
Abstract
Despite the existence of well-founded data around the relationship between reactive oxygen species (ROS) and glucose-6-phosphate dehydrogenase (G6PD), current research around G6PD-deficient patients with viral infections, and limitations as a result of their condition, are inadequate. Here, we analyze existing data around immunological risks, complications, and consequences of this disease, particularly in relation to COVID-19 infections and treatment. The relationship between G6PD deficiency and elevated ROS leading to increased viral load suggests that these patients may confer heightened infectivity. Additionally, worsened prognoses and more severe complications of infection may be realized in class I G6PD-deficient individuals. Though more research is demanded on the topic, preliminary studies suggest that antioxidative therapy which reduces ROS levels in these patients could prove beneficial in the treatment of viral infections in G6PD-deficient individuals.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glucose-6-phosphate dehydrogenase (G6PD) is an enzyme responsible for reducing NADP+ to NADPH via oxidation byglucose-6 phosphate. Produced NADPH is then utilized by glutathione reductase to reduce oxidized glutathione back into its active state. Thereby, the reduced glutathione works as an antioxidant to neutralize reactive oxidative species (ROS) via electron donation [1]. The production of ROS, such as hydrogen peroxide or superoxide and hydroxyl radicals, is an integral part of human metabolism. Moreover, inflammation or adverse medication effects may lead to elevated ROS within organisms [1, 2]. G6PD deficiency is one of the most common enzymopathies with more than 400 million individuals affected worldwide, mostly men [3]; it is an X-linked recessive genetic disorder characterized by the markedly reduced enzymatic activity of G6PD as a result of defective production. Low G6PD levels lead to an increase in oxidized and non-functional glutathione. The consequent ROS accumulation in the body is inevitably associated with tissue injury [1], since ROS are known to damage cell components, particularly DNA, leading to loss of function, induction of apoptosis, or even carcinogenesis [2]. Most patients with G6PD deficiency are asymptomatic until exposed to environmental triggers that increase ROS production; potential triggers include viral infections, certain foods—notoriously fava beans—or various medications: sulfa drugs or particular antibiotics [4]. Following trigger exposure, hemolysis, anemia, jaundice, or even renal failure may be classically present, though occurring symptoms may differ between variants (Table 1). Regardless of variant, ROS-mediated erythrocytic hemolysis may drive the patient into an anemic state. Furthermore, hemoglobin damaged by ROS may accumulate and form Heinz bodies within erythrocytes [1, 5].
An early in vitro study has illustrated that a deficiency in G6PD may increase human susceptibility to infection by coronavirus 229E [6]. Though similar studies on severe acute respiratory syndrome coronavirus 2 (SARS CoV-2) have not yet been performed, due to existing similarities in these pathogens and the immunological response they trigger, recent studies have suggested that G6PD deficiency may potentially impact the prognosis, clinical outcomes, and severity of SARS-CoV-2 infection; this may be due to the nature of the condition or via the limitation of therapeutic options available to these patients [7,8,9,10,11,12]. Elevated levels of oxidative stress as a result of G6PD deficiency cultivate a favorable environment for viral replication, consequently deteriorating the course of infection [12]. Additionally, the activity of neutrophils, cytokines, and inflammasomes is typically impaired in those with G6PD deficiency, which may cause increased susceptibility to coronavirus disease 2019 (COVID-19) and other viral infections [13,14,15,16,17,18]. Furthermore, hydroxychloroquine, a medication previously used to treat COVID-19 despite being rapidly abandoned as a therapeutic option, increases oxidative stress in patients which may incidentally trigger hemolytic anemia; this effect may drastically exacerbate symptoms in patients with G6PD deficiency. Current FDA recommendations suggest remdesivir as a treatment for COVID-19 patients; however, those with G6PD deficiency may exhibit more severe side effects than those with in-tact G6PD as a result of a decreased threshold for processing ROS in the liver. These examples illustrate some limitations of pharmacological agents which may be utilized in treating COVID-19 among this demographic of patients.
Altogether, the aim of this paper is to review existing literature regarding the impact of G6PD deficiency on the course and management of COVID-19 infections. In the following sections of the manuscript, we emphasize the role of immunologic disturbances associated with G6PD deficiency, such as neutrophil dysfunction, impaired inflammasome activation, and disruption in the NF-κB signaling pathway, which may contribute to increased ROS production, viral replication, and contagiousness in SARS-CoV-2 infection. Furthermore, we pay attention to COVID-19 complications potentially associated with ROS accumulation, including hemolysis, thrombosis, and elevated cardiovascular risk. Moreover, we show limitations on COVID-19 treatment in G6PD-deficient individuals resulting from the pathophysiological background of the underlying disease. Finally, we discuss current findings and suggest directions for further research regarding the relationship between COVID-19 and G6PD deficiency.
Impaired immune response against COVID-19 in G6PD-deficient patients.
