
Abstract
This study aims to retrospectively analyze the clinical characteristics, treatments, and prognosis of aggressive peripheral T cell lymphoma (PTCL) patients with a lymphoma-associated hemophagocytosis syndrome (LAHS). We compared the clinical features and the overall survival (OS) rates of 159 PTCL patients with and without LAHS as well as the treatment outcomes of these patients with CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) or intensive chemotherapy regimens. We observed that in 23 % (36/159) patients PTCL was associated with LAHS. Different subtypes of PTCL in LAHS patients were diagnosed and peripheral T cell lymphoma, not otherwise specified (PTCL-NOS) was the main subtype (78 %). The median survival rates of the LAHS and non-LAHS groups were 3 and 16 months, respectively. The elevated rates of serum β2-microglobulin, ferritin, fasting triglycerides, and hypofibrinogen levels were higher in the LAHS group, so were bone marrow involvement, liver dysfunction, hepatosplenomegaly, and B symptoms. Three patients who were treated with a plasma exchange had a longer survival time. There was no statistically significant difference in the OS rates between the intensive chemotherapy and CHOP regimen groups (P > 0.05). PTCL patients with LAHS had a poorer prognosis. Awareness of the clinical symptoms and laboratory findings are crucial in order to diagnose LAHS in an early stage and repeated biopsies of multiple bone marrows from different locations in those patients without enlargement of superficial lymph nodes are necessary to improve the diagnosis. Intensive chemotherapy due to its severe toxicity was not obviously advantageous for the OS rate compared to the CHOP regimen.
Similar content being viewed by others


Introduction
Aggressive peripheral T cell lymphomas (PTCLs) account for about 7–10 % of non-Hodgkin's lymphomas (NHLs) in Western countries, compared to 20–30 % NHLs in East Asia [1, 2]. They are usually characterized by a diffuse disease process in 68 %, systemic symptoms in nearly half (45 %), bone marrow involvement (BMI) in a quarter (26 %), and extra nodal diseases in a third (37 %) of the patients [3]. Hemophagocytic lymphohistiocytosis (HLH) is an occasional but severe complication of PTCL in which T cells induce the uncontrolled activation of phagocytosing macrophages, leading to the lymphoma-associated hemophagocytosis syndrome (LAHS) with a high fatality rate [3, 4], and no particular mode of therapy has been satisfactory enough yet for the treatment of PTCL complicated with the LAHS. HLH is a potentially fatal hyperinflammatory condition. It includes genetic (primary) HLH, which is due to mutations in genes important for the cytolytic secretory pathway and causes perforin and granzymes to induce apoptosis in target cells and acquired (secondary) HLH, which occurs as a secondary disorder in association with severe infections, malignancies, rheumatologic disorders, and some metabolic diseases [5]. In non-immunocompromised patients, the main malignancy to be found associated with HLH is T cell lymphoma, leading to the LAHS [6]. However, reports of the clinical features of the LAHS are relatively rare, and only some case reports and author's experiences are available [7, 8]. Thus, we performed a retrospective analysis of clinical features from a large series of patients diagnosed with PTCLs by the criteria of the World Health Organization (WHO) Classification and analyzed the clinical features of LAHS, including the relationship between LAHS and some sub-types of PTCL, the clinical manifestations, some laboratory findings, and overall survival (OS) rates of different treatments in order to elucidate the clinical characteristics, treatments, and prognosis of LAHS patients.
