
Abstract
Diffuse large B-cell lymphoma (DLBCL) constitutes most primary central nervous system (CNS) lymphoma (PCNSL), whereas T-cell, low-grade and Burkitt’s lymphomas (BL) are rarely encountered. Due to the paucity of cases, little is known about the clinical features and treatment outcomes of PCNSL other than DLBCL. The objective of this study was to describe the clinical characteristics and outcomes for patients with PCNSL other than DLBCL. Fifteen patients, newly diagnosed with PCNSLs other than DLBCL between 2000 and 2010, were included. The male to female ratio was 0.67:1 with a median age of diagnosis of 31 years (range 18–59). Pathologic distributions were as follows: peripheral T-cell lymphoma (PTCL; n = 7), marginal zone B-cell lymphoma (MZBCL; n = 1), lymphoplasmacytic lymphoma (LPL; n = 2), Burkitt’s lymphoma (n = 1), other unspecified (T-cell lineage, n = 2; B-cell lineage, n = 2). Thirteen patients (87%) showed Eastern Cooperative Oncology Group performance score (ECOG PS) 1–2. The remaining two were one PTCL patient and one Burkitt’s lymphoma patient. Of the nine patients with T-cell lymphoma, five (56%) had multifocal lesions, and one (20%) with LPL of the five patients with B-cell lymphoma showed a single lesion. Leptomeningeal lymphomatosis was identified in two patients (one with Burkitt’s lymphoma and one with unspecified B-cell lymphoma). Two patients (22%) with T-cell lymphoma died 7.7 and 23.3 months later, respectively, due to disease progression, despite HD-MTX-based therapy. Six patients with T-cell lymphoma (6/9, 66.7%) and four patients with low-grade B-cell lymphoma (4/5, 80%) achieved complete response and have survived without relapse (Table 3). One patient with Burkitt’s lymphoma showed poor clinical features with ECOG PS 3, deep structure, multifocal, and leptomeningeal lymphomatosis, and died 7.6 months after the initiation of treatment. In comparison with previously reported DLBCLs (median OS 6.4 years, 95% CI 3.7–9.1 years), T-cell lymphoma showed equivocal or favorable clinical outcomes and low-grade B-cell lymphomas, such as MZBCL and LPL, had a good prognosis. However, primary CNS Burkitt’s lymphoma presented poor clinical outcomes and showed a comparatively aggressive clinical course. In conclusion, primary CNS lymphoma other than DLBCL occurred more in younger patients and showed a generally good prognosis, except for Burkitt’s lymphoma. Further research on treatment strategies for Burkitt’s lymphoma is needed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary central nervous system (CNS) lymphoma (PCNSL) is a rare variant of extranodal non-Hodgkin’s lymphoma that occurs in the brain, leptomeninges, spinal cord, or eyes without evidence of systemic disease. It is an aggressive disease with a median survival time of 1.5–3.3 months in untreated patients [20, 23]. PCNSL accounts for approximately 4% of all primary brain tumors and 4–6% of extranodal lymphomas [23]. Diffuse large B-cell lymphoma (DLBCL) is the most common type (>90%) of PCNSL, whereas approximately 10% are T-cell lymphomas (<5%), poorly characterized low-grade lymphomas and Burkitt’s lymphoma (BL). Few case reports have been described in the literature [1, 2, 17]. According to the World Health Organization (WHO) classification [30], primary DLBCL of the CNS was categorized as a subclassification of the DLBCL rather than as a separate entity due to indistinguishable morphology from systemic DLBCL. However, the remaining T-cell or other B-cell lymphomas of the CNS are not mentioned, and little is known about their clinical features and treatment outcomes because of their rarity. Therefore, we reviewed our experiences to analyze clinical features and treatment outcomes of 15 patients with rare primary CNS lymphoma other than DLBCL.
