
Abstract
Rituximab, gemcitabine and oxaliplatin (R-GemOx) has demonstrated to be effective and safe in lymphoma patients. We aimed to determine the maximum tolerated dose (MTD) of oxaliplatin in combination with rituximab and gemcitabine and to explore the efficacy and safety of R-GemOx in relapsed or refractory (r/r) indolent and mantle cell lymphoma (MCL). In this single-arm, phase I/II trial, we enrolled 55 patients with r/r indolent lymphoma and MCL not suitable for autologous stem-cell transplantation. Patients received 4 cycles of R-GemOx. In the dose escalation group, 70 mg/m2 of oxaliplatin was applied and interindividually increased by 10 mg/m2 until the MTD was reached together with fixed doses of rituximab and gemcitabine. At the oxaliplatin MTD, an extension cohort was opened. Primary aim was to detect an overall response rate (ORR) greater than 65% (α = 0.05). Oxaliplatin 70 mg/m2 (MTD) was chosen for the extension cohort after 3 of 6 patients experienced a DLT at 80 mg/m2. Among 46 patients evaluable for the efficacy analysis ORR was 72% (33/46), missing the primary aim of the study (p = 0.21). After a median follow-up of 7.9 years, median PFS and OS were 1.0 and 2.1 years. Most frequent grade ≥ 3 adverse events were cytopenias. R-GemOx induces decent response rates in r/r indolent lymphoma and MCL, though novel targeted therapies have largely replaced chemotherapy in the relapse setting. Particularly in MCL, R-GemOx might be an alternative option in late relapses or as bridging to CAR-T-cells. This study was registered with ClinicalTrials.gov on Aug 4th, 2009, number NCT00954005.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Indolent lymphoma account for approximately 35–40% of all malignant lymphoma and are generally characterized by slow progression and reccurring relapses [1]. Follicular lymphoma (FL) represents the most common subtype of indolent lymphoma. Most patients are diagnosed in advanced stage III or IV according to Ann Arbor. When requiring treatment, these patients are managed with immunochemotherapy [2,3,4,5]. The prognosis is usually favorable with an overall survival of almost two decades although advanced indolent lymphoma is not considered curable [6, 7]. On the other hand, mantle cell lymphoma (MCL) typically shows a more aggressive course of disease and has the worst prognosis among B-cell lymphoma [8]. Furthermore, early disease progression within 24 months (POD24) is associated with a poor outcome in advanced stage FL and MCL [9,10,11,12]. While conventional immunochemotherapy can be repeated in relapses after long-term remission, early relapses require alternative therapy regimens. High-dose therapy (HDT) followed by autologous stem-cell transplantation (ASCT) has been considered standard in first-line treatment for younger MCL patients [13,14,15]. Interestingly, recently published data from the TRIANGLE trial suggests that the addition of ibrutinib to the induction treatment might substitute ASCT in the majority of MCL patients [16]. ASCT is also effective in patients with FL [17, 18], but is only considered superior in early relapses of younger patients without comorbidities [19, 20].
Lately, new approaches such as rituximab-lenalidomide (R2) in FL or ibrutinib in MCL have expanded the therapeutic landscape in relapsed NHL [16, 21,22,23]. However, conventional chemotherapy still remains an option after long-term remissions. Rituximab in combination with fludarabine, cyclophosphamide and mitoxantrone (R-FCM) demonstrated a good efficacy in relapsed FL and MCL patients with an overall response rate of 79% [24]. Gemcitabine as well as oxaliplatin demonstrated a promising single-agent activity in multiple lymphoma entities [25,26,27,28,29,30,31]. Both substances were also capable of inducing responses in refractory disease [25, 30]. Particularly interesting is the good efficacy in MCL [25, 30]. As both substances exhibit a different side-effect profile, they seem very suitable for combination therapy. Additionally, as compared to cisplatin, oxaliplatin features an improved safety profile regarding renal toxicity making this substance highly attractive for elderly and comorbid patients [30, 32, 33]. In aggressive NHL, the combination of gemcitabine and oxaliplatin together with Rituximab (R-GemOx) showed promising activity and an acceptable toxicity profile with predominantly hematological toxicities in several phase II trials [34,35,36]. As CHOP- or bendamustine-based regimens are widely applied in first-line, gemcitabine and oxaliplatin represent cytostatic agents without known cross-resistance to these drugs.