Materials and methods
Patients
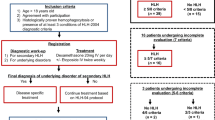
We consecutively reviewed 159 patients diagnosed with PTCL and who were hospitalized between January 2005 and December 2011 in the Zhejiang University First Affiliated Hospital of the Medical School. All diagnoses were confirmed by histopathological hematoxylin and eosin staining as well as determination of the immunophenotypes according to the WHO Classification 2008 [9]. Complete clinical profiles of all 159 patients who received chemotherapy and finished the follow-up were obtained. Clinical staging and diagnostic methods included a clinical history and physical examination, chest, abdominal, and pelvic computed tomography (CT) scans, full-digital full-body color Doppler ultrasonic diagnostic analyzer examinations as well as bone marrow aspirate and biopsy investigations. Clinical data collected for each patient included lactate dehydrogenase (LDH) and β2-microglobulin (β2-MG) levels, immunoglobulin (IG), blood biochemical functions, liver function, blood routine, coagulation functions, tumor markers, ferritin, bone marrow smear, and chemotherapy regimens. We analyzed the differences in laboratory findings and OS rates between PTCL patients with or without LAHS. In addition, we also compared the outcome of the patients who received CHOP or intensive chemotherapy regimens in LAHS patients. Informed consents were obtained from all patients before the collection of patients' information and serum samples for analyses.
Immunohistochemical staining
Sections of all patients were stained with T cell (CD2, CD3, and CD45) and B cell (CD20 and CD79a) markers by using mouse monoclonal antibodies, and disease specificity was confirmed in case that positive staining was observed only with one or more T cell-specific markers without any positive B cell-specific marker staining. Moreover, CD30 and anaplastic leukemia kinase-1 immunostaining were further used for the differential diagnosis of systemic anaplastic large cell lymphoma, and CD56, CD57, TIA-1, and granzyme B were used as marker for NK/T cell lymphoma diagnosis.
Diagnostic criteria for LAHS
The diagnostic criteria for LAHS are listed in Table 1 [10].
Clinical staging
Disease grades were evaluated according to the Ann Arbor Staging System [11] and performance status evaluation based on the Eastern Cooperative Oncology Group scale (0–4) [12]. The patients' international prognostic index [13] scores were determined and used in the survival rate analyses.
Treatment
Chemotherapy regimens including CHOP (cyclophosphamide 750 mg/m2, day 1, epirubicin 60 mg/m2 day 1, vincristine 1.4 mg/m2 day 1, and prednisolone 100 mg days 1–5) or CHOP-like regimens (idamycin (8 mg/m2 day 1), mitoxantrone (8 mg/m2 day 1), or liposomal doxorubicin (40 mg/m2 day 1) substituting for epirubicin) were employed. Intensive chemotherapies included ECHOP (CHOP with etoposide 100 mg days 1–3), CHOP with Ara-C (cytosine arabinoside 200 mg days 1–7), MINE (mitoxantrone 8 mg/m2 days 1–3, isophosphamide 2.0 g days 1–3, mesna 500 mg/m2 days 1–3 4 h after each isophosphamide, etoposide 100 mg days 1–3), ESHAP (etoposide 40 mg/m2 days 1–4, solumedrol 250–500 mg days 1–5, Ara-C 2 g/m2 day 5, platinol 25 mg/m2 days 1–4), DHAP (cisplatin 100 mg/m2 day 1, dexamethasone 40 mg days 1–4, cytarabine 2.0/m2 q12h day 2), and hyper CVAD (cyclophosphamide 300 mg/m2 q12h days 1–3, doxorubicin 50 mg/m2 day 4, vincristine 2 mg days 4 and 11, dexamethasone 40 mg days 1–4 and 11–14).
Statistics
OS rates were calculated beginning from the date of diagnosis to death or the last follow-up date. Survival curves were analyzed by the Kaplan–Meier method and log-rank test was used for comparisons between individual clinical features and survival. χ 2 test was used to compare the clinical and laboratory data of T cell lymphomas with and without LAHS. Statistical significance was defined as a P-value <0.05. All data were processed with the Statistical Package for the Social Sciences 16 software.