Recent studies have suggested that G6PD plays an important role in immune response and that G6PD deficiency may increase susceptibility to infections [12, 16]. Neutrophils are the most abundant leukocytes in the system and play a major role in the innate immune response. They are the first cells to arrive at the infection site and are responsible for the neutralization of pathogens and the recruitment of additional immune cells. Although the role of neutrophils in bacterial infection is well understood, the mechanism of the neutrophilic response in viral infection has not yet been extensively studied [15, 19, 20]. There are several studies suggesting that neutrophils have the ability to phagocytose various viruses, including Influenza, Cytomegalovirus, and Herpes simplex viruses [20,21,22,23,24]. However, present understanding regarding the role of neutrophils in COVID-19 infection is notably limited.
Neutrophil extracellular traps (NETs) are comprised of modified neutrophilic chromatin that is expelled into the system to neutralize and prevent the dissemination of microbes, whilst concurrently alerting the immune system of the infection [25,26,27]. Early studies have confirmed that G6PD-deficient patients may display impeded neutrophilic function and impaired NETs, potentially affecting the immune system’s ability to clear infections [15,16,17,18]. Studies and case reports have revealed that NET formation may increase dramatically in COVID-19 infections, suggesting that neutrophils and NETs play a substantial role in immunity against SARS-CoV-2 [28, 29]. Within these studies, increased NET formation was also specifically associated with certain complications of COVID-19 infections, including vascular occlusion and pneumocyte damage, allowing NETs to be labelled as multi-purposed in the immune response against SARS-CoV-2 and other viruses [26, 28,29,30,31].
Interleukin-1β (IL-1β) is a mediator of immunity produced by monocytes and macrophages during infection and is essential to the host response against pathogens [32, 33]. Studies have shown that IL-1β and NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome activation is crucial in inhibiting viral replication via maintenance of optimal interferon and immune response [34, 35]. The disruption of IL-1β and NLRP3 can lead to impairment of the innate cellular immune response, which may have varying clinical implications during infections [13]. IL-1β binds to the interleukin-1 receptor (IL-1R) to activate myeloid differentiation primary response 88 (MyD88) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB); these upregulate the expression of genes specific for immune-mediated inflammation, adaptive immunity, and antiviral response [33, 34]. Type I Interferons (IFNs) and IL-1β are known to work together to inhibit viral replication [34, 35]. One study supports that the absence of IL-1R signaling results in a reduction of type I IFNs, thus leading to an increase in viral load and potentially increasing host mortality rates as well [34]. Furthermore, studies suggest that the reduction of IL-1β and inhibition of the NLRP3 inflammasome could impair the anti-viral immune response, exposing patients with IL-1β and NLRP3 inhibition to a more severe course of a viral infection than those without. A recent study revealed a significant decrease in IL-1β expression and defective NLRP3 inflammasome activation in G6PD-deficient patients [13]. This supports that G6PD-deficient patients may be more susceptible to viral and bacterial infections, which may also encompass COVID-19 infections. Elevated levels of IL-1β and the activation of NLRP3 inflammasome were observed in SARS-CoV-2 infected patients [36, 37], implying that they are involved in immunologic defence mechanisms against COVID-19 infection.
Elevated ROS in G6PD-deficient patients favors viral replication.
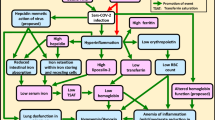
It has been shown that viral infections may trigger NF-κB activation, leading to the inhibition of viral replication [6, 38]; the NF-κB signaling pathway is also involved in regulating oxidative stress in the human body [39, 40]. However, in G6PD-deficient cells, the ability to activate any NF-κB-mediated immune response is impaired due to an imbalance in reduction and oxidation mechanisms (Fig. 1) [38]. In these cells, an increased viral load was detected with concomitant ROS elevation. Meanwhile, the subsequent introduction of antioxidant agents like lipoic acid led to the alleviation of these conditions [6, 41, 42]. The results of these in vitro studies suggest that elevated ROS facilitates the replication process in an array of viruses, including coronaviruses, especially in G6PD-deficient cells where the level of ROS is dysregulated [6, 38, 41, 42]. As long as the in vitro studies translate to human physiology, these results further support the positive feedback loop between ROS concentration and viral load, resulting in a higher risk of severe infection in G6PD-deficient patients. This increased viral load can therefore be potentially attributed to the absence of specific and efficient antiviral medication targeting SARS-CoV-2. Consequently, the management of ROS levels seems to be beneficial in the treatment of COVID-19 patients. If an increased viral load corresponds with increased infectivity, COVID-19 patients with G6PD deficiency could therefore be more contagious than patients with in-tact G6PD enzymes [43].

Pathways that increase viral replication [1, 6, 13, 38, 41, 42]. The diagram iterates the suggested pathways that could lead to increased viral replication in G6PD deficiency. Interleukin-1β and NLRP3 inflammasome were found to be impaired in G6PD deficiency, which results in impeded type 1 interferons level. The low level of G6PD enzymes and viral infection gives rise to the accumulation of ROS. Both pathways favor viral replication
Risk of cardiovascular and hemolytic disease in G6PD-deficient patients.
