
Abstract
The Janus kinase inhibitor ruxolitinib is approved for the treatment of myelofibrosis (MF) and improved overall survival (OS) versus control therapy in the phase 3 COMFORT trials. The aim of this retrospective analysis was to examine the real-world impact of ruxolitinib on OS in patients with MF. The US Medicare Fee-for-Service claims database (parts A/B/D) was used to identify patients with ≥ 1 inpatient or ≥ 2 outpatient claims with an MF diagnosis (January 2010–December 2017). Eligible patients with MF were ≥ 65 years old (intermediate-1 or higher risk based on age). Patients were divided into 3 groups based on ruxolitinib approval status at diagnosis and ruxolitinib exposure: (1) preapproval, ruxolitinib-unexposed; (2) post-approval, ruxolitinib-unexposed; and (3) post-approval, ruxolitinib-exposed. In total, 1677 patients with MF were included (preapproval [all ruxolitinib-unexposed], n = 278; post-approval, n = 1399 [ruxolitinib-unexposed, n = 1127; ruxolitinib-exposed, n = 272]). Overall, median age was 78 years, and 39.8% were male. Among patients with valid death dates (preapproval, n = 119 [42.8%]; post-approval, ruxolitinib-unexposed, n = 382 [33.9%]; post-approval ruxolitinib-exposed, n = 54 [19.9%]), 1-year survival rates were 55.6%, 72.5%, and 82.3%, and median OS was 13.2 months, 44.4 months, and not reached, respectively. Risk of mortality was significantly lower post- versus preapproval regardless of exposure to ruxolitinib (ruxolitinib-unexposed: adjusted hazard ratio [HR], 0.67; ruxolitinib-exposed: adjusted HR, 0.36; P < 0.001 for both); post-approval, mortality risk was significantly lower in ruxolitinib-exposed versus ruxolitinib-unexposed patients (adjusted HR, 0.61; P = 0.002). Findings from this study complement clinical data of ruxolitinib in MF by demonstrating a survival benefit in a real-world setting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myelofibrosis (MF) is a chronic myeloproliferative neoplasm characterized by hyperproliferation of myeloid cells, bone marrow fibrosis, and burdensome constitutional symptoms [1,2,3]. MF can develop de novo (primary MF) or may occur in patients with antecedent polycythemia vera (PV) or essential thrombocythemia (ET; secondary MF) [1]. Patients with MF have reduced overall survival (OS) compared with the general population [4, 5]. Based on recent population-based studies, the estimated incidence of primary MF in the USA is approximately 0.3 per 100,000 person-years and median OS is < 5 years from the time of diagnosis [6, 7]. When stratified by risk, median survival in patients with primary MF ranges from 2 years from diagnosis for high-risk MF to 11 years in low-risk MF by the International Prognostic Scoring System (IPSS) [8], which was designed to assess risk at the time of MF diagnosis.
Ruxolitinib, a Janus kinase (JAK) 1 and JAK2 inhibitor, was approved by the US Food and Drug Administration in November 2011 for the treatment of adult patients with intermediate- to high-risk primary or secondary MF based on data from the phase 3 COMFORT-I and COMFORT-II trials [9, 10]. Ruxolitinib significantly reduced spleen volume and improved MF-related symptoms and quality of life compared with placebo or best available therapy in the COMFORT studies [11, 12]. Furthermore, in the pooled analysis of COMFORT data, patients treated with ruxolitinib demonstrated improved OS compared with patients in the control arm (placebo, COMFORT-I; best available therapy, COMFORT-II; 5.3 versus 3.8 years, respectively; hazard ratio [HR], 0.70; 95% CI, 0.54–0.91; P = 0.007), and when patients were censored at crossover, the survival advantage was even greater (5.3 versus 2.4 years, respectively; HR, 0.53; 95% CI, 0.36–0.78; P = 0.001) [10]. In a phase 3b expanded access study (JUMP), reductions from baseline in spleen volume and MF symptom burden were observed in a large cohort of patients with MF receiving ruxolitinib treatment (N = 2233), including in patients ineligible for the COMFORT studies due to low-risk disease (i.e., low risk or intermediate-1) or low platelet count (i.e., < 100 × 109/L) [13, 14]. Median OS for patients with MF enrolled in JUMP was 4.9 and 2.8 years among patients with intermediate-2 and high-risk MF, respectively [14].
