
Abstract
The 21-kD protein Ras of the low-molecular-weight GTP-binding (LMWG) family plays an important role in transduction of extracellular signals. Ras functions as a ‘molecular switch’ in transduction of signals from the membrane receptors of many growth factors, cytokines, and other second messengers to the cell nucleus. Numerous studies have shown that in multiple malignant tumors and hematopoietic malignancies, faulty signal transduction via the Ras pathway plays a key role in tumorigenesis. In this work, a non-radioactive assay was used to quantify Ras activity in hematologic malignancies. Ras activation was measured in six different cell lines and 24 patient samples, and sequence analysis of N- and K-ras was performed. The 24 patient samples comprised of seven acute myelogenous leukemia (AML) samples, five acute lymphocytic leukemia (ALL) samples, four myeloproliferative disease (MPD) samples, four lymphoma samples, four juvenile myelomonocytic leukemia (JMML) samples, and WBC from a healthy donor. The purpose of this study was to compare Ras activity determined by percentage of Ras-GTP with the mutational status of the Ras gene in the hematopoietic cells of the patients. Mutation analysis revealed ras mutations in two of the seven AML samples, one in codon 12 and one in codon 61; ras mutations were also found in two of the four JMML samples, and in one of the four lymphoma samples (codon 12). We found a mean Ras activation of 23.1% in cell lines with known constitutively activating ras mutations, which was significantly different from cell lines with ras wildtype sequence (Ras activation of 4.8%). Two of the five activating ras mutations in the patient samples correlated with increased Ras activation. In the other three samples, Ras was probably activated through “upstream” or “downstream” mechanisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mutations in the ras proto-oncogenes (H-ras, N-ras, K-ras) are the most common genetic aberrations in human tumors, occurring in 20–30% of all tumors; although this figure varies considerably depending on the tumor entity. The most frequently observed ras mutations occur in codons 12, 13, and 61, with these mutations disrupting the GTPase function of Ras. Although the mutated Ras still forms a complex with regulatory GTPase activating proteins (GAPs), conversion of guanosine-5′-triphosphate (GTP) to guanosine-5′-diphosphate (GDP), i.e., Ras inactivation, is attenuated. This blockade leads to a longer half-life of the active GTP-binding form of Ras. Besides mutations in codons 12, 13, and 61, sporadic mutations have been described in codons 116, 117, 119, and 146. These mutations decrease the protein’s affinity towards nucleotides, leading to an exchange in favor of cytosolic GTP, of which there is a ten-fold higher level than GDP. In addition, the binding of hydrolysis-enhancing GAPs is inhibited. Moreover it has been shown that mutations in codons 59 and 63 activate the transforming potential of the H-ras gene [1] but are of little clinical relevance. Hence, all known mutations lead to an accumulation of constitutively active Ras and uninterrupted signal transfer. In malignant hematopoietic diseases, activating Ras mutations occur most commonly in myeloid disorders, in myelodysplastic syndrome (MDS) and AML, with the level of Ras activation ranging from 6 to 37%. The mutations occur largely in N-ras codons 12, 13, and 61, and in K-ras codon 12; H-ras mutations are rarely seen in this entity. The most common mutation is a G→A mutation in codon 12 [2, 3]. Activated Ras mutations appear to play an important part in the transformation from MDS to AML, and a high percentage of patients with MDS-based AML develop Ras mutations in the course of their disease [4]. Furthermore, a prospective study showed that MDS patients with a confirmed ras mutation have a significantly higher risk of developing AML [5].
In addition to mutations in the ras gene, Ras can be activated by constitutively active proto-oncogenes and inactivated by tumor suppressor genes [6]. Several receptor tyrosine kinases and non-receptor tyrosine kinases signal via the Ras transduction pathway. Point mutations in the CSF-1 receptor (c-FMS) are found in 10–20% of all AML and MDS patients [7]. Another common defect in AML and MDS is tandem duplications in the FLT3 receptor, which occur in 20–25% of all AML cases. Simultaneous mutations in N-ras and FLT3 are very rare [8]. Members of the c-Kit/c-FMS receptor kinase family (e.g., c-Kit, c-FMS, FLT3) associate with proteins of the Ras/MAPK signal transduction pathway (e.g., Grb-2 and Shc). Hence, their activation could activate Ras [9].