It is well understood that patients with G6PD deficiency are more prone to thrombotic and hemolytic events [10]. To elucidate this concept, clinical cases have revealed that COVID-19 infection in G6PD-deficient patients could further elevate the risk of such events, leading to more severe clinical outcomes such as intravascular and extravascular hemolysis [1, 10, 44, 45]. In G6PD-deficient patients, acute hemolysis occurs when an elevation of oxidative stress is triggered by viral infection, certain medications, or even fava beans. Typically, increased NADPH, which reduces glutathione, is able to compensate for elevated levels of oxidative stress caused by such triggers. Nevertheless, the presence of impaired G6PD activity may result in ROS accumulation, causing severe hemolysis; Fig. 1 illustrates how G6PD concentration correlates with ROS levels. When ROS levels exceed the metabolic capacity of active glutathione, cell damage, thrombosis, and acute hemolytic anemia may occur; concomitant viral infection may exacerbate existing symptoms of infection and cause the failure of multiple organs [3, 9, 10, 46]. Moreover, this condition could be further exacerbated in G6PD-deficient patients who are elderly, as increased age is correlated with reduced G6PD expression [14].
An increased incidence of venous thromboembolism in patients with G6PD deficiency has been addressed in previous literature, attributed to various potential causes [45, 47]. Factor V Leiden is a prothrombotic condition where the degradation of clotting factor V is impaired, resulting in an increased risk of venous thrombosis [47]. One study report that Factor V Leiden is present in 11% of G6PD-deficient patients, a significantly higher incidence than the 2.4% reported in the normal population of Dalmatia [47].
Additionally, the advanced progression and rupture of atherosclerotic plaques may contribute to an increased incidence of thrombosis. The combination of low NADPH concentration and high oxidative stress may lead to the progression and pathogenesis of atherosclerosis [48]. Higher levels of ROS and inflammation within atherosclerotic lesions favor the loss of collagen and suppress its production; this is associated with thinning of the fibrous cap leading to decreased stability of atherosclerotic plaques and an increased risk of plaque rupture [49]. Rupture of atherosclerotic plaques is inherently associated with endothelial disruption, one element of the Virchow Triad, and thereby one of three main factors triggering thrombosis; static blood flow and hypercoagulable states like Factor V Leiden comprise the other two elements [49, 50]. Thus, elevated oxidative stress does not merely accelerate the progression of atherosclerosis, but can also increase the risk of plaque rupture, which may directly lead to a thrombotic event.
In addition to an increased incidence of hemolytic and thrombotic events, increased risk of cardiovascular disease development has been observed in previous studies; this includes coronary heart disease, cerebrovascular disease, peripheral arterial disease [51, 52]. One study specifically identified the infection as a risk factor of cardiovascular disease development in elderly G6PD-deficient patients [51]. Although the study focused on a specific bacterial infection, it is fair to assume that other pathogens may elicit a similar response [51]. Further studies could be done to evaluate this assumption. Moreover, it is consequently logical to think that G6PD-deficient patients are more prone to COVID-19-induced myocardial injury and other cardiovascular complications [48]. The aforementioned impaired ability to neutralize ROS may also increase the risk of reperfusion injury in this patient population. Therefore, continual monitoring of cardiac function in early stages of infection could prove beneficial in patients with G6PD deficiency.
Limitations on COVID-19 treatment in G6PD-deficient patients.
Historically, chloroquine and hydroxychloroquine have been utilized to induce oxidative stress in order to kill malarial parasites [9, 10, 53]. However, at the start of the COVID-19 pandemic, both drugs were used to treat COVID-19 infections due to their ability to increase endosomal pH, inhibiting both the fusion of SARS-CoV-2 and the angiotensin-converting enzyme 2 (ACE2) receptor presented on the host cell membrane [54, 55]. In either case, the mechanism of these drugs cause increased systemic oxidative stress; this fairly establishes their use as contraindicated in patients with G6PD deficiency [4]. In multiple case reports, hydroxychloroquine administration for the treatment of COVID-19 in G6PD-deficient patients revealed a dramatic drop in hemoglobin and haptoglobin, indicating erythrocyte breakdown [56,57,58,59,60]. Since COVID-19 infection in patients with G6PD may independently promote hemolysis, prescribing hydroxychloroquine or chloroquine may exacerbate this hemolytic effect as a result of increased oxidative stress. Although recent studies have proven chloroquine and hydroxychloroquine inefficient in treating COVID-19, it is still worthwhile to evaluate whether these drugs are safe for G6PD-deficient patients [61].