Aim of this study was to evaluate a novel non-cross resistant treatment option for relapsed or refractory (r/r) indolent lymphoma and MCL. At the time of the study design, no phase I study existed which had investigated the combination of gemcitabine and oxaliplatin in indolent and mantle cell lymphoma. Based on these considerations, the German Low Grade Lymphoma Study Group (GLSG) initiated a phase I/II trial to determine the maximum tolerated dose (MTD) of oxaliplatin in combination with gemcitabine and to explore the efficacy of the R-GemOx regimen with the prespecified MTD of oxaliplatin in r/r indolent and mantle cell lymphoma patients.
Patients and methods
Study design and patients
The R-GO study (NCT00954005) was a prospective, single-arm, multicenter national phase I/II trial conducted at 21 centers in Germany initiated by the GLSG. R-GO was designed to explore safety, tolerability and efficacy of rituximab, gemcitabine and oxaliplatin. Eligible patients were ≥ 18 years of age and had relapsed or refractory histologically confirmed indolent B-cell non-Hodgkin’s lymphoma or MCL according to the World Health Organization (WHO) classification from 1997 [37], were in need of treatment, had Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 2, an estimated life expectancy of 12 weeks or more and at least one bi-dimensionally measurable lesion. Patients must not have received cytotoxic treatment within 4 weeks before study entry. We excluded patients suitable for high-dose chemotherapy, with transformation in high grade lymphoma, HIV-infection, and hematopoietic insufficiency with leucocyte counts < 1.5 × 109 cells per L or thrombocyte counts < 100 × 109 cells per L unless caused by lymphoma. Key eligibility criteria are listed in the supplemental Appendix.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2008. All patients provided written informed consent.
Treatment
The initial study design intended to treat patients with the combination of gemcitabine and oxaliplatin (GemOx). After demonstrating a significant advantage in PFS for the addition of rituximab in combination with FCM in r/r lymphoma, an amendment was implemented on 17.11.2004 [24]. From this time, all patients received rituximab-gemcitabine and oxaliplatin (R-GemOx).
The study treatment comprised four 28-day cycles of R-GemOx. Rituximab was administered at a dose of 375 mg/m2 by intravenous infusion on day 0 or 1. Gemcitabine was applied intravenously at a dose of 1000 mg/m2 on days 1 and 15.
In the phase I dose escalation cohort, oxaliplatin was given in a 3 + 3 design on days 1 and 15 by intravenous infusion, starting at a dose of 70 mg/m2 and increased by steps of 10 mg/m2 until the maximum tolerated dose (MTD) was reached. If one out of three patients from one dose level experienced a DLT, the cohort was expanded to six patients. The MTD was defined as the dose at which a further dose step led to dose-limiting toxicities (DLTs) in ≥ 2 out of three, or ≥ 2 out of six patients after expansion of the cohort.
DLT was defined as one of the following side effects in the first cycle: Neutropenia < 0.5 × 109 cells per L, thrombocytopenia < 25 × 109 cells per L or bleeding with thrombocytopenia, neutropenia < 1.5 × 109 cells per L or thrombocytopenia < 100 × 109 cells per L on day 29 of the first cycle, non-hematological toxicity WHO grade 3 with exception of alopecia and nausea or emesis, or persistence of any toxicity WHO grade ≥ 2 until day 29 of the first cycle. No intraindividual dose increases were made. In case of a DLT on day 15, treatment was delayed until the absolute neutrophil count (ANC) recovered to > 0.5 × 109 cells per L, platelets to > 25 × 109 cells per L or bleeding was stopped.
In the phase II cohort, oxaliplatin was intravenously applied at a fixed dose of 70 mg/m2 (the MTD) on days 1 and 15.
An interim staging was performed after 2 cycles. In case of complete remission (CR), partial remission (PR), objective response (OR) or stable disease (SD), treatment should be completed with another 2 cycles. In case of progressive disease (PD) or non-hematological WHO toxicity grade 4, therapy was discontinued. Cycles were postponed for a maximum of 21 days, until ANC reached ≥ 1.5 × 109 cells per L and the platelet count reached ≥ 100 × 109 cells per L or any non-hematological response resolved to grade ≤ 2 (except for alopecia and emesis).