Results
Patient characteristics
Thirty-six of the 159 PTCL patients were diagnosed with LAHS. In the LAHS group, 28 (78 %) patients had peripheral T cell lymphoma, not otherwise specified (PTCL-NOS), while two (6 %) were diagnosed with subcutaneous panniculitis-like T cell lymphoma (SCPTCL), three (8 %) with angioimmunoblastic T cell lymphoma (AILT), and three (8 %) with nasal NK/T cell lymphoma (NKTCL). In the non-LAHS group, 64 (52 %) patients were diagnosed with PTCL-NOS, five (4 %) with SCPTCL, 18 (15 %) with AILT, eight (7 %) with anaplastic large cell lymphoma (ALCL), four (3 %) with enteropathic-type T cell lymphoma, and 24 (20 %) with NKTCL. The demographical and clinical characteristics of the patients are listed in Table 2. In the LAHS group the median age was 48 years (range, 16–78 years) and 46 years (range, 10–77 years) in the non-LAHS group. All of the patients with LAHS had B symptoms. Most of them were classified into the Ann Arbor III/IV stage with hepatosplenomegaly and cytopenia. Fifty-six percent (20/36) of the LAHS patients showed combined BMI, which was higher than the BMI in patients without LAHS (28 %, 34/123).
Laboratory findings are listed in Table 3. The elevated rates of β2-MG, ferritin, fasting triglycerides, prothrombin time, and CA125 levels were higher in the LAHS group than in the non-LAHS group (P < 0.05), so were hypofibrinogenemia (P < 0.05) and liver dysfunction (P < 0.05). In contrast, the increased levels of LDH, IG, and renal dysfunctions were not significantly different between the LAHS and the non-LAHS group.
Therapeutic results and survival analysis
With the median follow-up of 11 months (range, 1–79 months), the overall OS rate of the 159 patients was 36 %. Statistically significant differences of the OS rates were observed between the LAHS (11 %) and non-LAHS (44 %) group (log-rank P < 0.001; Fig. 1). The median survival times of the two groups were 3 and 16 months, respectively. Moreover, we compared the OS rates of PTCL-NOS patients with and without LAHS and noted a significant difference (log-rank P < 0.001; Fig. 2). In the LAHS group, statistically significant OS rate differences were not observed comparing elevated IG and normal IG levels (log-rank P =0.504; Fig. 3). The OS rate of LAHS patients treated with a CHOP regimen (12 %) was lower than the one of patients treated with an intensive chemotherapy (21 %), but the difference was not statistically significant (log-rank P = 0.545; Fig. 4). In our study, three LAHS patients were in critical conditions with high fever, hepatosplenomegaly, severe liver dysfunctions, and cytopenia and were given one to three times of plasma exchange before they received chemotherapies. The results showed that the median survival time of these three patients was 5 months, which was longer than that of all other LAHS patients. In summary, LAHS led to worse outcomes not only for all PTCL-affected but also for the PTCL-NOS-affected patients and elevated IG levels were not a significant prognostic factor for LAHS patients.