Currently, according to the Food and Drug Administration (FDA), remdesivir is the first medication to be approved for the treatment of COVID-19 [61]. Remdesivir is a nucleoside analog that inhibits the RNA-dependent RNA polymerase (RdRp) of SARS-CoV-2, which results in impaired viral replication [62]. Since adverse hepatotoxic effects are common with remdesivir, severe impairment of hepatic function is a strict contraindication of its use [63, 64]. In G6PD-deficient patients, liver vulnerability is often expected due to the low concentration of G6PD enzymes and high oxidative stress in hepatocytes. It has been shown that liver enzymes, including alanine transaminase (ALT) and aspartate transaminase (AST), are significantly higher in G6PD-deficient patients than in unaffected individuals [65]. This finding supports the idea that the liver in G6PD-deficient patients is more susceptible to damage and drug-related toxicity; therefore, medications with hepatotoxic effects could prove injurious to the already vulnerable G6PD-deficient liver. At present, there are no clinical studies evaluating the impact of medication use in G6PD-deficient patients. Thus, the safety of remdesivir utilization in G6PD-deficient subpopulations needs to be elucidated and requires further research. Additionally, other antiviral medications, such as molnupiravir and Paxlovid (combination of nirmatrelvir and ritonavir), were evaluated and authorized for emergency use in the treatment of COVID-19 [66,67,68]. Given the immense potential benefit to G6PD-deficient patients if new and safe treatment options arise, rigorous clinical studies must be performed to determine the safest choice of drugs for these patients.
Recent studies and directions for further research
Despite an array of laboratory findings suggesting that COVID-19 patients with G6PD deficiency may suffer a worse prognosis, one clinical study concluded that G6PD-deficient patients might experience less severe symptoms, requiring reduced ventilatory support and an overall lower case-fatality rate than patients with in-tact G6PD [11]. This could be potentially explained by the nature of the G6PD enzyme. In addition to its well-known antioxidative activity, G6PD exhibits a pro-inflammatory mechanism of action [69]; activated G6PD may enhance oxidative inflammation in acute lung injury during infection, possibly exacerbating clinical symptoms [70]. It is understood that complications due to aggressive host immune response—such as dramatic apoptosis as a result of fulminant inflammation in lungs and other organs—may prove challenging in COVID-19 infections [30, 71]. The slightly immunocompromised condition of G6PD-deficient patients may prevent severe inflammation, which may be consequently beneficial to patients. However, it is important to note that this only applies to patients with less severe forms of G6PD deficiency; in severe class I G6PD deficiencies, G6PD levels may be too low to even adequately clear viral infections [72, 73]. However, the sample size of G6PD-deficient patients analyzed in the clinical study mentioned is too small to establish high study power. Additionally, the clinical presentation of G6PD-deficient patients may vary, so a well-designed large-scale clinical study or an animal model scalable to the human immune system would be required in order to provide sufficient evidence of this point.
Multiple articles and original research have concluded that oxidative stress plays a key role in COVID-19 and virally induced acute lung injuries [44, 74]. Exacerbation of acute lung injury via elevated ROS has been observed in animal models [74]. The reduction of oxidative stress has also been shown to inhibit viral replication [6, 41]. Herein, after reviewing existing data around ROS and viral infections, we suggest that the use of antioxidant or redox-modulating agents to control viral infection should be evaluated further. Particular attention should be paid to use in infections that could cause severe lung injury, including SARS-CoV-2. Polydatin, a specific redox-modulating agent, was promoted in existing literature, as it has potential to suppress oxidative inflammation induced by G6PD while working concurrently as an antioxidant [69]. Although the use of antioxidative agents like lipoic acid show a positive benefit at the cellular level, the results of cellular studies may not translate clinically due to the inherent complexities of human physiology [6, 41, 42]. Therefore, monitored clinical trials should be performed to examine whether adding antioxidative agents to standard treatment is safe and if doing so may improve the prognosis or reduce hospitalization in COVID-19 patients.
At present, clinical studies regarding the relationship between G6PD deficiency and COVID-19 infection are sparse. However, in vitro studies of the interaction between G6PD knocked-out cells and several viruses, including human coronavirus 229E, have been conducted; wild type and G6PD knocked-out cells were separately cultured with coronavirus 229E, and the number of viral genes was subsequently measured [6]. Similar studies could be performed with SARS-CoV-2 samples to evaluate whether G6PD knocked-out cells are more susceptible to SARS-CoV-2 infection at the cellular level. Additionally, further ex vivo studies could be done to assess the immune response against SARS-CoV-2 in G6PD-deficient animals. Clinical studies including the comparison of the SARS-CoV-2 viral load in infected G6PD-deficient patients versus normal patients could also be conducted. Such studies allow us to identify whether COVID-19 patients with G6PD deficiency are more contagious. Further testing of antioxidative agents on these models would help to evaluate the safety and effectiveness of such treatment. Though there is no guarantee that the actual host immune response will be analogous to that of the models, the suggested studies will allow us to have an overview of what could potentially happen in G6PD-deficient patients during SARS-CoV-2 infection; these results could serve as a foundation for future clinical studies [75]. Moreover, the physiological reaction of G6PD-deficient patients treated with remdesivir should be analyzed as to its use in treating COVID-19 patients expands [76]. When considering the findings of these studies and the potential of future research, physicians should be well-informed of the G6PD status of their COVID-19 infected patients, using remdesivir or other antiviral medication with great caution in positive patients, particularly those within class I [77].
