
Abstract
This study compared the efficacy and safety of CsA monotherapy with eltrombopag (E-PAG) + CsA combined treatment in children with severe aplastic anemia (SAA). The study including 30 children had SAA. Ten were a retrospective cohort treated with CsA monotherapy. The other 20 were prospective cohort received E-PAG + CsA. All patients were evaluated for partial (PR) and complete (CR) hematological response at 3, 6, and 12 months. overall response (OR), overall survival rates (OS), and treatment safety. OR for the E-PAG patients was 40% after 3 months of therapy. At 6 months, this had increased to 75% with significantly higher CR rate (40%) than in the CsA group (p = 0.0001). After a year of treatment, the CR for the E-PAG + CsA regimen had increased to 50% and the OR to 85%, compared to 20% in the CsA group (p = 0.0001). The OS at 12 months was 100% in the E-PAG+ CsA group compared to 80% in the CsA cohort. At 24 months, the OS in the E-PAG + CsA group was 90%. In conclusion, E-PAG+ CsA was found to be a safe and effective alternative treatment for children with SAA particularly in countries with limited resources.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aplastic anemia (AA) is a life-threatening condition characterized by pancytopenia and hypocellular bone marrow but without major dysplastic symptoms or marrow fibrosis [1, 2]. The incidence of acquired AA is about two children per million each year in Europe and North America but this number is 2–3 times higher in East Asia [2, 3]. AA affects both genders equally and can occur at any age. However, it is slightly more common during childhood, and 50% of cases occur in the first three decades of life [4, 5]. The pathogenesis of AA is multifactorial and may involve an abnormal hematopoietic microenvironment, hematopoietic stem cell/progenitor cell deficiencies, immunity disorders, or mutation of the genes responsible for hematopoiesis. Any of these factors can cause damage or primary defects of the stem cells or marrow microenvironment [6].
It can be difficult to differentiate between the acquired and inherited forms of this disease. Inherited causes are responsible for about 25–30% of pediatric cases of AA5. Acquired aplastic anemia may be idiopathic (>80%), post-infection (15% [particularly after hepatitis, Epstein-Barr virus, human immune deficiency virus, parvovirus, and mycobacteria]), or toxin/ drug-induced (4%) [7, 8].
Hematopoietic stem cell transplantation (HSCT) from a human leukocyte antigen (HLA)-matched sibling donor is the definitive curative therapy for AA [9, 10]. The major drawback of HSCT is that only 30% of patients have a suitably matched donor. Also, there is a risk of graft-versus-host disease (GVHD), which can cause mortality or morbidities with long-term effects on quality of life [11]. Unfortunately, allogeneic transplantation is not possible in most developing countries. The alternative treatment for AA in about two-thirds of cases is immunosuppressive therapy (IST) consisting of anti-thymocyte globulin (ATG) (horse or rabbit) and cyclosporine (CsA) [12, 13]. IST carries a satisfactory long-term response but 30–40% of patients do not respond and pancytopenia [9 ]or thrombocytopenia can continue after therapy, even in cases with improvements in life-threatening neutropenia [14]. HSCT and IST regimens can control the manifestation of AA effectively but both have limitations. HSCT is very expensive and requires a suitable donor. Many patients do not meet the requirements for HSCT. Yet, IST can leave a significant number of patients with persistent cytopenia.