Patients discontinued study treatment if the next cycle was delayed by more than 21 days. However, these patients remained evaluable for the final analysis. Dose adjustments were recommended for patients who developed febrile neutropenia and WHO grade 4 hematotoxicity, with a 25% reduction in the planned doses of oxaliplatin and gemcitabine. For non-hematological toxicity of grade 3 (grade 4 for nausea and vomiting) a dose reduction by 25%, for non-hematological toxicity of grade 4 (except for nausea and vomiting) a dose reduction by 50% was foreseen.
Study endpoints
Primary endpoints
In the phase I dose escalation cohort, the primary end point was DLT of oxaliplatin in combination with gemcitabine and rituximab.
In the phase II cohort, the primary end point was overall response rate (ORR), defined by CR and PR. Patients who achieved PR or CR at the interim staging but progressed or died before the completion of the therapy were assessed as treatment failures.
Secondary endpoints
Secondary endpoints were safety, progression-free survival (PFS), time-to-treatment failure (TTF), overall survival (OS) and the rate of CR. Parameters were also reported in an intention-to-treat analysis of all registered patients. The efficacy analysis comprised all evaluable patients of the phase I and phase II. Three patients of the phase I that were excluded due to incorrect dosage were included in the response and survival analysis.
Progression-free survival was defined by the time of study registration until progression or death from any cause. Time-to-Treatment failure was the time from initiation of chemotherapy to its premature discontinuation. Overall survival was calculated from the time of study registration until death. The rate of complete remission was assessed after completion of study therapy.
Response to treatment was assessed by clinical examination, laboratory testing, bone marrow examinations and computed tomography scan of the neck, chest and abdomen at baseline, after 2 treatment cycles and at the end of therapy (after 4 cycles). Repeated bone marrow biopsies were only performed when lymphoma infiltration was present at baseline. Adverse events were assessed according to WHO toxicity grades (WHO Handbook for reporting results of cancer treatment, No. 48 (1979)).
Sample size and statistical analysis
In a previous trial of the GLSG, the FCM regimen (fludarabine, cyclophosphamide and mitoxantrone) induced an ORR of 58% [46%, 71%] [24]. The combination of FCM with rituximab significantly improved the ORR to 79% [67%, 88%] in the randomized trial [24]. A remission rate of 65% or less would certainly not be adequate for a randomized comparison with an antibody chemotherapy combination. A remission rate in the range of 75-85% could compete with previous standard therapies.
The study was powered to detect with probability 95% an improvement of 20% points in overall response rate (ORR) to 85% for rituximab, gemcitabine and oxaliplatin compared to an ORR of ≤ 65% (null hypothesis). Therefore, a one-sided binomial test with a significance level of 0.05 was performed to test the primary outcome.
To evaluate the MTD, a minimum of 9–24 patients needed to be enrolled to the phase I part of the study. According to the primary objective of the phase II trial, the null hypothesis should be rejected with a probability of 95% if the actual remission rate is 85%. Based on these considerations, 48 evaluable observations were required for the dose expansion cohort. Assuming a drop-out rate of 10% (e.g. change in histology, treatment not started), it was planned to enroll 53 patients to the phase II part.
It was planned to prematurely terminate enrollment as soon as more than 10 patients of the planned evaluable 48 patients in the phase II part failed to achieve a remission as the null hypothesis cannot be rejected any more at this point.
All secondary endpoints were analyzed in a descriptive manner, numeric estimates were reported with two-sided 95% confidence intervals (CI). Time-to-event variables were described by Kaplan-Meier-estimates.
Cut-off date was 16.10.2015, the latest available date for medically reviewed study data.
Results
Patient characteristics
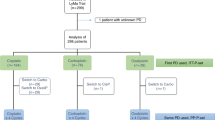
Between March 2004 and June 2009, the study enrolled 55 patients at 21 centers in Germany, 14 in the phase I and 41 in the phase II part.
In the phase I trial period, 13 of 14 patients enrolled have received the full 4 cycles of treatment. Nine patients were evaluable for primary analysis (Fig. 1).
A total of 41 patients were enrolled in the phase II trial period, whereof 34 remained evaluable for primary analysis (Fig. 1). Twenty-nine patients (71%) received all 4 treatment cycles.

Primary analysis population
Clinical characteristics of evaluable patients are shown in Table 1. The most common histological subtypes were FL (n = 18) and MCL (n = 14). Patients received a median of 2 prior lines of therapy. Baseline characteristics of enrolled patients are listed in supplemental Table 4.