Juneja et al. demonstrated that advanced age, male gender, diabetes, and abnormal hematological profile are associated with moderate to severe course of COVID-19 infection in a general population [78]. Given currently inadequate evidence to support that COVID-19 patients with G6PD deficiency have a worse prognosis in terms of mortality, severity, and rate of hospitalization [11], a larger scale study should be performed that encompasses different ages, ethnicity, gender, and more importantly the disease-alleles (hemizygous, homozygous or heterozygous) with consideration of G6PD deficiency variants. Furthermore, the incidence of G6PD-deficient individuals with SARS-CoV-2 infections should also be calculated; the viral load could also be compared between patients with wild-type G6PD and G6PD deficiency. The result of such studies will aid in defining whether G6PD deficiency is one of the factors of infection risk and whether such patients may experience heightened contagiousness when compared with unaffected individuals.
In conclusion, patients with G6PD deficiency are notorious for elevated levels of ROS in response to classic triggers including viral infections such as COVID-19. The slightly immunocompromised status of these patients is shown to favor viral replication; this may potentially result in increased viral load and infectivity within affected patients. Additionally, due to inherent proclivity to hemolytic, thrombotic, and other medically threatening events, G6PD-deficient patients may be limited in treatment options available to them, particularly in the case of COVID-19 infection. Though more research is demanded on the topic, preliminary studies suggest that antioxidative therapy that reduces ROS levels in these patients could prove beneficial in the treatment of viral infections in G6PD-deficient individuals.
Data availability
Not applicable.
References
Berg JM, Tymoczko JL, Stryer L (2002) Glucose 6-Phosphate Dehydrogenase Plays a Key Role in Protection Against Reactive Oxygen Species. Biochemistry 5th edition
Efferth T, Schwarzl SM, Smith J, Osieka R (2006) Role of glucose-6-phosphate dehydrogenase for oxidative stress and apoptosis. Cell Death Differ 13:527–528. https://doi.org/10.1038/sj.cdd.4401807
Cappellini MD, Fiorelli G (2008) Glucose-6-phosphate dehydrogenase deficiency. Lancet 371:64–74. https://doi.org/10.1016/S0140-6736(08)60073-2
Bubp J, Jen M, Matuszewski K (2015) Caring for Glucose-6-Phosphate Dehydrogenase (G6PD)–Deficient Patients: Implications for Pharmacy. P T 40:572–574
Herman TF, Javaid MU (2021) Heinz Body. In: StatPearls. StatPearls Publishing, Treasure Island (FL)
Wu Y-H, Tseng C-P, Cheng M-L et al (2008) Glucose-6-phosphate dehydrogenase deficiency enhances human coronavirus 229E infection. J Infect Dis 197:812–816. https://doi.org/10.1086/528377
Vick DJ (2021) Evaluation of glucose-6-phosphate dehydrogenase (G6PD) status in US military and VA patients with COVID-19 infection. BMJ Mil Health 167:144–144. https://doi.org/10.1136/bmjmilitary-2020-001706
Vick DJ (2020) Glucose-6-Phosphate Dehydrogenase Deficiency and COVID-19 Infection. Mayo Clin Proc 95:1803–1804. https://doi.org/10.1016/j.mayocp.2020.05.035
Youssef JG, Zahiruddin F, Youssef G et al (2021) G6PD deficiency and severity of COVID19 pneumonia and acute respiratory distress syndrome: tip of the iceberg? Ann Hematol 100:667–673. https://doi.org/10.1007/s00277-021-04395-1
Aydemir D, Dağlıoğlu G, Candevir A et al (2021) COVID-19 may enhance risk of thrombosis and hemolysis in the G6PD deficient patients. Nucleosides Nucleotides Nucleic Acids 40:505–517. https://doi.org/10.1080/15257770.2021.1897457
Kumar N, AbdulRahman A, AlAwadhi AI, AlQahtani M (2021) Is glucose-6-phosphatase dehydrogenase deficiency associated with severe outcomes in hospitalized COVID-19 patients? Sci Rep 11:19213. https://doi.org/10.1038/s41598-021-98712-3
Buinitskaya Y, Gurinovich R, Wlodaver CG, Kastsiuchenka S (2020) Centrality of G6PD in COVID-19: The Biochemical Rationale and Clinical Implications. Front Med (Lausanne) 7:584112. https://doi.org/10.3389/fmed.2020.584112
Yen W-C, Wu Y-H, Wu C-C et al (2019) Impaired inflammasome activation and bacterial clearance in G6PD deficiency due to defective NOX/p38 MAPK/AP-1 redox signaling. Redox Biol 28:101363. https://doi.org/10.1016/j.redox.2019.101363