Patients and methods
Patients
Fifteen patients diagnosed with PCNSL other than DLBCL from January 2000 to July 2010 were included in this analysis. No patients had been in immunocompromised states brought about by, for example, organ transplantation or HIV infection. The pathologic diagnosis of PCNSL was based on the WHO classification after surgical resection. To evaluate the T- or B-cell lineage of PCNSL, immunohistochemical analysis was performed; and to confirm the T- or B-cell lineage of the tumors, TCR-γ gene and immunoglobulin heavy chain gene rearrangements were examined. The clinical information of patients, including age, sex, symptoms, physical findings, diagnostic procedures, and treatment was retrieved from our electronic medical records. Laboratory findings at diagnosis, such as blood cell counts, chemistry profile, serum protein electrophoresis, serum lactate dehydrogenase (LDH), serology of hepatitis B, C and HIV, and CSF laboratory findings, such as cell counts, protein, glucose, LDH and cytology, and bone marrow examinations were reviewed. Through meticulous physical examination including Waldeyer’s ring, chest and abdomino-pelvic computed tomography (CT), and positron emission tomography-CT, patients who had other primary sites of lymphoma were excluded.
Treatment
Data regarding treatment regimens and clinical courses for each patient were extracted from the review of medical records. The treatment course and response of each patient are summarized in Table 3. Treatment cohorts were generally divided into two groups in accordance with the chemotherapy regimen of our institution. Cohort 1[3], diagnosed prior to December 2002, was treated with 1 g/m2 of intravenous MTX (HD-MTX 1 g/m2 regimen) on days 1 and 8, in addition to 12 mg of intrathecal MTX via an Ommaya reservoir on days 1, 4, 8, 11, 15, and 18. This was followed by whole-brain irradiation (WBRT) and an additional 7.2–14.4 Gy boost to primary tumors. All patients received WBRT, regardless of age. WBRT is a parallel-opposing technique using 4- or 6-MV photons. The median dose was 40 Gy (range 18–50 Gy) in a daily fraction of 2 Gy. After radiotherapy, two cycles of intravenous cytosine arabinoside (HiDAC) at a dose of 3 g/m2 on two consecutive days was administered. Leucovorin (25 mg intravenously or orally, every 6 h) was given starting 24 h after intravenous MTX, until the plasma MTX concentration fell below 10−7 M. Cohort 2 [4], diagnosed from January 2003, was treated with five cycles of 3.5 g/m2 of intravenous MTX (HD-MTX 3.5 g/m2 regimen) and vincristine 1.4 mg/m2 at 2-weekly intervals, with/without procarbazine 100 mg/m2 for 7 days on cycles 1, 3, and 5. Intrathecal MTX (12 mg) was given via an Ommaya reservoir at 2-weekly intervals on the alterative weeks of intravenous MTX for five cycles. After the intravenous and intrathecal MTX schedules, a total dose of 30–45 Gy (median 45 Gy) was delivered to the whole brain with the same technique used in cohort 1. Patients with ocular involvement received additional ocular radiotherapy at 30–40 Gy. After completion of WBRT, the majority of patients were treated with HiDAC, as in cohort 1. For patients 60 years and older, WBRT was deferred.
Response evaluation
We reassessed the treatment response in accordance with the International Primary CNS Lymphoma Collaborative Group Guidelines for Response Assessment for Clinical Trials [1]. Complete response (CR) was defined as non-enhancing disease with normal eye examination and negative CSF cytology. Unconfirmed complete response was defined as minor-enhancing disease or minor retina pigment epithelial abnormality with negative CSF cytology. Partial response was defined as a 50% decrease in enhancement regardless of minor retina pigment epithelial abnormality with negative CSF cytology. Progressive disease was defined as a 25% increase in enhancement/new site or recurrent/new disease on eye examination or recurrent/positive CSF cytology. Stable disease (SD) was defined as other scenarios not covered by the above criteria. Assessment of treatment response was evaluated at least twice by brain magnetic resonance imaging at the time point between the completion of intravenous and intrathecal MTX and the start of WBRT and/or high-dose cytarabine, and after completion of the planned treatment scheme. CSF cytology was assessed at every intrathecal treatment.