Despite the established survival benefit of ruxolitinib in the clinical trial setting, limited real-world data exist. The objective of this analysis was to examine the real-world impact of ruxolitinib on patients with MF by comparing survival of patients newly diagnosed with MF before ruxolitinib approval with patients who were ruxolitinib-unexposed versus ruxolitinib-exposed after ruxolitinib approval.
Methods
Study design and patients
A retrospective analysis was performed of the US Medicare Fee-for-Service claims database (parts A/B/D) to identify patients with ≥ 1 inpatient or ≥ 2 outpatient claims with a diagnosis of MF from January 2010 through December 2017. The index date was the date of MF diagnosis as indicated by the first qualifying MF claim. Eligible patients with MF were ≥ 65 years old (and therefore intermediate-1 or higher risk based on age) with a minimum of 12 months of pre-index continuous medical and pharmacy enrollment. Patients with evidence of MF diagnosis ≤ 12 months before the index date were excluded; additionally, patients with a diagnosis of myelodysplastic syndrome; other hematologic malignancies (i.e., leukemias, multiple myeloma, lymphomas); or solid tumors either ≤ 12 months before, on, or any time after index were excluded in a stepwise manner (Fig. 1).

Patient attrition with exclusions. aExcluding AML. AML, acute myeloid leukemia; FFS, Fee-for-Service; MDS, myelodysplastic syndrome; MF, myelofibrosis; RUX, ruxolitinib
Patients were divided into 3 groups based on ruxolitinib approval status at the time of diagnosis and subsequent patient exposure to ruxolitinib: (1) preapproval, ruxolitinib-unexposed (index year 2010–2011); (2) post-approval, ruxolitinib-unexposed (index year 2012–2017); and (3) post-approval, ruxolitinib-exposed (index year 2012–2017). Ruxolitinib exposure during the post-index period was determined based on patient prescription fill history; patients categorized as ruxolitinib-unexposed did not receive ruxolitinib at any time following the index event.
Statistical analyses
Patient demographics and clinical characteristics were summarized using descriptive statistics. One- and 2-year survival rate and risk of mortality were estimated using Kaplan–Meier and Cox proportional hazards regression analyses, adjusting for baseline demographic and clinical characteristics. One-year mortality was evaluated from the index date through the end of 1 year. OS was evaluated from the index date through the entire length of follow-up until death or end of data availability. Patients without a death date were censored at disenrollment or at the end of study period, whichever occurred first.
Results
Baseline patient characteristics
The analysis included 1677 eligible patients with an MF diagnosis, with 278 patients diagnosed pre-ruxolitinib approval (all ruxolitinib-unexposed) and 1399 diagnosed post-ruxolitinib approval (ruxolitinib-unexposed, n = 1127; ruxolitinib-exposed, n = 272; Fig. 1). Overall, median age was 78 years, 39.8% of patients were male, and 84.1% were White. The preapproval group was the oldest (median age, 80.7 years) and had the highest proportion of female patients (70.1%; Table 1). History of PV and ET also varied among the 3 groups, with the highest proportion of patients with previous history in the post-approval ruxolitinib-exposed group (PV, 20.2%; ET, 19.5%). The Charlson Comorbidity Index was lowest among post-approval ruxolitinib-exposed patients (mean [SD], 2.2 [2.2] versus 3.7 [2.7] for the preapproval group and 3.2 [2.9] for the post-approval ruxolitinib-unexposed group). Median duration of follow-up was 12.5 months for the preapproval group, 10.2 months for post-approval ruxolitinib-unexposed group, and 14.0 months for the post-approval ruxolitinib-exposed group.