Primary outcome
Phase I - dose finding
In the phase I population, 7 patients were treated with GemOx and 7 with R-GemOx. Three patients at the first dose level of 70 mg/m2 and 6 patients at the second dose level of 80 mg/m2 were evaluable. No patient on dose level 1 received rituximab. None of the patients at the first dose level had a DLT. At the second dose level, three out of six patients had a DLT. In this group, all patients received rituximab as provided for by the protocol amendment. One patient developed hypotension, one neurotoxicity grade 3 and one had neutropenia as well as thrombocytopenia grade 4. Accordingly, dose level 1 was chosen for the phase II trial period.
Efficacy
In the second study period, all patients received R-GemOx (n = 41). Twenty-eight patients (68%) completed all 4 cycles. In 9 patients (22%) a dose reduction was performed. After observing 11 patients who did not achieve a CR or PR, the recruitment was terminated prematurely in November 2009 for futility as per protocol because the null hypothesis could no longer be rejected (p = 0.45).
A total of 34 patients of 41 recruited patients were evaluable for the primary outcome analysis of the phase II cohort (Fig. 1).
Among 46 patients that were evaluable for the efficacy analysis, an overall response rate of 72% (33/46) was observed. One patient (2%) achieved CR and 32 patients PR (70%) (supplemental Table 2). Minimal remission (MR) and stable disease (SD) was observed in 4 patients (9%) and 5 patients (11%). Three patients (7%) progressed, and 1 patient (2%) died upon treatment with R-GemOx.
Among 22 patients with FL, ORR was 68% (14/22). Eleven out of 16 patients with MCL had CR or PR (ORR 69%).
Secondary outcomes
The secondary intention-to treat (ITT) analysis of all recruited patients in the phase II population revealed an ORR of 64% (25/39, one-sided 95% CI: [50%; 100%]) (supplemental Table 5). Among all 55 recruited patients, ORR was 65% (31/48).
After a median follow-up of 7.9 years and 7.7 years, median PFS of all evaluable patients (n = 46) was 1.0 years (Fig. 2A) and median OS was 2.1 years (Fig. 2B). Median PFS and OS of all 55 recruited patients was 0.8 and 1.9 years (supplemental Fig. 4).

Progression-free (A) and Overall Survival (B) of evaluable patients (n = 46*). Kaplan-Meier estimates of PFS (A) and OS (B) among patients evaluable for the efficacy analysis from the phase I and II population. Censoring is indicated by crosses *n = 3 patients from the phase I population that were excluded for the dose finding analysis due to incorrect dosage were included for the efficacy analysis
Median PFS and OS in FL patients was 1.3 (Fig. 3A) and 2.1 years, while MCL patients had a shorter PFS and OS of 0.7 (Fig. 3B) and 1.5 years.

Progression-free survival of Follicular Lymphoma (A) (n = 22*) and Mantle Cell Lymphoma patients (B) (n = 16*) among evaluable patients (n = 46*) Kaplan-Meier estimates of PFS among patients with Follicular Lymphoma (A) and Mantle Cell Lymphoma (B). Censoring is indicated by crosses. *n = 3 patients from the phase I population that were excluded for the dose finding analysis due to incorrect dosage were included for the efficacy analysis
Safety
Overall, hematological toxicity was the most frequent grade ≥ 3 adverse event (AE) (supplemental Table 3). Among patients evaluable for safety analysis (n = 43), grade ≥ 3 leukopenia was observed in 19 (44%), granulocytopenia in 17 (40%), thrombocytopenia in 16 (37%) and anemia in 10 (23%). Peripheral neurotoxicity grade ≥ 3 was reported in 2 (5%) patients. Grade 3 or 4 fever, infection, nausea and vomiting, and mucositis only occurred in 1 patient (2%) each. There was no treatment-related death.
Discussion
This single-arm, multicenter phase I/II clinical trial evaluated the maximum tolerated dose of oxaliplatin in combination with (rituximab-)gemcitabine and subsequently the efficacy of R-GemOx in recurrent or refractory MCL and indolent NHL. In this study, 70 mg/m2 of oxaliplatin was determined as MTD for the further conduct of the study. Remarkably, significantly higher doses of oxaliplatin ranging from 100 to 120 mg/m2 were applied in several published phase II studies investigating R-GemOx in lymphoma patients [34,35,36, 38]. However, at least in some of these trials G-CSF has been added to the regimen. In our study, the use of G-CSF was only optional. Despite the higher dosage, in studies that applied R-GemOx in the relapse setting, neutropenia grade ≥ 3 (47–73%), thrombocytopenia grade ≥ 3 (17–44%) and neurotoxicity grade ≥ 3 (0–8%) was comparable with the reported toxicity profile in our trial [34, 36, 38].