Results
Patient characteristics
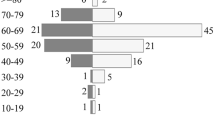
The main clinical characteristics of patients at diagnosis are summarized in Table 1. There were six male and nine female patients with a median age of diagnosis of 31 years (range 18–59). According to the WHO classification, histopathologic distribution is shown in Table 3 and as follows: peripheral T-cell lymphoma (PTCL; n = 7), marginal zone B-cell lymphoma (MZBCL; n = 1), lymphoplasmacytic lymphoma (LPL; n = 2), BL (n = 1), and other unspecified (T-cell lineage, n = 2/B-cell lineage, n = 2). PTCL was characterized by a population of predominantly small-sized cells admixed with occasional medium-sized cells positive for polyclonal CD3 antibody, representing a T-cell character of the tumor. The tumors of MZBCL were identical to those of MZBCLs in other sites. They were composed of sheets of small- to medium-sized lymphocytes with moderate amounts of cytoplasm and irregular nuclei, bearing a close resemblance to the small-cleaved cells or centrocytes of the lymphoid follicles. The neoplastic cells were immunoreactive for CD20 and CD79a and did not express CD3, CD5, CD10, or Cyclin D1. No evidence of amyloid deposition was present. Immunoglobulin light chain restriction was detected. No definitive features of transformation to a more aggressive histology were evident. The characters of LPL were immunoreactive for B-cell markers CD20, and negative for the T-cell marker CD3 and bcl. Immunoreactivity for the kappa chain was positive in the one patient. These pathologic findings were consistent with a lymphoplasmacytic lymphoma. Histopathologic examination of BL showed a distinct starry-sky pattern caused by interspersed tangible body macrophages and a diffuse monotonous infiltrate of medium-sized cells with a slight molding pattern. Immunohistochemical analysis revealed the tumor cells expressed LCA, CD10, CD20, and CD79a, but not CD3, CD45RO, CD30, panCK, CD117, BCL2, BCL6, synaptophysin, chromagranin, S-100 protein, and CD68. The tumor cells also displayed strong and diffuse nuclear positivity of Ki-67.
Most patients initially presented with neurologic symptoms such as headache/vomiting (31.3%), extremity weakness (40%), and facial nerve palsy (13.3%), but without B-cell symptoms such as fever, night sweats, and weight loss. Most patients showed good performance status of ECOG PS 1–2 and two patients had ECOG PS grade 3–4. Eight patients (53.3%) had the disease confined to only the cerebral hemisphere. Six patients (40%) were diagnosed with deep structure lesions such as the medulla (n = 2), medulla with spinal cord involvement (n = 1), basal ganglia with hippocampus (n = 1), thalamus (n = 1), and corpus callosum (n = 1) and one patient had only a single spinal cord lesion. Multiple lesions (more than two lesions) were detected in six patients (40%). Positive tumor cells in the CSF were found in two patients, including one with leptomeningeal enhancement of brain MRI. Seven patients (47%) had an elevated concentration of CSF protein, and eight patients (53.3%) had high CSF LDH. Three patients (20%) had an elevated serum LDH level. To facilitate comparison with DLBCL, brief characteristics are shown in Table 2 alongside those of the 50 primary CNS DLBCL patients reported by Uhm et al. [32]. PCNSL other than DLBCL showed a tendency to occur in younger patients. More than half of those with T-cell lymphoma developed multifocal lesions. However, among nine cases of T-cell lymphoma, none had positive CSF cytology, compared with two B-cell lymphoma patients with positive CSF cytology (one with Burkitt’s lymphoma and one with low-grade lymphoma).