Overall survival
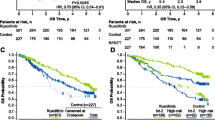
Valid death dates were available for 119 (42.8%) patients in the pre-ruxolitinib approval group and 436 (31.2%) in the post-ruxolitinib approval group (ruxolitinib-unexposed, n = 382 [33.9%]; ruxolitinib-exposed, n = 54 [19.9%]; Table 2). Among patients with valid death dates, the 1-year survival rate (95% CI) was 55.6% (49.4–61.3%) for the pre-ruxolitinib approval group, 72.5% (69.5–75.2%) for the post-approval ruxolitinib-unexposed group, and 82.3% (76.7–86.7%) for the post-approval ruxolitinib-exposed group. Median (95% CI) OS was 13.2 months (10.2–17.1) for the pre-ruxolitinib approval group, 44.4 months (37.3–62.0) for post-approval ruxolitinib-unexposed patients, and not reached (51.0–not reached) for post-approval ruxolitinib-exposed patients. Compared with the pre-ruxolitinib approval group, risk of mortality was significantly lower in the post-approval groups regardless of exposure to ruxolitinib (ruxolitinib-unexposed: adjusted HR, 0.67; 95% CI, 0.56–0.80; P < 0.001; ruxolitinib-exposed: adjusted HR, 0.36; 95% CI, 0.26–0.50; P < 0.001; Fig. 2). Among the patients in the post-approval groups, those who were exposed to ruxolitinib had a significantly lower risk of mortality than those who were never exposed to ruxolitinib (HR, 0.61; 95% CI, 0.45–0.83; P = 0.002).

OS for patients newly diagnosed with intermediate- to high-risk MF. One-year survival rate and risk of mortality estimated using Kaplan–Meier and Cox proportional hazards regression analyses. HR, hazard ratio; MF, myelofibrosis; OS, overall survival
Discussion
In this real-world study of US patients diagnosed with intermediate- to high-risk MF, OS was significantly longer for patients who received ruxolitinib than for those who did not receive ruxolitinib, providing real-world data to complement the survival benefit observed in the COMFORT studies. Few other real-world studies have evaluated the effect of ruxolitinib or diagnosis period (i.e., before or after ruxolitinib approval) on survival in patients with MF. A study from Sweden and Norway reported that the OS rate of patients with MF treated with ruxolitinib was estimated at 80% at 1 year and 52% at 4 years [15]. A study from Turkey reported 1- and 3-year OS estimates after ruxolitinib treatment initiation of 90% and 72%, respectively [16]. A single-institution retrospective analysis of medical charts from patients with MF demonstrated improved survival among patients with myelofibrosis since 2010 compared with patients diagnosed prior to 2010 (HR [95% CI], 0.7 [0.59–0.82]; P < 0.001) [17]; patients treated with ruxolitinib had superior survival to those who did not receive ruxolitinib irrespective of diagnosis time period. In contrast, another study of population-based registry data from the USA showed no improvement in survival among patients with primary MF diagnosed pre-ruxolitinib approval (2007–2011; 4-year relative survival, 55%) versus post-ruxolitinib approval (2012–2016; 4-year relative survival, 56%) [18]. Of note, ruxolitinib exposure was not captured in that study and survival data post-2016 were not reported; furthermore, only patients with primary MF were included, and risk status was not provided.