CsA is sometimes used alone as a monotherapy in countries with poor resources [15, 16]. It is an effective immunosuppressant, inexpensive, accessible, can be administered to outpatients and is less toxic than combined treatment with ATG. The response rate to CsA monotherapy is between 30% and 50% [17, 18]. Thrombopoietin (TPO) is a glycoprotein class 1 hematopoietic cytokine, primarily manufactured in the liver [19]. It is an important regulator of hematopoiesis [20]. It acts through c-Mpl TPO receptors expressed in hematopoietic stem cells and progenitor cells. TPO causes signal transduction events that prevent apoptosis, improve cell viability, promote growth and possibly increase differentiation [21]. Eltrombopag (E-PAG) is an oral thrombopoietin mimetic that selectively binds to c-Mpl at the transmembranes and juxtamembranes of TPO receptors. It can circumvent the inhibitory effect of interferon-γ on HSCs and signal the c-MPL to yield a stimulatory effect. Noteworthy, interferon-γ found to have an inhibitory effect on the endogenous TPO by forming a heterodimer-hindering signalling through c-MPL and consequently
E-PAG promotes thrombopoiesis, the release of platelets from mature megakaryocytes [22], and other forms of hematopoietic stem cell differentiation [19,20,21, 23, 24]. E-PAG was approved by the Drug Administration (FDA) in 2008 as the first oral platelet growth factor treatment for adults with chronic immune thrombocytopenic purpura (ITP). In 2015, this approval was extended to include children aged 1 to 17 with chronic ITP. It has recently demonstrated excellent results as a treatment for AA, with trilineage responses in some patients and transfusion independence in many [25, 26]. It was licensed by the European Medicines Agency for AA in 2015. In 2017, the National Institutes of Health made E-PAG a standard of care in AA [27].
To the best of our knowledge, no previous research has compared the efficacy and safety of E-PAG + CsA with that of CsA alone in pediatric patients with SAA. We designed this study to explore the effectiveness and safety of eltrombopag added to CsA in pediatric severe aplastic anemia (SAA)
Methods
Design
This was a prospective, single-center clinical trial conducted at Assiut University Children’s Hospital in Egypt. The study enrolled 30 SAA patients between 1 and 18 years old. Our sample comprised two groups. Ten patients (CsA group) were a historical cohort who received cyclosporine (CsA) monotherapy from August 2016 to February 2018 because ATG was unavailable for financial reasons and E-PAG had not yet been approved by health insurers when these patients were treated. This cohort was our comparison group. Twenty patients fulfilled the eligibility criteriax were recruited prospectively in the study from October 20 19 to August 2021 and treated with E-PAG + CsA (E-PAG + CsA group). All patients were evaluated for their hematological response to treatment, and to determine complete response (CR), partial response (PR), and Overall response (OR) rates and treatment safety after 3, 6, and 12 months of treatment. Pre-treatment evaluations included a complete medical history and physical examination, complete blood count (CBC) with differential, serum chemistry, bone marrow aspiration and biopsy, viral serology, immunological tests, flow cytometric tests, a diepoxybutane clastogenic stress assay, and inherited bone marrow failure panel and HLA typing. Patient follow-ups were performed every 2–4 weeks and included a CBC and monitoring of kidney and liver function.
Eligibility criteria
Children with newly diagnosed and previously untreated SAA and adequate hepatic and renal functions who met the standard guidelines for the diagnosis and treatment of pediatric AA [28} and the modified Camitta criteria for SAA were eligible for inclusion in this study [28]. According to these criteria, a diagnosis of SAA may be made if bone marrow cellularity is <25% and/or at least two of the following criteria are met: (i) the absolute neutrophil count is below 0.5 × 109/L, (ii) the platelet count is below 20 × 109/L, (iii) the reticulocyte count is below 20 × 109/L.
The exclusion criteria were inherited bone marrow failure, myelodysplasia, AA secondary to infection or organ failure, underproduction anaemias secondary to B12, folate or iron deficiency, or with other reversible causes. Patients with documented hypersensitivity to any of the component medications were also excluded. The study was approved by Assiut University’s Ethical Committee for Clinical Research and informed consents were obtained from the guardians of trial participants before the study.
Treatment plan
Patients aged 1–5 years received an initial oral dose of E-PAG of 25 mg once daily. Those aged >5 years received an initial daily dose of 50 mg/day [29]. Dose was escalated by 25 mg every two weeks in all patients, and then maintained at the maximum dose when it was reached. In patients aged 1–5, the maximum dose was 75 mg; in patients over five, it was 150 mg. Adjustments and reductions of the E-PAG dose were made where necessary based on the pharmacokinetic data for ITP [30]. Patients experienced distinct adverse events in response to treatment were excluded from the study.