Treatments and outcomes
The median follow-up time was 39 months (range 1–123 months). PCNSL other than DLBCL did not reach the median survival to date. Data of treatments and clinical outcomes of all patients are shown in Table 3. Of the nine patients with T-cell lymphoma, six patients (66.7%) achieved CR and one patient was alive at 2.5 months with SD. Of the five patients with low-grade B-cell lymphoma, three (60%) achieved CR and one patient was alive at 2.6 months with SD. One patient with LPL of one spinal cord lesion confined to T4 had surgical corpectomy and achieved CR without adjuvant chemotherapy. One patient with Burkitt’s lymphoma died at 7.6 months despite HD-MTX-based chemotherapy.
Discussion
We analyzed PCNSL other than DLBCL, which is the most common type of PCNSL, in a single institution. There are few reports about primary CNS T-cell lymphoma, anaplastic large cell lymphoma, low-grade lymphoma, and Burkitt’s lymphoma [6, 9, 14, 21, 24, 26, 27, 31]. The incidence of those lymphomas is unknown precisely because of its rarity. It is known that in immunocompetent patients, the median age at diagnosis of PCNSL is 53–57 years. The highest risk group is those 60 years and older [25], and the male to female ratio is 1.2–1.7:1. However, in this study, the median age of PCNSL other than DLBCL was 31 years (range 18–59) with a male-to-female ratio of 0.67:1. In Western countries, TPCNSL has been reported to constitute less than 4% of all PCNSLs, but in a nationwide survey of PCNSL in Japan, 8% of all cases were of T-cell origin [5, 12, 28]. Out of 121 consecutive PCNSL patients that we have followed up in our center during the last 10 years, nine PCNSL patients had T-cell lymphoma (7.4%), and most of them were PTCL, including two patients with unspecified T-cell lineage. The incidence of TPCNSL in our study is more comparable with Japanese than Western data. These data may correlate with a generally higher incidence of T-cell lymphomas in Far Eastern regions [16]. Shenkier et al. reported that among 45 patients, involvement of deep brain structures was seen in 16 patients (36%) and multiple lesions in 13 patients (29%) at diagnosis [27]. In our study, of the nine patients with TPCNSL, three (30%) had deep structures and five (56%) had multiple lesions. Three patients (#6, 7, 8) had deep locations of disease sites and presented with multiple lesions at the same time. The prognosis for T-cell lymphoma is controversial. There are several reports about primary TPCNSL with poor prognosis [18, 19, 29], and several with good response rates and overall survival time [10, 27]. Of our sample of nine patients, two died due to progressive disease after HD-MTX-based chemotherapy. The remaining six patients are alive in CR except one patient with SD at the 2.5-month follow-up. Although we could not draw conclusions on the prognosis of TPCNSL because of retrospective data and small patient numbers, our data showed equivocal or favorable clinical outcomes when compared with patients with DLBCL. It is meaningful that our study population was treated in a similar manner with HD-MTX-based treatment plus radiotherapy.
Uncommonly, MZBCL may present as a slowly growing localized mass arising from meningothelial cells of the dura (primary dural lymphoma), especially of the cerebral convexity, with rare cerebral parenchymal infiltration [13]. The origin site was not the dura but a single lesion of the thalamus. It is known that MZBCL is very radiosensitive. Treatment options for primary CNS MZBCL have ranged from local therapy to a combination with systemic chemotherapy [22]. Iwamoto et al. reported that eight patients with CNS MZBCL achieved complete response to treatment and no patient developed CNS recurrence during a median follow-up of 2 years (range 0.7–8.9 years). However, three patients developed systemic relapse after a median of 6.8 years (range 0.9–7.5 years). Bayraktar et al. showed that of six patients with primary CNS MZBCL, four achieved complete remission, and one relapsed at the same site after 5 years [6]. The results show that primary CNS MZBCLs display indolent clinical behavior and have a generally favorable clinical outcome with long-term disease control and survival. In our study, one patient (#10) received systemic chemotherapy and radiotherapy and is alive in complete remission at 39.4 months. Our study also showed that primary CNS MZBCL had a good prognosis despite involvement of a deep structure (thalamus). A review of the literature indicates that local therapy with radiation alone shows favorable outcomes (Table 4). Given these results, local treatment alone can be considered to be an effective modality for primary CNS MZBCL.