Abdel Hafez SMN (2020) Glucose-6-phosphate dehydrogenase deficiency enhances Covid-19 infection in elderly people. Bratisl Lek Listy 121:786–788. https://doi.org/10.4149/BLL_2020_128
Yang H-C, Ma T-H, Tjong W-Y, et al G6PD deficiency, redox homeostasis, and viral infections: implications for SARS-CoV-2 (COVID-19). Free Radic Res 1–11. https://doi.org/10.1080/10715762.2020.1866757
Siler U, Romao S, Tejera E et al (2017) Severe glucose-6-phosphate dehydrogenase deficiency leads to susceptibility to infection and absent NETosis. J Allergy Clin Immunol 139:212-219.e3. https://doi.org/10.1016/j.jaci.2016.04.041
Ardati KO, Bajakian KM, Tabbara KS (1997) Effect of glucose-6-phosphate dehydrogenase deficiency on neutrophil function. Acta Haematol 97:211–215. https://doi.org/10.1159/000203685
Lakshman R, Finn A (2001) Neutrophil disorders and their management. J Clin Pathol 54:7–19. https://doi.org/10.1136/jcp.54.1.7
Naumenko V, Turk M, Jenne CN, Kim S-J (2018) Neutrophils in viral infection. Cell Tissue Res 371:505–516. https://doi.org/10.1007/s00441-017-2763-0
Galani IE, Andreakos E (2015) Neutrophils in viral infections: Current concepts and caveats. J Leukoc Biol 98:557–564. https://doi.org/10.1189/jlb.4VMR1114-555R
Grundy JE, Lawson KM, MacCormac LP et al (1998) Cytomegalovirus-infected endothelial cells recruit neutrophils by the secretion of C-X-C chemokines and transmit virus by direct neutrophil-endothelial cell contact and during neutrophil transendothelial migration. J Infect Dis 177:1465–1474. https://doi.org/10.1086/515300
van Strijp JA, van Kessel KP, Miltenburg LA et al (1988) Attachment of human polymorphonuclear leukocytes to herpes simplex virus-infected fibroblasts mediated by antibody-independent complement activation. J Virol 62:847–850. https://doi.org/10.1128/JVI.62.3.847-850.1988
Ratcliffe D, Migliorisi G, Cramer E (1992) Translocation of influenza virus by migrating neutrophils. Cell Mol Biol 38:63–70
Hashimoto Y, Moki T, Takizawa T et al (2007) Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J Immunol 178:2448–2457. https://doi.org/10.4049/jimmunol.178.4.2448
Hidalgo A, Libby P, Soehnlein O, et al (2021) Neutrophil extracellular traps: from physiology to pathology. Cardiovasc Res cvab329. https://doi.org/10.1093/cvr/cvab329
Papayannopoulos V (2018) Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18:134–147. https://doi.org/10.1038/nri.2017.105
Agraz-Cibrian JM, Giraldo DM, Mary F-M, Urcuqui-Inchima S (2017) Understanding the molecular mechanisms of NETs and their role in antiviral innate immunity. Virus Res 228:124–133. https://doi.org/10.1016/j.virusres.2016.11.033
Leppkes M, Knopf J, Naschberger E et al (2020) Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine 58:102925. https://doi.org/10.1016/j.ebiom.2020.102925
Torres-Ruiz J, Absalón-Aguilar A, Nuñez-Aguirre M et al (2021) Neutrophil Extracellular Traps Contribute to COVID-19 Hyperinflammation and Humoral Autoimmunity. Cells 10:2545. https://doi.org/10.3390/cells10102545
Veras FP, Pontelli MC, Silva CM et al (2020) SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med 217:20201129. https://doi.org/10.1084/jem.20201129
Kaplan MJ, Radic M (2012) Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol 189:2689–2695. https://doi.org/10.4049/jimmunol.1201719
Lopez-Castejon G, Brough D (2011) Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev 22:189–195. https://doi.org/10.1016/j.cytogfr.2011.10.001
Dinarello CA (1996) Biologic Basis for Interleukin-1 in Disease. Blood 87:2095–2147. https://doi.org/10.1182/blood.V87.6.2095.bloodjournal8762095
Aarreberg LD, Wilkins C, Ramos HJ, et al Interleukin-1β Signaling in Dendritic Cells Induces Antiviral Interferon Responses. mBio 9:00342–18. https://doi.org/10.1128/mBio.00342-18
Ramos HJ, Lanteri MC, Blahnik G et al (2012) IL-1β signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog 8:1003039. https://doi.org/10.1371/journal.ppat.1003039
Vora SM, Lieberman J, Wu H (2021) Inflammasome activation at the crux of severe COVID-19. Nat Rev Immunol 21:694–703. https://doi.org/10.1038/s41577-021-00588-x
Pan P, Shen M, Yu Z et al (2021) SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat Commun 12:4664. https://doi.org/10.1038/s41467-021-25015-6
Wu Y-H, Chiu DT-Y, Lin H-R et al (2015) Glucose-6-Phosphate Dehydrogenase Enhances Antiviral Response through Downregulation of NADPH Sensor HSCARG and Upregulation of NF-κB Signaling. Viruses 7:6689–6706. https://doi.org/10.3390/v7122966