In the current study, OS was longer among patients diagnosed after ruxolitinib approval compared with those diagnosed before ruxolitinib approval. Many factors may have contributed to this observed improvement, including increased disease awareness and improved patient management for individuals with MF over time. Within the time frame of this study, 2010–2017, guidelines for the management of MF became available from the European LeukemiaNet in 2011 [19], the World Health Organization updated their guidelines in 2016 (last revised in 2008) [20], and the National Comprehensive Cancer Network® (NCCN®) Guidelines for Myeloproliferative Neoplasms became available in 2017 [21]. In addition, risk stratification tools for MF have expanded, and their uses have been refined over the years [22]. Prognostic modeling for MF started with the development of IPSS in 2009, which categorized risk as low, intermediate-1, intermediate-2, or high using factors such as age, constitutional symptoms, and blood counts [8]. Currently, NCCN® guidelines recommend stratification of patients into higher or lower risk, using scores from Mutation-Enhanced IPSS (MIPSS-70) or MIPSS-70 + version 2.0 (preferred), Dynamic IPSS (DIPSS)-Plus (if molecular testing is not available), DIPSS (if karyotyping is not available), or MF Secondary to PV and ET-Prognostic Model (MYSEC-PM) [23]. These newer prognostic systems take into account additional factors, such as molecular testing results, transfusion dependence, and karyotype [2], which may contribute to improved disease management compared with the IPSS.
The limitations of this study are consistent with the retrospective nature of real-world claims-based analyses. For example, some clinical characteristics (e.g., hemoglobin level, circulating blast percentage, symptoms) were not available in the claims database. Furthermore, no comparisons of patient characteristics were performed at index, so differences between groups may have contributed to observed outcomes.
In conclusion, this real-world study demonstrated improved survival among patients with MF in the years since ruxolitinib approval in 2011. Patients who were exposed to ruxolitinib had the best survival outcomes of the studied cohorts. The findings generally complement those from clinical studies demonstrating a survival benefit with ruxolitinib; however, additional real-world studies are needed to better understand the impact of ruxolitinib on survival among patients with MF.
Data availability
The data described in this paper are sourced from Centers for Medicare and Medicaid Services (CMS) Medicare FFS claims and enrollment data. The analytic file constructed for this analysis cannot be shared due to restrictions set forth in the governing Data Use Agreement with CMS. Researchers may request use of CMS data through the Research Data Assistance Center.
Code availability
Not applicable.
References
O’Sullivan JM, Harrison CN (2018) Myelofibrosis: clinicopathologic features, prognosis, and management. Clin Adv Hematol Oncol 16(2):121–131
Tefferi A (2018) Primary myelofibrosis: 2019 update on diagnosis, risk-stratification and management. Am J Hematol 93(12):1551–1560
Emanuel RM, Dueck AC, Geyer HL, Kiladjian JJ, Slot S, Zweegman S, te Boekhorst PAW, Commandeur S, Schouten HC, Sackmann F, Kerguelen Fuentes A, Hernández-Maraver D, Pahl HL, Griesshammer M, Stegelmann F, Doehner K, Lehmann T, Bonatz K, Reiter A, Boyer F, Etienne G, Ianotto JC, Ranta D, Roy L, Cahn JY, Harrison CN, Radia D, Muxi P, Maldonado N, Besses C, Cervantes F, Johansson PL, Barbui T, Barosi G, Vannucchi AM, Passamonti F, Andreasson B, Ferarri ML, Rambaldi A, Samuelsson J, Birgegard G, Tefferi A, Mesa RA (2012) Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol 30(33):4098–4103. https://doi.org/10.1200/JCO.2012.42.3863
Hultcrantz M, Kristinsson SY, Andersson TM, Landgren O, Eloranta S, Derolf AR, Dickman PW, Björkholm M (2012) Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol 30(24):2995–3001. https://doi.org/10.1200/JCO.2012.42.1925