We found two reports of primary CNS LPL [7, 15]. Carrasco et al. reported a pituitary LPL patient maintained complete remission for 4 years after transsphenoidal surgery, chemotherapy, and radiotherapy [7]. We experienced one patient with a spinal cord LPL at T4 level. The patient (#12) received only surgical resection (corpectomy) without neurologic sequela and has lived without relapse for 51 months. Another patient, with LPL at the corpus callosum and left frontal lobe, is receiving the cohort 2 regimen. The interim evaluation is SD at 2.6 months. For these two patients, the prognosis of LPL seems to be satisfactory. Two patients did not show systemic symptoms and signs such as IgM paraproteinemia, neuropathy, and hyperviscosity.
Since Valsamis described the first case of primary CNS Burkitt’s lymphoma in an infant in 1976 [33], there have been several case reports of primary CNS Burkitt’s lymphoma [11]. The reported data showed poor clinical outcome. In our institution, one patient was diagnosed with primary CNS Burkitt’s lymphoma at the medulla and a leptomeningeal lesion with positive tumor cells in the CSF. Although the patient received HD-MTX-based chemotherapy, the final response was poor and the patient died at 7.6 months with progressive disease. Given other reports that show a poor outcome of primary CNS Burkitt’s lymphoma, HD-MTX-based chemotherapy does not appear to have an efficacious outcome. Therefore, more effective and aggressive treatment approaches for CNS Burkitt’s lymphoma are needed. In the literature review with our data, low-grade B-cell lymphoma, such as MZBCL and LPL, showed good prognosis, whereas Burkitt’s lymphoma showed an unfavorable clinical outcome (Table 4).
Reported 5-year survival rates after conventional treatment with HD-MTX, either as a single agent or in combination, followed by WBRT, is close to 40% [8]. Although heterogeneous and small number of cases does not warrant that these disease entities has the good prognosis than DLBCL, primary CNSLs, T-cell or B-cell lymphoma, other than DLBCL showed satisfactory treatment outcome in our series except for Burkitt’s lymphoma.
In this study, we described our experiences of primary CNS lymphoma other than DLBCL and made a historical comparison with previously reported DLBCL data from our institute. T-cell lymphoma showed equivocal or favorable clinical outcomes, and low-grade B-cell lymphomas such as MZBCL and LPL showed a good prognosis. However, CNS Burkitt’s lymphoma is more aggressive and presents a poor clinical outcome. Further research about treatment strategies for Burkitt’s lymphoma is needed. Additional analysis with a larger study population, including patients from other institutes, should be warranted.
