
Kuenen BC, Giaccone G, Ruijter R, Kok A, Schalkwijk C, Hoekman K, Pinedo HM (2005) Dose-finding study of the multitargeted tyrosine kinase inhibitor SU6668 in patients with advanced malignancies. Clin Cancer Res 11:6240–6246
Kitamura R, Yamamoto Y, Nagayama S, Otagiri M (2007) Decrease in plasma concentrations of antiangiogenic agent TSU-68 ((Z)-5-[(1, 2-dihydro-2-oxo-3H-indol- 3-ylidene)methyl]-2, 4-dimethyl-1H-pyrrole-3-propanoic acid) during oral administration twice a day to rats. Drug Metab Dispos 35:1611–1616
Kitamura R, Asanoma H, Nagayama S, Otagiri M (2008) Identification of human liver cytochrome P450 isoforms involved in autoinduced metabolism of the antiangiogenic agent (Z)-5-[(1, 2-dihydro-2-oxo-3H-indol-3-ylidene)methyl]-2, 4-dimethyl-1H-pyrrole-3-propanoic acid (TSU-68). Drug Metab Dispos 36:1003–1009
Brahmer JR, Kelsey S, Scigalla P, Hill G, Bello C, Elza-Brown K, Donehower R (2002) A phase I study of SU6668 in patients with refractory solid tumors. Proc Am Soc Clin Oncol 21:abstr 335
Ueda Y, Shimoyama T, Murakami H, Yamamoto N, Yamada Y, Tamura T (2002) Phase I study of TSU-68, VEGF receptor tyrosine kinase inhibitor, by twice daily oral administration between meals in patients with advanced solid tumors. Proc Am Soc Clin Oncol 21:abstr 443
Acknowledgments
We are also grateful to Yutaka Ariyoshi, Nagahiro Saijo, and Yuh Sakata for extramural review. We thank Terumi Sakamoto, Tsuneo Suzuki, Taro Furuie, Kenzo Iizuka, Toyomitsu Sato, Junichi Yonezawa, Kumio Aoyagi, Masahito Komuro, Kentaro Nagai, Hiroshi Nakayama, Yoshio Yamamoto, Ryuichi Kitamura, and Kiyoshi Eshima for assistance in data collection and analysis.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Murakami, H., Ueda, Y., Shimoyama, T. et al. Phase I, pharmacokinetic, and biological studies of TSU-68, a novel multiple receptor tyrosine kinase inhibitor, administered after meals with solid tumors. Cancer Chemother Pharmacol 67, 1119–1128 (2011). https://doi.org/10.1007/s00280-010-1405-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-010-1405-y