With an ORR of 72% the primary aim of the study was missed. Corazzelli and colleagues reported an ORR of 78% for R-GemOx in a study population that also included patients with DLBCL besides indolent lymphoma [38]. The response rate was higher in aggressive (79%) compared to indolent lymphoma (70%) (without MCL) [38]. Similar results were observed in another trial investigating R-GemOx in a mixed cohort of aggressive and indolent B-cell lymphoma with an ORR of 83% [34]. Notably, the addition of rituximab has substantially improved the response rate in the relapsed B-cell lymphoma [24, 38]. The disclosure of this data during the conduct of the study had prompted us to incorporate rituximab in this trial, nowadays being the reference treatment alongside a chemotherapy backbone.
The PFS of around 1 year is consistent with the results from the R-FCM trial without maintenance therapy [24]. In contrast, bendamustin-rituximab (BR) has been proven to be more effective [39].
Particularly in MCL, small subgroup analyses indicated decent response rates to R-GemOx [34, 38]. In our study, 69% of 16 MCL patients achieved PR or CR. However, as novel substances like ibrutinib have proven to be superior to conventional chemotherapy in early relapses of MCL, R-GemOx might be an alternative option for later relapses occurring after > 24 months [10]. In the era of cellular therapy, it might be used as bridging to CAR-T cell therapy.
In r/r FL, several novel treatments such as rituximab-lenalidomide, mosunetuzumab or CAR-T-cells have recently been approved, making R-GemOx obsolete in this indication [21, 40, 41].
Hematological toxicity constitutes by far the most common grade 3/4 adverse events in this trial, being in line with previously reported trials evaluating GemOx in r/r B-cell lymphoma patients [34, 36, 38]. However, except for one grade 4 infection no cases of severe infections or bleeding were reported.
Interestingly, when R-GemOx was applied in a first-line setting, hematotoxicity was significantly lower suggesting limited bone marrow reserve poses an important risk for developing severe cytopenias [35]. Comparable toxicities were observed with other relapse regimens such as R-FCM and BMR [24, 42]. In contrast, BR offers a more favorable safety profile [39].
Peripheral neurotoxicity grade ≥ 3 occurred in 5% of the patients corresponding to the results in previously published trials with oxaliplatin and gemcitabine [35, 36, 38].
In conclusion, the study failed to meet its primary aim, though it could be demonstrated that the combination of rituximab, gemcitabine with oxaliplatin at a dose of 70 mg/m2 is safe and feasible in r/r indolent and mantle cell lymphoma. Although the reported ORR of 72% demonstrates a significant anti-lymphoma efficacy, other combinations such as BR offer a more favorable efficacy and safety profile [39]. The approval of bispecific antibodies, immunomodulatory drugs and CAR-T-cells in recent years has displaced the use of conventional chemotherapy to a large part in the therapeutic landscape of r/r indolent and mantle cell lymphoma. In our opinion, R-GemOx only represents an alternative option in late relapses or for bridging to CAR-T-cell therapy specifically in MCL.