References
Abrey LE, Batchelor TT, Ferreri AJ, Gospodarowicz M, Pulczynski EJ, Zucca E, Smith JR, Korfel A, Soussain C, DeAngelis LM, Neuwelt EA, O’Neill BP, Thiel E, Shenkier T, Graus F, van den Bent M, Seymour JF, Poortmans P, Armitage JO, Cavalli F (2005) Report of an international workshop to standardize baseline evaluation and response criteria for primary CNS lymphoma. J Clin Oncol 23:5034–5043
Abrey LE, Ben-Porat L, Panageas KS, Yahalom J, Berkey B, Curran W, Schultz C, Leibel S, Nelson D, Mehta M, DeAngelis LM (2006) Primary central nervous system lymphoma: the Memorial Sloan–Kettering Cancer Center prognostic model. J Clin Oncol 24:5711–5715
Abrey LE, DeAngelis LM, Yahalom J (1998) Long-term survival in primary CNS lymphoma. J Clin Oncol 16:859–863
Abrey LE, Yahalom J, DeAngelis LM (2000) Treatment for primary CNS lymphoma: the next step. J Clin Oncol 18:3144–3150
Bataille B, Delwail V, Menet E, Vandermarcq P, Ingrand P, Wager M, Guy G, Lapierre F (2000) Primary intracerebral malignant lymphoma: report of 248 cases. J Neurosurg 92:261–266
Bayraktar S, Stefanovic A, Montague N, Davis J, Murray T, Lossos IS (2010) Central nervous system manifestations of marginal zone B-cell lymphoma. Ann Hematol 89:1003–1009
Carrasco CA, Rojas ZD, Chiorino R, Gonzalez G (2011) Primary pituitary lymphoma in immunocompetent patient: diagnostic problems and prolonged follow-up. Pituitary (in press)
Ferreri AJ, Crocchiolo R, Assanelli A, Govi S, Reni M (2008) High-dose chemotherapy supported by autologous stem cell transplantation in patients with primary central nervous system lymphoma: facts and opinions. Leuk Lymphoma 49:2042–2047
George DH, Scheithauer BW, Aker FV, Kurtin PJ, Burger PC, Cameselle-Teijeiro J, McLendon RE, Parisi JE, Paulus W, Roggendorf W, Sotelo C (2003) Primary anaplastic large cell lymphoma of the central nervous system: prognostic effect of ALK-1 expression. Am J Surg Pathol 27:487–493
Gijtenbeek JM, Rosenblum MK, DeAngelis LM (2001) Primary central nervous system T-cell lymphoma. Neurology 57:716–718
Gu Y, Hou YY, Zhang XB, Hu F (2010) Primary central nervous system Burkitt lymphoma as concomitant lesions in the third and the left ventricles: a case study and literature review. J Neurooncol 99:277–281
Hayabuchi N, Shibamoto Y, Onizuka Y (1999) Primary central nervous system lymphoma in Japan: a nationwide survey. Int J Radiat Oncol Biol Phys 44:265–272
Iwamoto FM, DeAngelis LM, Abrey LE (2006) Primary dural lymphomas: a clinicopathologic study of treatment and outcome in eight patients. Neurology 66:1763–1765
Jahnke K, Korfel A, O’Neill BP, Blay JY, Abrey LE, Martus P, Poortmans PM, Shenkier TN, Batchelor TT, Neuwelt EA, Raizer JJ, Schiff D, Pels H, Herrlinger U, Stein H, Thiel E (2006) International study on low-grade primary central nervous system lymphoma. Ann Neurol 59:755–762
Kanavaros P, Mikol J, Nemeth J, Galian A, Dupont B, Thiebaut JB, Thurel C (1990) Stereotactic biopsy diagnosis of primary non-Hodgkin’s lymphoma of the central nervous system. A histological and immunohistochemical study. Pathol Res Pract 186:459–466
Ko YH, Kim CW, Park CS, Jang HK, Lee SS, Kim SH, Ree HJ, Lee JD, Kim SW, Huh JR (1998) REAL classification of malignant lymphomas in the Republic of Korea: incidence of recently recognized entities and changes in clinicopathologic features. Hematolymphoreticular Study Group of the Korean Society of Pathologists. Revised European–American lymphoma. Cancer 83:806–812
Miller DC, Hochberg FH, Harris NL, Gruber ML, Louis DN, Cohen H (1994) Pathology with clinical correlations of primary central nervous system non-Hodgkin’s lymphoma. The Massachusetts General Hospital experience 1958–1989. Cancer 74:1383–1397