Oral CsA treatment in both groups was initiated at 5–10 mg/kg/day and the dose adjusted to maintain trough levels of 170–270 ng/ml. CsA was continued for at least 12 months as tolerated and, in those who responded, continued at a fixed daily dose for at least an additional 6 months before weaning. Serum CsA levels were measured every 2 to 4 weeks while patients were receiving the drug [29].
Supportive therapy
Supportive therapy was allowed for both cohorts throughout the study when required. This included granulocyte colony-stimulating factor (G-CSF), iron chelation, or platelet transfusion (if the count was <10,000/μL with an apparent bleeding tendency or <20,000/μL with fever) and red blood cell (RBC) transfusion (if hemoglobin was <7 g/dL or in the presence of significant symptoms, such as exertional dyspnea or anemic heart failure).
Primary outcome measures
The primary outcomes were safety, hematological response either CR or PR, and OR rates of combined E-PAG + CsA treatment after 3, 6, and 12 months, using the standard guidelines for the diagnosis and treatment of pediatric AA [31].
Response criteria
A hematological response was defined as a platelet count increase of at least 20 000/μL and/or platelet transfusion independence for a minimum of 8 weeks, a hemoglobin level increase of at least 1.5 g/L or a reduction in the number of PRBCs units transfused by at least four for eight consecutive weeks (compared with transfusion requirements during the 8 weeks preceding study treatment onset) and an increase of absolute neutrophil count (ANC) of >500/μL in patients with a pre-treatment count <500/μl. A PR was defined as a blood count no longer meeting the Camitta criteria for SAA and no transfusion dependence for platelets or red blood cells . A CR was defined as Hb levels of ≥100 g/l, a platelet count ≥100 × 109/L, ANC of ≥1 × 109/L and transfusion and growth factor independence. OR rates included all PR and CR within each group.
Secondary outcome measures
Secondary outcomes were the tolerability and toxicity of the E-PAG + CsA combination.
Statistical analysis
Data analyses were carried out using SPSS version 20. Descriptive statistics were expressed as frequencies and percentages for categorical data. Continuous variables were expressed as medians and interquartile ranges (IQR Q1 to Q3) as the sample size was small. Categorical data were compared using z score tests when the expected frequencies were less than five. The Mann-Whitney U test was used to determine differences in continuous variables between groups. A p-value of <0.05 was deemed statistically significant.
Results
Patient characteristics
A total of 30 patients were enrolled in this study. All patients were negative for Fancony anemia by Chromosome breakage analysis (CBA) and no one had any genetic mutations on the inherited bone marrow failure panel. Also all were negative for paroxysmal nocturnal hemoglobinuria (PNH) by Flow cytometry.
Ten of these received CsA alone (CsA group) and the other 20 received E-PAG + CsA (E-PAG + CsA). All children achieved adequate serum levels of cyclosporine.
The demographic and clinical characteristics and baseline CBC of the groups are shown in Table 1. Age and sex were matched between groups.
There was no significant difference between the groups in the baseline CBC. Bone marrow biopsies showed <10% nucleated cell proliferation in eight patients, 10–20% in fourteen patients, and 20–30% in eight patients.
Hematological response
Summaries of the hematological responses of both groups before treatment and after 3, 6, and 12 months of treatment are provided in Tables 2 and 3. All 30 patients were dependent on platelet and RBC transfusions before the treatment regimen began.
Hematological response after 3 months of treatment
At the 3 months evaluation, two patients in the E-PAG + CsA group (10% ) fulfilled the hematological criteria for CR and no longer required transfusion of packed red blood cells (PRBCs) or platelets. Six more patients (30%) in this group achieved PR. All six were still PRBC transfusion-dependent but no longer required platelet transfusion The remaining twelve (60%) E-PAG + CsA patients did not meet any of the response criteria and were still transfusion-dependent. In contrast, none of the 10 patients in the CsA group fulfilled the criteria for hematological response and all (100%) continued to require regular PRBC and platelet transfusions Additionally, ANC, Hg and platelets (Table 4) were significantly higher in the E-PAG group than the CsA group (p = 0.04, p = 0.01, p = 0.009, respectively).