References
Teras LR et al (2016) US lymphoid malignancy statistics by World Health Organization subtypes CA Cancer J Clin, 2016. 66(6): p. 443–459
Dreyling M et al (2021) Newly diagnosed and relapsed follicular lymphoma: ESMO Clinical Practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 32(3):298–308
Zucca E et al (2020) Marginal zone lymphomas: ESMO Clinical Practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 31(1):17–29
Kastritis E et al (2019) Waldenström’s macroglobulinaemia: ESMO Clinical Practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 30(5):860–862
Dreyling M et al (2017) Newly diagnosed and relapsed mantle cell lymphoma: ESMO Clinical Practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 28(suppl4):iv62–iv71
Magnano L et al (2019) Life expectancy of follicular lymphoma patients in complete response at 30 months is similar to that of the Spanish general population. Br J Haematol 185(3):480–491
Provencio M et al (2017) Prognostic value of event-free survival at 12 and 24 months and long-term mortality for non-hodgkin follicular lymphoma patients: a study report from the Spanish Lymphoma Oncology Group. Cancer 123(19):3709–3716
Silkenstedt E, Dreyling M (2023) Mantle cell lymphoma-update on molecular biology, prognostication and treatment approaches. Hematol Oncol 41(Suppl 1):36–42
Sortais C et al (2020) Progression of disease within 2 years (POD24) is a clinically relevant endpoint to identify high-risk follicular lymphoma patients in real life. Ann Hematol 99(7):1595–1604
Visco C et al (2021) Outcomes in first relapsed-refractory younger patients with mantle cell lymphoma: results from the MANTLE-FIRST study. Leukemia 35(3):787–795
Casulo C et al (2022) Validation of POD24 as a robust early clinical end point of poor survival in FL from 5225 patients on 13 clinical trials. Blood 139(11):1684–1693
Visco C et al (2019) Time to progression of mantle cell lymphoma after high-dose cytarabine-based regimens defines patients risk for death. Br J Haematol 185(5):940–944
Hermine O et al (2016) Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL younger): a randomised, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. Lancet 388(10044):565–575
Dreyling M et al (2005) Early consolidation by myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission significantly prolongs progression-free survival in mantle-cell lymphoma: results of a prospective randomized trial of the European MCL Network. Blood 105(7):2677–2684
Zoellner AK et al (2021) Long-term survival of patients with mantle cell lymphoma after autologous haematopoietic stem-cell transplantation in first remission: a post-hoc analysis of an open-label, multicentre, randomised, phase 3 trial. Lancet Haematol 8(9):e648–e657
Dreyling M et al (2022) Efficacy and safety of Ibrutinib combined with Standard First-Line Treatment or as substitute for autologous stem cell transplantation in younger patients with Mantle Cell Lymphoma: results from the Randomized Triangle Trial by the European MCL Network. Blood 140(Supplement 1):1–3
Schouten HC et al (2003) High-dose therapy improves progression-free survival and survival in relapsed follicular non-hodgkin’s lymphoma: results from the randomized European CUP trial. J Clin Oncol 21(21):3918–3927
Lenz G et al (2004) Myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission prolongs progression-free survival in follicular lymphoma: results of a prospective, randomized trial of the German Low-Grade Lymphoma Study Group. Blood 104(9):2667–2674
Jurinovic V et al (2018) Autologous stem cell transplantation for patients with early progression of follicular lymphoma: a Follow-Up study of 2 randomized trials from the German low Grade Lymphoma Study Group. Biol Blood Marrow Transpl 24(6):1172–1179
Casulo C et al (2018) Autologous transplantation in Follicular Lymphoma with early therapy failure: a National LymphoCare Study and Center for International Blood and Marrow Transplant Research Analysis. Biol Blood Marrow Transpl 24(6):1163–1171
Leonard JP et al (2019) AUGMENT: a phase III study of Lenalidomide Plus Rituximab Versus Placebo Plus Rituximab in Relapsed or Refractory Indolent Lymphoma. J Clin Oncol 37(14):1188–1199
Wang ML et al (2013) Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 369(6):507–516
Dreyling M et al (2016) Ibrutinib versus Temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet 387(10020):770–778
Forstpointner R et al (2004) The addition of rituximab to a combination of fludarabine, cyclophosphamide, mitoxantrone (FCM) significantly increases the response rate and prolongs survival as compared with FCM alone in patients with relapsed and refractory follicular and mantle cell lymphomas: results of a prospective randomized study of the German low-Grade Lymphoma Study Group. Blood 104(10):3064–3071
Dumontet C et al (2001) Gemcitabine as a single agent in the treatment of relapsed or refractory low-grade non-hodgkin’s lymphoma. Br J Haematol 113(3):772–778