Kaplan-Meier estimates for the OS rates of 36 PTCL patients with LAHS compared to 123 PTCL patients without LAHS. A statistically significant difference was observed between the two groups (LAHS (11 %) and non-LAHS (44 %)), p < 0.001

Kaplan-Meier estimates for the OS rates of 28 PTCL-NOS patients with LAHS compared with 64 PTCL-NOS patients without LAHS. A statistically significant difference was observed between the groups PTCL-NOS patients with LAHS (7 %) and PTCL-NOS patients without LAHS (41 %), p < 0.001

Kaplan-Meier estimates for the OS rates of LAHS patients with elevated immunoglobulin (IG) levels at initial diagnosis compared to patients with normal IG levels. There was no statistically significant difference between the groups (P = 0.504)

Kaplan-Meier estimates for the OS rates of LAHS patients with intensive chemotherapy compared to those with a CHOP regimen. There was no statistically significant difference between the two groups (P = 0.545)
Discussion
HLH is a potentially fatal hyperinflammatory condition caused by a highly stimulated but ineffective immune response [14]. Acquired HLH is associated with malignant diseases, especially lymphomas (LAHS) [6, 15] and most cases of LAHS are associated with T cell or natural killer (NK)/T cell lymphoma, while LAHS secondary to B cell lymphomas are quite rare [15–19]. The frequency of LAHS varies greatly among different clinicopathological entities of PTCLs. Kobayashi et al. [19] reported in pediatric patients from Asia an incidence of 23 % from 22 PTCL patients with LAHS at initial diagnosis and the frequency of HLH was 33 % (3/9) in PTCL-NOS, followed by 20 % (2/10) in nasal NK/T cell lymphoma. Our data revealed 23 % of 159 PTCL patients with LAHS at initial diagnosis and the frequency of HLH was up to 30 % (28/92) in the PTCL-NOS group, 29 % (2/7) in the SCPTCL group, 14 % (3/21) in the AILT group, and 11 % (3/27) in the NKTCL group. In our study, the proportion of LAHS in the PTCL and PTCL-NOS groups was corresponding to Kobayashi's report [19], except that the frequency of HLH in the NKTCL group was lower. Because the number of pediatric patients is small in the literature, further research needs to be done.
We noted that in addition to β2-MG, ferritin and triglyceride increased more remarkably in the LAHS group than in the group without LAHS, which suggested a poor prognosis and further confirmed their importance as indicators for evaluating a T cell lymphoma prognosis. In contrast, renal dysfunction, increased levels of LDH and IG were irrelevant to whether LAHS was complicated or not. In our study, 56 % of the 36 patients had BMI and 72 % of the 36 patients had hepatosplenomegaly. The percentages in the LAHS group were clearly higher than that in the non-LAHS group (56 vs. 28 %, P = 0.002; 72 vs. 23 %, P < 0.001, respectively), which indicates that early BMI and hepatosplenomegaly are prominent features in T cell lymphomas with LAHS. Moreover, we detected peripheral lymphadenopathy only in 47 % of the 36 patients with LAHS. It is difficult to diagnose the disease accurately at an early stage without enlargement of superficial lymph nodes. Consequently, repeated biopsies of bone marrow from different locations are necessary and may increase the detection rates.
Unlike cytopenia and liver dysfunction being often mentioned, renal failure in LAHS patients was observed rarely. Renal failure was reported to occur at the advanced stages of HLH and is related to abnormally high concentrations of nephrotoxic IL-6 in the blood serum [20]. In our study, we found renal failures in 17 and 10 % (P = 0.264) of the PTCLs patients with and without LAHS, respectively. All of them were classified into Ann Arbor III/IV stages. Though the difference was not statistically significant, we need to be vigilant of creatinine and blood urea nitrogen elevation for early detection of renal failures.
CA125 has been widely used as a tumor marker in monitoring epithelial ovarian cancer. In aggressive NHL, CA125 was found to correlate with stage, tumor bulk, involvement of more than one extra-nodal site, and presence of effusion. Elevated levels of CA125 were found to predict decreased survival and also highly related to high levels of LDH and β2-MG [21–23]. Contrary to other serum markers such as β2-MG and LDH, CA125 apparently is not released by lymphoma cells and might be secreted by mesothelial cells stimulated via lymphokines derived from NHL [24]. Our data showed that the serum CA125 level increased in 82 % of the patients with LAHS and in 51 % of the patients without LAHS (P = 0.003). As it increased in 45 % of the total aggressive T cell lymphoma patients, we believe that the determination of the serum CA125 level is useful in predicting poor T cell lymphoma prognoses.