Lingappan K (2018) NF-κB in Oxidative Stress. Curr Opin Toxicol 7:81–86. https://doi.org/10.1016/j.cotox.2017.11.002
Djavaheri-Mergny M, Javelaud D, Wietzerbin J, Besançon F (2004) NF-kappaB activation prevents apoptotic oxidative stress via an increase of both thioredoxin and MnSOD levels in TNFalpha-treated Ewing sarcoma cells. FEBS Lett 578:111–115. https://doi.org/10.1016/j.febslet.2004.10.082
Sun R, Deng Z, Han X et al (2021) Porcine Circovirus 2 Manipulates the PERK-ERO1α Axis of the Endoplasmic Reticulum To Favor Its Replication by Derepressing Viral DNA from HMGB1 Sequestration within Nuclei. J Virol 95:0100921. https://doi.org/10.1128/JVI.01009-21
Chen X, Ren F, Hesketh J et al (2012) Reactive oxygen species regulate the replication of porcine circovirus type 2 via NF-κB pathway. Virology 426:66–72. https://doi.org/10.1016/j.virol.2012.01.023
Marc A, Kerioui M, Blanquart F, et al (2021) Quantifying the relationship between SARS-CoV-2 viral load and infectiousness. eLife 10:69302. https://doi.org/10.7554/eLife.69302
Cecchini R, Cecchini AL (2020) SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med Hypotheses 143:110102. https://doi.org/10.1016/j.mehy.2020.110102
L’Acqua C, Hod E (2015) New perspectives on the thrombotic complications of haemolysis. Br J Haematol 168:175–185. https://doi.org/10.1111/bjh.13183
Karki P, Malik S, Mallick B, et al (2016) Massive Hemolysis Causing Renal Failure in Acute Hepatitis E Infection. Journal of Clinical and Translational Hepatology 4:345–347. https://doi.org/10.14218/JCTH.2016.00042
Čikeš V, Abaza I, Krželj V et al (2004) Prevalence of factor V Leiden and G6PD 1311 silent mutations in dalmatian population. Arch Med Res 35:546–548. https://doi.org/10.1016/j.arcmed.2004.07.005
Patil M, Singh S, Henderson J, Krishnamurthy P (2021) Mechanisms of COVID-19-induced cardiovascular disease: Is sepsis or exosome the missing link? J Cell Physiol 236:3366–3382. https://doi.org/10.1002/jcp.30109
Burtenshaw D, Kitching M, Redmond EM et al (2019) Reactive Oxygen Species (ROS), Intimal Thickening, and Subclinical Atherosclerotic Disease. Front Cardiovasc Med 6:89. https://doi.org/10.3389/fcvm.2019.00089
Kushner A, West WP, Pillarisetty LS (2021) Virchow Triad. In: StatPearls. StatPearls Publishing, Treasure Island (FL)
Dore MP, Portoghese M, Pes GM (2021) The Elderly with Glucose-6-Phosphate Dehydrogenase Deficiency are More Susceptible to Cardiovascular Disease. J Atheroscler Thromb 28:604–610. https://doi.org/10.5551/jat.56531
Dore MP, Parodi G, Portoghese M, Pes GM (2021) The Controversial Role of Glucose-6-Phosphate Dehydrogenase Deficiency on Cardiovascular Disease: A Narrative Review. Oxidative Medicine and Cellular Longevity 2021:e5529256. https://doi.org/10.1155/2021/5529256
Klouda CB, Stone WL (2020) Oxidative Stress, Proton Fluxes, and Chloroquine/Hydroxychloroquine Treatment for COVID-19. Antioxidants (Basel) 9:894. https://doi.org/10.3390/antiox9090894
Wang M, Cao R, Zhang L et al (2020) Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 30:269–271. https://doi.org/10.1038/s41422-020-0282-0
Vincent MJ, Bergeron E, Benjannet S et al (2005) Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J 2:69. https://doi.org/10.1186/1743-422X-2-69
Beauverd Y, Adam Y, Assouline B, Samii K (2020) COVID-19 infection and treatment with hydroxychloroquine cause severe haemolysis crisis in a patient with glucose-6-phosphate dehydrogenase deficiency. Eur J Haematol. https://doi.org/10.1111/ejh.13432.10.1111/ejh.13432
Maillart E, Leemans S, Van Noten H et al (2020) A case report of serious haemolysis in a glucose-6-phosphate dehydrogenase-deficient COVID-19 patient receiving hydroxychloroquine. Infect Dis (Lond) 52:659–661. https://doi.org/10.1080/23744235.2020.1774644
Afra TP, VasudevanNampoothiri R, Razmi TM (2020) Doubtful precipitation of hemolysis by hydroxychloroquine in glucose-6-phosphate dehydrogenase-deficient patient with COVID-19 infection. Eur J Haematol 105:512–513. https://doi.org/10.1111/ejh.13460
Commons RJ, Simpson JA, Thriemer K et al (2019) The haematological consequences of Plasmodium vivax malaria after chloroquine treatment with and without primaquine: a WorldWide Antimalarial Resistance Network systematic review and individual patient data meta-analysis. BMC Med 17:151. https://doi.org/10.1186/s12916-019-1386-6