Hematological response after 6 months of treatment
At the 6-month assessment a CR was found in seven (35%) of the E-PAG + CsA patients and PR in another eight patients (40%), all were transfusion-independent but three still required transfusion of blood components.
The remaining five E-PAG+ CsA patients (25%) showed no response to the combination therapy. In the CsA group, only two patients (20%) met the PR criteria and seven showed no response to treatment (Table 3). One patient died of a severe infection in the fourth month of CsA. All of the remaining nine were still PRBC and platelet transfusion-dependent. The highest response rate in the E-PAG+ CsA group was associated with a significant increase in ANC, Hb and platelet counts (Table 4) compared to that in the CsA group (p = 0.01, 0.01 and 0.004, respectively).
Hematological response after 12 months of treatment
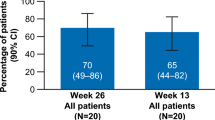
At 12 months, the number of patients in the E-PAG+ CsA group who respond to treatment increased to 17. Ten (50%) patients fulfilled the CR criteria, and seven (35%) patients had PR. All of them had become independent of platelet transfusion but three patients of PR still required PRBCs transfusion.
Three patient (15%) in the E-PAG+ CsA group had not responded to the therapy and was still dependent on transfusion support and waiting for a bone marrow transplant (BMT) (Table 2). In the CsA group, another patient died of a massive intracranial hemorrhage (Table 3). Two of the eight patients who had previously attained PR after 6 months of treatment achieved CR (25%) and were no longer transfusion-dependent. Another two patients displayed PR (25%), with one still dependent on PRBC and platelet transfusions every 8 weeks and the other requiring only platelet transfusion. Lastly, four patients in this group did not fulfil any of the hematological response criteria and were still transfusion-dependent. Furthermore, ANC, Hb and platelet were significantly higher in the E-PAG+ CsA group than the CsA group (p = 0.04, p = 0.047, and p = 0.01, respectively) (Table 4).
Overall response and survival rates
At 3 months, the OR rate in the E-PAG+ CsA group was 40% (10% CR and 30% PR) which differed significantly from 0% in the CsA group (p = 0.006). At 6 months, CRs in the E-PAG+ CsA had risen significantly to 35% (p = 0.0001), with an 75% OR rate. The OR rate in the CsA group had increased to 20% (Table 5).
Lastly, at the 1-year evaluation, the OR was 85% (50% CR and 35% PR) in the E-PAG + CsA group compared to 50% (25% CR and 25% PR) in the CsA group (p = 0.001). The overall survival rate at 1 year was 100% in the E-PAG+ CsA group and 80% in the CsA group.
Side effects and clonal evolution
Overall, both treatment arms had acceptable toxicities. None of the patients had to withdraw from the study due to adverse events. The most common adverse event in the E-PAG+ CsA group was indirect bilirubin elevation (n = 3, 15%). Two patients (10%) showed transient elevation of their liver enzyme levels and two patients experienced headaches. The abnormal levels of bilirubin and liver enzymes self-resolved or disappeared after transient E-PAG dose adjustment. Hirsutism, a known CsA side effect, occurred in two E-PAG+ CsA patient. Asymptomatic gum hypertrophy was noted in one patient but did not require a decrease in drug dosage (Table 2). In the CsA group, mild renal dysfunction was seen in one patient but this subsided after decreasing the CsA dose to 5mg/kg for two weeks. Hirsutism occurred in two patients (Table 3).
Long-term outcomes (at 24 months) of the E-PAG+ CsA group
At the 2-year evaluation of the E-PAG+ CsA group, eight ( 40%) of the patients who had responded completely still fulfilled the CR criteria without the need for transfusion support, while one patient (5%) had clinical signs of clonal evolution and one had relapse (5%). One of the seven PR patients now met the CR criteria while four patients (20%) had undergone BMT since the previous follow-up. The other two PR patients, their parents refused the BMT. The two E-PAG patient (10%) who had not responded to therapy had died of severe infection before reaching the top of the BMT waiting list while one had undergone BMT. There was no 2-year follow-up of the CsA group due to the retrospective data collection for that group.