Combination chemotherapy remains the main treatment for T cell lymphoma associated with LAHS. Our data showed that 17 patients received a CHOP regimen as initial therapy, while some other regimens such as ECHOP, CHOP+Ara-C, DHAP, hyper CVAD, MINE, and ESHAP were employed in 19 patients. The estimated OS rates of LAHS patients treated with a CHOP regimen (12 %) was lower than that of patients with intensive chemotherapy (21 %), but the difference was not statistically significant (log-rank P = 0.545). The possible explanation for this is that LAHS is usually accompanied by multiorgan and especially cardiac dysfunctions, and intensive chemotherapy might further aggravate organ damage, thereby affecting the survival time. Because intensive chemotherapy is more bone marrow- and immune-suppressive, the toxicity of chemotherapy agents should be considered when patients accept intensive chemotherapies.
The mechanism of LAHS is generally believed to be related to a cytokine storm [25], and based on this theory, some researchers reported that plasmapheresis could achieve good therapeutic outcomes in secondary HLH [26, 27]. Yanagiya et al. [28] noted that a patient with diffuse large B cell lymphoma-associated HLH had his TNF-a, IL-6, and IL-8 serum levels reduced to normal values after plasmapheresis. In our study, three LAHS patients who were given one to three times of plasma exchange before they received chemotherapies had longer survival times. However, like in our retrospective analysis, most of the related reports by now are limited to single cases, and thus more studies are needed to further investigate the efficacy of plasmapheresis.
In conclusion, PTCL patients with LAHS had poorer prognoses than those without and the therapeutic outcome was unsatisfactory. Awareness of the clinical symptoms such as fever, hepatosplenomegaly, cytopenia, and constantly increasing levels of serum β2-MG, ferritin, CA125, and triglyceride is crucial in order to diagnose LAHS early and start life-saving therapies on time. Repeated biopsies of multiple bone marrows from different locations in those without enlargement of superficial lymph nodes are advisable in order to improve the diagnosis. Plasmapheresis as initial therapy is worth trying if the overall outcome of other treatments is unsatisfactory. In general, the treatment of LAHS remains a challenge and an intensive chemotherapy did not led to obvious advantages for the OS rate than a CHOP regimen due to its severe toxicity. Because of the present unsatisfactory therapeutic outcomes, well-designed clinical studies are required to improve the therapeutic procedures.
References
Tomita N, Motomura S, Hyo R et al (2007) Comparison of peripheral T–cell lymphomas and diffuse large B–cell lymphoma. Cancer 109:1146–1151
Kojima H, Hasegawa Y, Suzukawa K et al (2004) Clinicopathological features and prognostic factors of Japanese patients with “peripheral T-cell lymphoma, unspecified” diagnosed according to the WHO classification. Leuk Res 28:1287–1292
Ascani S, Zinzani PL, Gherlinzoni F et al (1997) Peripheral T-cell lymphomas. Clinico-pathologic study of 168 cases diagnosed according to the R.E.A.L. Classification. Ann Oncol 8:583–592
Armitage JO, Vose JM, Linder J et al (1989) Clinical significance of immunophenotype in diffuse aggressive non-Hodgkin's lymphoma. J Clin Oncol 7:1783–1790
Weitzman S (2011) Approach to hemophagocytic syndromes. Hematology Am Soc Hematol Educ Program 2011:178–183
Jaffe ES, Costa J, Fauci AS et al (1983) Malignant lymphoma and erythrophagocytosis simulating malignant histiocytosis. Am J Med 75:741–749
Takahashi N, Miura I, Chubachi A et al (2001) A clinicopathological study of 20 patients with T/natural killer (NK)-cell lymphoma-associated hemophagocytic syndrome with special reference to nasal and nasal-type NK/T-cell lymphoma. Int J Hematol 74:303–308