Mohammad S, Clowse MEB, Eudy AM, Criscione-Schreiber LG (2018) Examination of Hydroxychloroquine Use and Hemolytic Anemia in G6PDH-Deficient Patients. Arthritis Care Res 70:481–485. https://doi.org/10.1002/acr.23296
Commissioner O of the (2020) FDA Approves First Treatment for COVID-19. In: FDA. www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-covid-19. Accessed 2 Dec 2021
Kokic G, Hillen HS, Tegunov D et al (2021) Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat Commun 12:279. https://doi.org/10.1038/s41467-020-20542-0
Zampino R, Mele F, Florio LL, et al (2020) Liver injury in remdesivir-treated COVID-19 patients. Hepatol Int 1–3. https://doi.org/10.1007/s12072-020-10077-3
Aleem A, Kothadia JP (2021) Remdesivir. In: StatPearls. StatPearls Publishing, Treasure Island (FL)
Dorgalaleh A, Shahzad MS, Younesi MR et al (2013) Evaluation of liver and kidney function in favism patients. Med J Islam Repub Iran 27:17–22
Commissioner O of the (2021) Coronavirus (COVID-19) Update: FDA Authorizes First Oral Antiviral for Treatment of COVID-19. In: FDA. www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-first-oral-antiviral-treatment-covid-19. Accessed 15 Jan 2022
Covid-19: Pfizer’s paxlovid is 89% effective in patients at risk of serious illness, company reports | The BMJ. www.bmj.com/content/375/bmj.n2713.long. Accessed 15 Jan 2022
Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients | NEJM. www.nejm.org/doi/full/https://doi.org/10.1056/NEJMoa2116044. Accessed 15 Jan 2022
Doustimotlagh AH, Eftekhari M (2021) Glucose-6-phosphate dehydrogenase inhibitor for treatment of severe COVID-19: Polydatin. Clin Nutr ESPEN 43:197–199. https://doi.org/10.1016/j.clnesp.2021.02.021
Nadeem A, Al-Harbi NO, Ahmad SF et al (2018) Glucose-6-phosphate dehydrogenase inhibition attenuates acute lung injury through reduction in NADPH oxidase-derived reactive oxygen species. Clin Exp Immunol 191:279–287. https://doi.org/10.1111/cei.13097
Mishra KP, Singh AK, Singh SB (2020) Hyperinflammation and Immune Response Generation in COVID-19. NIM 27:80–86. https://doi.org/10.1159/000513198
Lin H-R, Wu Y-H, Yen W-C et al (2016) Diminished COX-2/PGE2-Mediated Antiviral Response Due to Impaired NOX/MAPK Signaling in G6PD-Knockdown Lung Epithelial Cells. PLOS ONE 11:0153462. https://doi.org/10.1371/journal.pone.0153462
Lee J, Park J, Choi H et al (2017) Genetic Profiles of Korean Patients With Glucose-6-Phosphate Dehydrogenase Deficiency. Ann Lab Med 37:108–116. https://doi.org/10.3343/alm.2017.37.2.108
Imai Y, Kuba K, Neely GG et al (2008) Identification of Oxidative Stress and Toll-like Receptor 4 Signaling as a Key Pathway of Acute Lung Injury. Cell 133:235–249. https://doi.org/10.1016/j.cell.2008.02.043
van der Worp HB, Howells DW, Sena ES et al (2010) Can Animal Models of Disease Reliably Inform Human Studies? PLOS Medicine 7:1000245. https://doi.org/10.1371/journal.pmed.1000245
National Academies of Sciences E, Studies D on E and L, Research I for LA, Use R on S and W in LA (2018) Assessing Safety and Toxicology. National Academies Press (US)
Kassi EN, Papavassiliou KA, Papavassiliou AG (2020) G6PD and chloroquine: Selecting the treatment against SARS-CoV-2? J Cell Mol Med 24:4913–4914. https://doi.org/10.1111/jcmm.15312
Juneja R, Gadkari R, Meshram N, Selvaraj K (2022) Haematology audit of 801 COVID-19 patients’ basics and beyond- Prospective observational study. J Family Med Prim Care 11:4460–4466. https://doi.org/10.4103/jfmpc.jfmpc_44_22
Lacerda MVG, Llanos-Cuentas A, Krudsood S et al (2019) Single-Dose Tafenoquine to Prevent Relapse of Plasmodium vivax Malaria. N Engl J Med 380:215–228. https://doi.org/10.1056/NEJMoa1710775
Yoshida A, Beutler E, Motulsky AG (1971) Human glucose-6-phosphate dehydrogenase variants. Bull World Health Organ 45:243–253
Author information
Authors and Affiliations
Contributions
All authors contributed to the design and implementation of the research, to the analysis of the results, and to the writing of the manuscript.
Corresponding authors
Ethics declarations
Ethical approval
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Au, T.Y., Wiśniewski, O.W., Benjamin, S. et al. G6PD deficiency—does it alter the course of COVID-19 infections?. Ann Hematol 102, 1629–1636 (2023). https://doi.org/10.1007/s00277-023-05164-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-023-05164-y