Price GL, Davis KL, Karve S, Pohl G, Walgren RA (2014) Survival patterns in United States (US) Medicare enrollees with non-CML myeloproliferative neoplasms (MPN). PLoS One 9(3):e90299. https://doi.org/10.1371/journal.pone.0090299
Shallis RM, Wang R, Davidoff A, Ma X, Podoltsev NA, Zeidan AM (2020) Epidemiology of the classical myeloproliferative neoplasms: the four corners of an expansive and complex map. Blood Rev 42:100706. https://doi.org/10.1016/j.blre.2020.100706
Szuber N, Mudireddy M, Nicolosi M, Penna D, Vallapureddy RR, Lasho TL, Finke C, Begna KH, Elliott MA, Hook CC, Wolanskyj AP, Patnaik MM, Hanson CA, Ketterling RP, Sirhan S, Pardanani A, Gangat N, Busque L, Tefferi A (2019) 3023 Mayo Clinic patients with myeloproliferative neoplasms: risk-stratified comparison of survival and outcomes data among disease subgroups. Mayo Clin Proc 94(4):599–610. https://doi.org/10.1016/j.mayocp.2018.08.022
Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E, Vannucchi AM, Mesa RA, Demory JL, Barosi G, Rumi E, Tefferi A (2009) New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 113(13):2895–2901. https://doi.org/10.1182/blood-2008-07-170449
JAKAFI® (ruxolitinib). 2020 Full prescribing information, Incyte Corporation, Wilmington, DE, USA
Verstovsek S, Gotlib J, Mesa RA, Vannucchi AM, Kiladjian JJ, Cervantes F, Harrison CN, Paquette R, Sun W, Naim A, Langmuir P, Dong T, Gopalakrishna P, Gupta V (2017) Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J Hematol Oncol 10(1):156
Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, Catalano JV, Deininger M, Miller C, Silver RT, Talpaz M, Winton EF, Harvey JH Jr, Arcasoy MO, Hexner E, Lyons RM, Paquette R, Raza A, Vaddi K, Erickson-Viitanen S, Koumenis IL, Sun W, Sandor V, Kantarjian HM (2012) A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 366(9):799–807. https://doi.org/10.1056/NEJMoa1110557
Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, McQuitty M, Hunter DS, Levy R, Knoops L, Cervantes F, Vannucchi AM, Barbui T, Barosi G (2012) JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med 366(9):787–798. https://doi.org/10.1056/NEJMoa1110556
Passamonti F, Gupta V, Martino B, Foltz L, Zaritskey A, Al-Ali HK, Tavares R, Maffioli M, Raanani P, Giraldo P, Griesshammer M, Guglielmelli P, Bouard C, Paley C, Tiwari R, Vannucchi AM (2021) Comparing the safety and efficacy of ruxolitinib in patients with Dynamic International Prognostic Scoring System low-, intermediate-1-, intermediate-2-, and high-risk myelofibrosis in JUMP, a phase 3b, expanded-access study. Hematol Oncol. https://doi.org/10.1002/hon.2898
Al-Ali HK, Griesshammer M, Foltz L, Palumbo GA, Martino B, Palandri F, Liberati AM, le Coutre P, García-Hernández C, Zaritskey A, Tavares R, Gupta V, Raanani P, Giraldo P, Hänel M, Damiani D, Sacha T, Bouard C, Paley C, Tiwari R, Mannelli F, Vannucchi AM (2020) Primary analysis of JUMP, a phase 3b, expanded-access study evaluating the safety and efficacy of ruxolitinib in patients with myelofibrosis, including those with low platelet counts. Br J Haematol 189(5):888–903. https://doi.org/10.1111/bjh.16462
Schain F, Vago E, Song C, He J, Liwing J, Lofgren C, Bjorkholm M (2019) Survival outcomes in myelofibrosis patients treated with ruxolitinib: a population-based cohort study in Sweden and Norway. Eur J Haematol 103(6):614–619
Soyer N, Haznedaroglu IC, Comert M, Cekdemir D, Yilmaz M, Unal A, Cagliyan G, Bilgir O, Ilhan O, Ozdemirkiran F, Kaya E, Sahin F, Vural F, Saydam G (2017) Multicenter retrospective analysis of Turkish patients with chronic myeloproliferative neoplasms. Turk J Haematol 34(1):27–33. https://doi.org/10.4274/tjh.2016.0005
Masarova L, Bose P, Pemmaraju N, Zhou L, Pierce SA, Estrov ZE, Kantarjian HM, Verstovsek S (2020) Improved survival of patients with myelofibrosis in the last decade. Blood 136(suppl 1):50–51. https://doi.org/10.1182/blood-2020-142578