Discussion
CsA has been used to treat AA patients lacking a donor, the financial means, or medical eligibility for HSCT since the 1980s. CsA is a potent immunosuppressant. It is inexpensive, easily available, and non-myelotoxic. CsA exerts its effects by suppressing early T cell activation, inhibiting lymphokine production [32, 33]. Several studies have demonstrated the effectiveness of CsA in the treatment of SAA [34, 35]. It has a 50% response rate in SAA refractory to treatment with ATG or anti-lymphocyte globulin (ALG) [36, 37].
In the present study, we evaluated CsA monotherapy in 10 children with SAA. The response rates were 20% and 50% after 6 and 12 months, respectively, in eight of the 10 patients (due to a mortality rate of 20%). These figures support those found in a previous retrospective evaluation of CsA monotherapy in 66 children with AA, which found a 47% response rate in SAA cases over a period ranging from 2 to 34 weeks [38]. Another study of 44 children evaluated combined CsA + corticosteroid treatment of AA. They reported a mortality rate of 44.9% (18 patients). 42.3% of the surviving patients showed a CR but most had non-SAA. After 18 months, the response rate was 34.6% [39].
Our results differ from those found in studies of CsA treatment for adults with SAA. Shetty et al. recorded a 56.2% response rate to CsA in 20 adult patients with SAA after 3 months39, while Ghazaly et al. found a 50% response in adults with SAA at 6 months [15].
E-PAG is a thrombopoietin receptor agonist that has proven effective in adults with AA. When combined with IST, E-PAG is well-tolerated, and produces improved response rates, recovery of blood cell counts, and restoration of trilineage haematopoiesis, even after drug discontinuation. E-PAG has recently been approved for use as a first-line treatment for adult patients with SAA in combination with standard IST28.
Studies to evaluate the efficacy of E-PAG in pediatric SAA are rare and most assess its use of E-PAG with standard IST of ATG + CsA (Table 6) [29, 40,41,42].
To the best of our knowledge, this is the first prospective study to evaluate the efficacy and safety of E-PAG in combination with CsA alone in children with SAA.
In the present study, the OR rate of E-PAG patients was 40% after 3 months of therapy, with two patient achieving CR and six, PR. At 6 months, the OR was 75% with a CR of 35% and most of these patients were independent of transfusion support. In contrast, the CsA group experienced 20% PR at 6 months. After a year of regular treatment, the rate of complete responses to the E-PAG+ CsA regimen had increased to ten patients (50%), with an OR rate of 85%. The survival rate at 12 months was 100% compared to 80% in the CsA cohort. Additionally, the E-PAG+ CsA group experienced a 90% survival rate at 24-month follow-up, although one of the survivors underwent a hematological relapse. Elevated transaminase levels or indirect elevation of bilirubin levels occurred in five of the patients treated with E-PAG+ CsA. Renal insufficiency occurred in one CsA patient. The elevations and renal insufficiency were corrected with dose adjustments. Hirsutism, a known side effect of CsA, was observed in both cohorts.
Lesmana et al. [29] conducted a retrospective comparison of children with SAA treated with IST and E-PAG and those treated with standard IST. The CR rate in the IST and E-PAG group was 29% at 6 months and 58% at 12 months, while the OR rate was 77.7% at six and 100% 12 months. However, they recorded a significantly higher rate of renal insufficiency and elevated transaminase in the E-PAG cohort. These results were somewhat similar to our own, which may suggest that E-PAG and CsA exert similar effects to E-PAG and IST.
Another recent study of the efficacy and safety of E-PAG as a first-line treatment of pediatric AA found CR and OR rates at 6 months of 64.3% (9/14 case) and 78.6% (11/14 cases), respectively, with a 100% survival rate at [24] months and no relapse or intolerable side effects. They found a significantly higher rate of CR rate in SAA children treated with E-PAG + IST than that seen in the historical cohort [41]. This data shows a notable convergence between the results of the different treatment regimens used in that study and this one.