Fossa A et al (1999) Gemcitabine as a single agent in the treatment of relapsed or refractory aggressive non-hodgkin’s lymphoma. J Clin Oncol 17(12):3786–3792
Savage DG et al (2000) Gemcitabine for relapsed or resistant lymphoma. Ann Oncol 11(5):595–597
Santoro A et al (2000) Gemcitabine in the treatment of refractory Hodgkin’s disease: results of a multicenter phase II study. J Clin Oncol 18(13):2615–2619
Zinzani PL et al (2000) Value of gemcitabine treatment in heavily pretreated Hodgkin’s disease patients. Haematologica 85(9):926–929
Chau I et al (2001) An oxaliplatin-based chemotherapy in patients with relapsed or refractory intermediate and high-grade non-hodgkin’s lymphoma. Br J Haematol 115(4):786–792
Germann N et al (1999) Preliminary results on the activity of oxaliplatin (L-OHP) in refractory/recurrent non-hodgkin’s lymphoma patients. Ann Oncol 10(3):351–354
Machover D et al (2001) Dexamethasone, high-dose cytarabine, and oxaliplatin (DHAOx) as salvage treatment for patients with initially refractory or relapsed non-hodgkin’s lymphoma. Ann Oncol 12(10):1439–1443
Tixier F et al (2017) Comparative toxicities of 3 platinum-containing chemotherapy regimens in relapsed/refractory lymphoma patients. Hematol Oncol 35(4):584–590
El Gnaoui T et al (2007) Rituximab, gemcitabine and oxaliplatin: an effective salvage regimen for patients with relapsed or refractory B-cell lymphoma not candidates for high-dose therapy. Ann Oncol 18(8):1363–1368
Shen QD et al (2018) Gemcitabine-oxaliplatin plus rituximab (R-GemOx) as first-line treatment in elderly patients with diffuse large B-cell lymphoma: a single-arm, open-label, phase 2 trial. Lancet Haematol 5(6):e261–e269
Mounier N et al (2013) Rituximab plus Gemcitabine and oxaliplatin in patients with refractory/relapsed diffuse large B-cell lymphoma who are not candidates for high-dose therapy. A phase II Lymphoma Study Association trial. Haematologica 98(11):1726–1731
Harris NL et al (1999) World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee Meeting-Airlie House, Virginia, November 1997. J Clin Oncol 17(12):3835–3849
Corazzelli G et al (2009) Long-term results of gemcitabine plus oxaliplatin with and without rituximab as salvage treatment for transplant-ineligible patients with refractory/relapsing B-cell lymphoma. Cancer Chemother Pharmacol 64(5):907–916
Robinson KS et al (2008) Phase II multicenter study of bendamustine plus rituximab in patients with relapsed indolent B-cell and mantle cell non-hodgkin’s lymphoma. J Clin Oncol 26(27):4473–4479
Budde LE et al (2022) Safety and efficacy of mosunetuzumab, a bispecific antibody, in patients with relapsed or refractory follicular lymphoma: a single-arm, multicentre, phase 2 study. Lancet Oncol 23(8):1055–1065
Jacobson CA et al (2022) Axicabtagene ciloleucel in relapsed or refractory indolent non-hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol 23(1):91–103
Weide R et al (2007) High anti-lymphoma activity of bendamustine/mitoxantrone/rituximab in Rituximab pretreated relapsed or refractory indolent lymphomas and mantle cell lymphomas. A multicenter phase II study of the German Low Grade Lymphoma Study Group (GLSG). Leuk Lymphoma 48(7):1299–1306
Acknowledgements
The authors thank all patients who participated in this clinical trial and their families; all the personnel at the 21 sites across Germany who cared for the patients and made this trial possible; members of the Study Center for Hematology of the Department of Internal Medicine III of the LMU University Hospital, LMU Munich; Jörn Ohnesorge for contributions to data curation. A complete list of the NHL 2003 09 − 03 GO trial investigators is provided in the Supplementary Appendix.
Funding
The study received funding support by Eli Lilly, Roche and Sanofi.
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Consortia
Contributions
Conceptualization of the trial: Martin Hoffmann, Martin Dreyling, Wolfgang Hiddemann, Eva Hoster, Michael Unterhalt. Formal analysis: Michael Unterhalt, Eva Hoster, Vindi Jurinovic. Assembly of data, writing – original draft: Gabriel Scheubeck. Writing – review and editing: Martin Hoffmann, Eva Hoster, Martin Dreyling, Luca Fischer, Christian Schmidt. Collection of clinical data: Martin Hoffmann, Hans-Peter Böck, Ulrich Dührsen, Joachim Kaesberger, Stephan Kremers, Hans-Walter Lindemann, Luisa Mantovani. All authors approved the final version of the manuscript and are accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Scheubeck, G., Hoffmann, M., Jurinovic, V. et al. Rituximab, gemcitabine and oxaliplatin in relapsed or refractory indolent and mantle cell lymphoma: results of a multicenter phase I/II-study of the German Low Grade Lymphoma Study Group. Ann Hematol (2024). https://doi.org/10.1007/s00277-024-05689-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00277-024-05689-w