Thomas JW, Shah MV, Vachhani P, Jamy O, Go RS, Goyal G (2020) Risk of mortality and leukemic transformation in primary myelofibrosis before and after ruxolitinib approval. Blood 136(suppl 1):28–28. https://doi.org/10.1182/blood-2020-134161
Barbui T, Barosi G, Birgegard G, Cervantes F, Finazzi G, Griesshammer M, Harrison C, Hasselbalch HC, Hehlmann R, Hoffman R, Kiladjian JJ, Kröger N, Mesa R, McMullin MF, Pardanani A, Passamonti F, Vannucchi AM, Reiter A, Silver RT, Verstovsek S, Tefferi A, European LeukemiaNet (2011) Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol 29(6):761–770. https://doi.org/10.1200/JCO.2010.31.8436
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127(20):2391–2405. https://doi.org/10.1182/blood-2016-03-643544
NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Myeloproliferative Neoplasms, version 2.2017. © National Comprehensive Cancer Network, Inc. 2016. All rights reserved. Accessed 24 June 2020
Michaelis LC (2017) Risk stratification in myelofibrosis: the quest for simplification. Haematologica 102(1):2–3
NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for myeloproliferative neoplasms, version 1.2020. © National Comprehensive Cancer Network, Inc. 2020. All rights reserved. Accessed February 5, 2021.
Acknowledgements
Support for this study was provided by Incyte Corporation (Wilmington, DE, USA). Writing assistance was provided by Tania Iqbal, PhD, an employee of ICON (North Wales, PA), and was funded by Incyte Corporation.
Funding
Support for this study was provided by Incyte Corporation (Wilmington, DE).
Author information
Authors and Affiliations
Contributions
S.V., S.P., J.Y., A.S., S.K., A.X., and C.H. designed the study, analyzed the data, and participated in the interpretation of the data, as well as drafting and revising the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
The study did not require approval from an institutional review board, as all analyzed data were from secondary sources and no personally identifying information was collected or reported.
Consent to participate
Informed consent was not required owing to the retrospective study design.
Consent for publication
Not applicable. No personally identifying information was collected or reported.
Conflict of interest
SV received research support from AstraZeneca, Blueprint Medicines Corp, Celgene, CTI BioPharma Corp, Genentech, Gilead, Incyte Corporation, ItalPharma, Novartis, NS Pharma, PharmaEssentia, Promedior, Protagonist Therapeutics, Roche, and Sierra Oncology; and is a paid consultant for Celgene, Incyte Corporation, Novartis, and Sierra Oncology. SP and JY are employees and shareholders of Incyte Corporation. AS, SK, and AX are employees of Avalere Health, a paid consultant of Incyte Corporation. CH served on speakers bureaus for Celgene, CTI BioPharma Corp, Gilead Sciences, Incyte Corporation, Janssen, Novartis, and Shire; has received research funding (institutional) from Celgene and Novartis; and has received honoraria from AOP Orphan Pharmaceuticals, Celgene, CTI BioPharma Corp, Gilead Sciences, Novartis, Promedior, Roche, Shire, and Sierra Oncology.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Disclosure of previous presentation:
This work was previously presented, in part, at the 25th European Hematology Association (EHA) Annual Congress (Virtual), June 11–14, 2020; the Society of Hematologic Oncology 2020 Annual Meeting (Virtual), September 9–12, 2020; the European School of Haematology 2nd How to Diagnose and Treat CML/MPN E-Conference, February 25–28, 2021; and the British Society for Haematology 2021 Virtual Annual Scientific Meeting, April 25–28, 2021.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Verstovsek, S., Parasuraman, S., Yu, J. et al. Real-world survival of US patients with intermediate- to high-risk myelofibrosis: impact of ruxolitinib approval. Ann Hematol 101, 131–137 (2022). https://doi.org/10.1007/s00277-021-04682-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-021-04682-x