Fang et al., [40 ] found IST + E-PAG to be more effective than IST alone in children with SAA. The CR and OR were significantly higher in their IST + E-PAG group than their IST group after 6 months (CR: 17.9% vs. 50%; p < 0.05, OR: 69.2% vs. 94.4%, p < 0.05). The present study found roughly equivalent, significantly higher CRs and ORs, in children treated CsA + E-PAG to the outcomes obtained with children treated with IST + E-PAG.
In a study of Scheinberg et al., [42] 54 treatment-naïve adults with SAA treated with E-PAG + CsA for 6 months, the goal OR rate was met by 46.3% of patients with 5.4% achieving CR. The OR rate at 3 months was 40.7%. Townsley et al. [27] conducted a prospective study of a cohort of 92 patients, 19 of whom were children, treated with IST + E-PAG. This study divided patients into three cohorts according to the day of E-PAG treatment initiation. The third cohort in whom E-PAG treatment was initiated on day one reported an OR rate of 80% and 87% after 3 and 6 months, respectively, while the CR was 30% at 3 and 6 months. They found the beneficial effects of E-PAG to be directly proportionate to the length of exposure to the drug. In addition to the hematologic response, bone marrow was found to be highly enriched with hematopoietic stem cells and multipotent progenitors after three and 6 months of therapy. Moreover, Hwang et al. [43], investigated E-PAG in AA patients on a non-trial all-comer basis over a 4.5-year period. They concluded that E-APG in AA patients was feasible, safe, and associated with very good responses. Instead, Groarke et al. evaluated whether the addition of EPAG to IST enhanced ORR in pediatric SAA. There was no significant difference in survival between the pediatric EPAG group and either the pediatric IST group. They concluded that, addition of eltrombopag to IST did not afford any clear therapeutic advantage to pediatric patients with SAA [44].
IST in the form of ATG and CsA is well-established as an alternative treatment for patients with SAA when an HLA-matched familial donor is not available [10, 12, 13, 45]. However, in healthcare systems with inadequate resources, few patients can afford such an expensive combination. E-PAG + CsA is an available, safe, easily monitored treatment option for pediatric SAA in developing nations where economic considerations are paramount. Combined cyclosporine + eltrombopag was found to be an effective and safe alternative treatment for pediatric SAA, particularly in countries with limited resources. This study was limited by its small sample size and the lack of similar studies in pediatric groups. A larger prospective study with longer follow-up is essential to evaluate response stability
References
Guinan EC (2009) Acquired aplastic anemia in childhood. Hematol Oncol Clin North Am 23(2):171–191
Young NS, Bacigalupo A, Marsh JC (2010) Aplastic anemia Pathophysiology and treatment. Biol Blood Marrow Transplant J Am Soc Blood Marrow Transplant 16(1):19–25
Issaragrisil S, Kaufman DW, Anderson T et al (2006) The epidemiology of aplastic anemia in Thailand. Blood 107(4):1299–1307
Montané E, Ibáñez L, Vidal X et al (2008) Catalan Group for Study of Agranulocytosis and Aplastic Anemia. Epidemiology of aplastic anemia: A prospective multicenter study. Haematologica 93(4):518–523
Helge D, Hartung M, Timothy S et al (2013) Acquired Aplastic Anemia in Children. Pediatr Clin North Am 60:1311–1336
Scheinberg P, Young NS (2011) How I treat aplastic anemia. Blood 120(6):1185–1196
Rauff B, Idrees M, Shah SA et al (2011) Hepatitis associated aplastic anemia: A review. Virol J 28(8):87
Zhang J, Yang T (2015) Meta-analysis of association between organo phosphorus pesticides and aplastic anemia. Zhonghua Liu Xing Bing Xue Za Zhi 36(9):1005–1009