Mineura K, Sawataishi J, Sasajima T, Kowada M, Sugawara A, Ebina K (1993) Primary central nervous system involvement of the so called ‘peripheral T-cell lymphoma’. Report of a case and review of the literature. J Neurooncol 16:235–242
Nitta T, Uda K, Ebato M, Ikezaki K, Fukui M, Sato K (1995) Primary peripheral-postthymic T-cell lymphoma in the central nervous system: immunological and molecular approaches to diagnosis. J Neurosurg 82:77–82
Panageas KS, Elkin EB, DeAngelis LM, Ben-Porat L, Abrey LE (2005) Trends in survival from primary central nervous system lymphoma, 1975–1999: a population-based analysis. Cancer 104:2466–2472
Park I, Huh J, Kim JH, Lee SW, Ryu MH, Kang YK (2008) Primary central nervous system marginal zone B-cell lymphoma of the basal ganglia mimicking low-grade glioma: a case report and review of the literature. Clin Lymphoma Myeloma 8:305–308
Puri DR, Tereffe W, Yahalom J (2008) Low-dose and limited-volume radiotherapy alone for primary dural marginal zone lymphoma: treatment approach and review of published data. Int J Radiat Oncol Biol Phys 71:1425–1435
Reni M, Ferreri AJ, Garancini MP, Villa E (1997) Therapeutic management of primary central nervous system lymphoma in immunocompetent patients: results of a critical review of the literature. Ann Oncol 8:227–234
Rupani A, Modi C, Desai S, Rege J (2005) Primary anaplastic large cell lymphoma of central nervous system—a case report. J Postgrad Med 51:326–327
Schabet M (1999) Epidemiology of primary CNS lymphoma. J Neurooncol 43:199–201
Shehu BB (2003) Primary central nervous system Burkitt’s lymphoma presenting with proptosis. Ann Trop Paediatr 23:319–320
Shenkier TN, Blay JY, O’Neill BP, Poortmans P, Thiel E, Jahnke K, Abrey LE, Neuwelt E, Tsang R, Batchelor T, Harris N, Ferreri AJ, Ponzoni M, O’Brien P, Rubenstein J, Connors JM (2005) Primary CNS lymphoma of T-cell origin: a descriptive analysis from the international primary CNS lymphoma collaborative group. J Clin Oncol 23:2233–2239
Shibamoto Y, Ogino H, Suzuki G, Takemoto M, Araki N, Isobe K, Tsuchida E, Nakamura K, Kenjo M, Suzuki K, Hosono M, Tokumaru S, Ishihara S, Kato E, Ii N, Hayabuchi N (2008) Primary central nervous system lymphoma in Japan: changes in clinical features, treatment, and prognosis during 1985–2004. Neuro Oncol 10:560–568
Stark AM, Tiemann M, Dorner L, Melnikowa E, Mehdorn HM, Blomer U (2004) Primary peripheral T-cell lymphoma of the central nervous system. Zentralbl Neurochir 65:191–194
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (2008) World health organization classification of tumours. Pathology and genetics of tumours of haematopoietic and lymphoid tissues, 4th edn. IARC Press, Lyon, pp 236–237
Toren A, Mandel M, Shahar E, Rimmoni E, Roizin H, Neuman Y, Brok-Simoni F, Mark Z, Biniaminov M, Rosenthal E et al (1994) Primary central nervous system Burkitt’s lymphoma presenting as Guillain–Barre syndrome. Med Pediatr Oncol 23:372–375
Uhm JE, Kim KH, Yi SY, Chang MH, Park KW, Kong DS, Lee JI, Nam do H, Park W, Lim do H, Kim SJ, Kim K, Ko YH, Kim WS (2009) A retrospective study to compare two methotrexate-based regimens for primary central nervous system lymphoma. Leuk Lymphoma 50:1110–1118
Valsamis MP, Levine PH, Rapin I, Santorineou M, Shulman K (1976) Primary intracranial Burkitt’s lymphoma in an infant. Cancer 37:1500–1507
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lim, T., Kim, S.J., Kim, K. et al. Primary CNS lymphoma other than DLBCL: a descriptive analysis of clinical features and treatment outcomes. Ann Hematol 90, 1391–1398 (2011). https://doi.org/10.1007/s00277-011-1225-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-011-1225-0