Gutierrez A, Solano C, Ferrandez A et al (2003) Peripheral T-cell lymphoma associated consecutively with hemophagocytic lymphohistiocytosis and hypereosinophilic syndrome. Eur J Haematol 71:303–306
Swerdlow SH, Campo E, Harris NL et al (2008) WHO classification of tumours of haematopoietic and lymphoid tissues, 4th edn. IARC, Lyon
Janka GE, Schneider EM (2004) Modern management of children with haemophagocytic lymphohistiocytosis. Br J Haematol 124:4–14
Carbone PP, Kaplan HS, Musshoff K et al (1971) Report of the Committee on Hodgkin's Disease Staging Classification. Cancer Res 31:1860–1861
Oken MM, Creech RH, Tormey DC et al (1982) Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 5:649–655
(1993) A predictive model for aggressive non-Hodgkin's lymphoma. The International Non-Hodgkin's Lymphoma Prognostic Factors Project. N Engl J Med 329:987-994
Janka G (2009) Hemophagocytic lymphohistiocytosis: when the immune system runs amok. Klin Padiatr 221:278–285
Falini B, Pileri S, De Solas I et al (1990) Peripheral T-cell lymphoma associated with hemophagocytic syndrome. Blood 75:434–444
Cheung MM, Chan JK, Lau WH et al (1998) Primary non-Hodgkin's lymphoma of the nose and nasopharynx: clinical features, tumor immunophenotype, and treatment outcome in 113 patients. J Clin Oncol 16:70–77
Florena AM, Iannitto E, Quintini G et al (2002) Bone marrow biopsy in hemophagocytic syndrome. Virchows Arch 441:335–344
Yamaguchi T, Ashihara E, Inaba T et al (1999) B-cell lymphoma associated with hemophagocytic syndrome. Gan No Japanese 45:439–443
Kobayashi R, Yamato K, Tanaka F et al (2010) Retrospective analysis of non–anaplastic peripheral T–cell lymphoma in pediatric patients in Japan. Pediatr Blood Cancer 54:212–215
Weber J, Yang JC, Topalian SL et al (1993) Phase I trial of subcutaneous interleukin-6 in patients with advanced malignancies. J Clin Oncol 11:499–506
Abd El Gawad IA, Shafik HE (2009) CA 125, a new prognostic marker for aggressive NHL. J Egypt Natl Canc Inst 21:209–217
Zidan J, Hussein O, Basher W et al (2004) Serum CA125: a tumor marker for monitoring response to treatment and follow-up in patients with non-Hodgkin's lymphoma. Oncologist 9:417–421
Zacharos ID, Efstathiou SP, Petreli E et al (2002) The prognostic significance of CA 125 in patients with non–Hodgkin's lymphoma. Eur J Haematol 69:221–226
Abd E, Gawad IA, Shafik HE (2009) CA125, a new prognostic marker for aggressive NHL. J Egypt Natl Canc Inst 21:209–217
Tsuda H (1997) Hemophagocytic syndrome (HPS) in children and adults. Int J Hematol 65:215–226
Sanada S, Ookawara S, Shindo T et al (2004) A case report of the effect of plasma exchange on reactive hemophagocytic syndrome associated with toxic shock syndrome. Ther Apher Dial 8:503–506
Coman T, Dalloz MA, Coolen N et al (2003) Plasmapheresis for the treatment of acute pancreatitis induced by hemophagocytic syndrome related to hypertriglyceridemia. J Clin Apher 18:129–131
Yanagiya N, Takahashi N, Nakae H et al (2002) Plasma exchange and continuous hemodiafiltration as an initial treatment for diffuse large B-cell lymphoma-associated hemophagocytic syndrome. Rinsho Ketsueki 43:35–40
Acknowledgments
This work was supported by the Special Funds for International Cooperation from the National Natural Science Foundation of China (81120108018), the Major Research Plan of the Chinese National Natural Science Foundation (91029740), and the Cultivation Program for Distinguished Talented Persons of Health of Zhejiang, China.
Conflict of interest
All authors declare that there is no conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Xie, W., Hu, K., Xu, F. et al. Clinical analysis and prognostic significance of lymphoma-associated hemophagocytosis in peripheral T cell lymphoma. Ann Hematol 92, 481–486 (2013). https://doi.org/10.1007/s00277-012-1644-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-012-1644-6