Scheinberg P (2012) Aplastic anemia: therapeutic updates in immunosuppression and transplantation. Hematology Am Soc Hematol Educ Program 2012(1):292–300
Marsh JC, Ball SE, Cavenagh J et al (2003) British Committee for Standards in Haematology. Guidelines for the diagnosis and management of aplastic anaemia. Br J Haematol 147(1):43–70
Gupta V, Eapen M, Brazauskas R, , et al (2010).Impact of age on outcomes after bone marrow transplantation for acquired aplastic anemia using HLA-matched sibling donors. Haematologica 95(12):2119–2125
Dezern AE, Brodsky RA (2011) Clinical management of aplastic anemia. Expert. Rev Hematol 4(2):221–230
Scheinberg P, Nunez O, Weinstein B, Scheinberg P et al (2011) Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med 365(5):430–438
Marsh JC, Mufti GJ (2014) Eltrombopag: a stem cell cookie? Blood 123(12):1774–1775. https://doi.org/10.1182/blood-2014-02-553404
Al-Ghazaly J, Al-Dubai W, Al-Jahafi AK et al (2005) Cyclosporine monotherapy for severe aplastic anemia: A developing country experience. Ann Saudi Med 25(5):375–379
Mahapatra M, Singh PK, Agarwal M, Prabhu M et al (2015) Epidemiological clinico-haematological profile and management of aplastic anaemia: Aiims experience. J Assoc Physicians India 63:30–35
Varma N, Malhotra P, Singh S, Sharma DR (1999) Cyclosporine: A monotherapy in young Indian aplastic anaemia patients. J Indian Med Assoc 97(7):505–506
Hanif S, Naz F, Siddiqui E, Raza J (2007) Acquired aplastic anaemia, treatment in a developing country. Pak J Med Sci 23(3):370–374
Garnock-Jones KP, Keam SJ (2009) Eltrombopag. Drugs 69(5):567–576
Qian H, Buza-Vidas N, Hyland CD et al (2007) Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell 1:671–684
Kaushansky K (2008) Historical review: megakaryopoiesis and thrombopoiesis. Blood 111(3):981–986
Alvarado LJ, Andreoni A, Huntsman HD et al (2017) Heterodimerization of TPO and IFNγ impairs human hematopoietic stem/progenitor cell signaling and survival in chronic inflammation [abstract]. Blood 130:4
Bussel JB, Cheng G, Saleh MN, Psaila B et al (2007) Eltrombopag for the treatment of chronic idiopathic thrombocyte openic purpura. N Engl J Med 357(22):2237–2247
De Laval B, Pawlikowska P, Petit-Cocault L (2013) Thrombopoietin-increased DNA-PK-dependent DNA repair limits hematopoietic stem and progenitor cell mutagenesis in response to DNA damage. Cell Stem Cell 12(1):37–48
Olnes MJ, Scheinberg P, Calvo KR, Desmond R, Tang Y, Dumitriu B, Parikh AR, Soto S, Biancotto A, Feng X, Lozier J (2012) Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med 367(1):11–19
Desmond R, Townsley DM, Dumitriu B et al (2014) Eltrombopag restores trilineage hematopoiesis in refractory severe aplastic anemia that can be sustained on discontinuation of drug. Blood 123(12):1818–1825
Townsley DM, Scheinberg P, Winkler T et al (2017) Eltrombopag added to standard immunosuppression for aplastic anemia. N Engl J Med 376(16):1540–1550
Camitta B, Rozman C, Marin P, Nomdedeu B, Montserrat E et al (1988) Criteria for severe aplastic anaemia. Lancet 331:303–304
Lesmana H, Jacobs T, Boals M, Gray N et al (2021) Eltrombopag in children with severe aplastic anemia. Pediatr Blood Cancer 68(8):e29066
Wire MB, Li X, Zhang J, Sallas W, Aslanis V, Ouatas T (2018) Modeling and simulation support eltrombopag dosing in pediatric patients with immune thrombocytopenia. Clin Pharmacol Ther 104(6):1199–1207
Barone A, Lucarelli A, Onofrillo D, Verzegnassi F, Bonanomi S, Cesaro S, Fioredda F, Iori AP, Ladogana S, Locasciulli A, Longoni D (2015) Diagnosis and management of acquired aplastic anemia in childhood. Guidelines from the Marrow Failure Study Group of the Pediatric Haemato-Oncology Italian Association (AIEOP). Blood Cells Mol Dis 55(1):40–47
Tichelli A, Schrezenmeier H, Bacigalupo A (2000) Immunosuppressive treatment of aplastic anemia. In: Schrezenmeier H, Bacigalupo A (eds) Aplastic anemia: pathophysiology and treatment. Press Cambridge University Book Company, Cambridge UK, pp 154–196
Guinan EC (1997) Clinical aspects of aplastic anemia. Hematol Oncol Clin North Am 11(6):1025–1044
Mackenzie IL, Manoharan A (1988) Cyclosporine A inthein the treatment of aplastic anemia. Am J Hematol 28(3):211
Tötterman TH, Höglund M, Bengtsson MA et al (1989) Treatment of pure red cell aplasia and aplastic anemia with cyclosporine. Long term clinical effects. Eur J Haematol 42(2):126–133
Leonard EM, Raefsky E, Griffith P et al (1989) Cyclosporin therapy of aplastic anemia, congenital and acquired red cell aplasia. Br J Haematol 72(2):278–284
Hinterberger‐Fischer M, Höcker P, Lechner K et al (1989) Oral cyclosporin-A is effective treatment foruntreatedfor untreated and also for previously immunosuppressed patients with severe bone marrow failure. Eur J Haematol 43:136–142
Maschan A, Bogatcheva N, Kryjanovskii O et al (1999) Results at a single centre of immunosuppression with cyclosporine-A in 66 children with aplastic anaemia. Br J Haematol 106(4):967–970
Shetty M, Narendra AM, Adiraju KP et al (2016) Study of Aplastic Anaemia with Cyclosporine in Resource Poor Setting. J Clin Diagn Res 10(6):OC15–OC18
Fang M, Hua Song H, Zhang J et al (2021) Efficacy and safety of immunosuppressive therapy with or without eltrombopag in pediatric patients with acquired aplastic anemia: A Chinese retrospective study. Pediatr Pediatr Hematol Oncol 38(7):633–646
Jie M, Fu L, Li S, He Y et al (2021) Efficacy and safety of eltrombopag in the first-line therapy of severe aplastic anemia in children. Pediatr Hematol Oncol 38(7):647–657
Scheinberg P, Maier J, Arikan OO et al (2017) Eltrombopag and Cyclosporine as First-Line Therapy in Patients with Severe Acquired Aplastic Anemia: A Two-part, 5-year, Single-Arm, Multicenter, Open-label, Phase 2 Trial (SOAR). Clin Lymphoma Myeloma Leuk 17:s389–s390
Hwang YY, Gill H, Chan TS, Leung GM, Cheung CY, Kwong YL (2018) Eltrombopag in the management of aplastic anaemia: real-world experience in a non-trial setting. Hematology 23(7):399–404
Rogers ZR, Nakano TA, Olson TS et al (2019) Immunosuppressive therapy for pediatric aplastic anemia: a North American Pediatric Aplastic Anemia Consortium study. Haematologica 104(10):1974–1983
Patel BA, Groarke EM, Lotter J et al (2019) Long-term outcomes in patients with severe aplastic anemia treated with immunosuppression and eltrombopag: a phase 2 study. Blood 139(1):34–43
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
Mervat A M Youssef, coordinated the research, conducted literature searches and wrote the manuscript. Mai A Abdelfattah designed, collected date and reviewed and revised the manuscript, Mohammed H ghazaly reviewed and revised the manuscript. and contributed with the data collection-
Corresponding author
Ethics declarations
Ethics approval
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2008
The study was approved by Assiut University’s Ethical Committee for Clinical Research.
Informed consent
Signed statements of informed content to participation and publication were obtained from the guardians of trial participants before the study. The consent requirement was waived for retrospective participants by the above-named ethics committee
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Youssef, M.A.M., Ghazaly, M.H. & Abdelfattah, M.A. Alternative treatment modality for severe aplastic anemia in a resource-limited setting: a single-institution prospective cohort study from Upper Egypt. Ann Hematol 102, 2997–3006 (2023). https://doi.org/10.1007/s00277-023-05440-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-023-05440-x