The Bcr-Abl fusion protein occurs in 95% of chronic myelogenous leukemia (CML) cases and in 10% of ALL cases. Although ras mutations in CML are rare, several experiments have shown that Ras activation via the constitutively active Bcr-Abl protein plays an important role. Inhibition of Ras activity by antisense oligonucleotides, microinjection of a blocking monoclonal antibody, or overexpression of dominant-negative Ras-GAP block transformation by Bcr-Abl [10, 11]. Another Abl fusion protein, Tel-Abl, is found in AML featuring a t(12;9) translocation [12, 13]. Other fusion proteins that activate the Ras/MAPK transduction pathway are: (1) the Npm-Alk fusion protein in anaplastic large cell non-Hodgkin’s lymphoma featuring a t(2;5) translocation [14, 15]; and (2) Tel-PDGFR—a fusion protein comprising the transcription factor Tel (translocation, Ets, leukemia) and PDGFR, which occurs in a subpopulation of chronic myelomonocytic leukemia (CMML) and features a t(5;12) translocation [12].
A number of tumor repressor genes trigger dysregulation of Ras activity. These include the Ras-GAP NF-1, which is mutated in the autosomal dominant form of neurofibromatosis type 1 [16, 17]. In adolescents, neurofibromatosis type 1 is associated with an increased risk of developing myeloproliferative disorders, particularly JMML. In 15% of children with JMML not showing clinical symptoms of neurofibromatosis, inactivating NF-1 mutations have been found [18]. Furthermore, 15–30% of JMML cases that lack the NF-1 mutation present with activating ras mutations [19]. Mutations in both the ras and NF-1 genes are very rare and seem to be mutually exclusive [20].
Somatic PTPN11 mutations occur in approximately 35% of JMML patients [18, 21–22]. PTPN11 encodes the tyrosine phosphatase SHP-2 which transmits signals of growth factor receptors to Ras and other effectors. Thus, Ras can be activated by either an activating mutation in the ras gene or by mutations in proteins that signal through Ras. Gain-of-function mutations of RAF1, a downstream Ras effector, cause Noonan syndrome in patients who do not have a mutation of Ras or upstream effectors [23, 24].
In this study, Ras activity determined by the ratio of Ras-GTP to Ras-GDP has been compared with the mutational status of the Ras gene in the hematopoietic cells of the patients.
Materials and methods
Patient samples from bone marrow aspirates or peripheral blood were collected between 1999 and 2002 after informed consent had been obtained. Approximately 10 ml of peripheral blood or 1 ml of bone marrow was obtained from normal subjects and from children with B-ALL according to a procedure approved by the UFMC IRB and from children with T-ALL according to approved protocols. Samples were shipped overnight with the bone marrow samples diluted 1:2 in RPMI medium. To generate peripheral WBCs and nucleated bone marrow cells, red cells were lysed in 0.9% ammonium acetate according to standard procedures, and to generate peripheral blood lymphocytes, whole blood was applied to a Ficoll gradient as previously described. Some of the samples were cells isolated on a Ficoll gradient and frozen in 10% DMSO; these samples were washed once in PBS before further processing. As discussed later in Results, none of these isolation procedures altered the activation state of Ras.
Samples from 23 patients were used: seven patients with AML, four patients with MPS (one osteomyelofibrosis (OMF), three CML), four patients with JMML, four patients with ALL and four with lymphoma including plasma cell leukemia. Except for the four patients with JMML, all patients were being treated in the Department of Medicine I at the University of Freiburg Medical Center.
Because of problems associated with using 32PO4 to assess Ras activation, we used biochemical non-radioactive methods for measuring GTP and GDP bound to Ras family proteins as described before [25, 26]. Ras was immunoprecipitated with the rat monoclonal antibody Y13-259. Absolute amounts of GTP and GDP were measured in coupled enzymatic reactions. GTP was measured by converting it to ATP using the enzyme nucleoside diphosphate kinase (NDPK, Sigma St. Louis, MO) in the presence of ADP. The resulting ATP was measured by firefly luciferase (Calbiochem, San Diego, CA) in a photon counting luminometer. The firefly luciferase system is extremely sensitive, allowing detection of 1 fmol of GTP. To measure GDP, we converted it to GTP using the enzyme pyruvate kinase (PK, Sigma St. Louis, MO) with phosphoenolpyruvate serving as a source of high-energy phosphate.
As positive controls, HL-60 cells (AML FAB M2) with a known N-ras mutation in codon 61 (CAA→CTA) were used. As negative controls with known wildtype ras, the myelogenous cell line U937 and the pre-B-ALL cell line 697 were used. Murine 3T3 fibroblasts were used both as a negative and a positive control, the latter after being transfected with an activated H-Ras Val12 vector (Table 1).
Genomic DNA was isolated using the commercially available “QIAamp DNA Blood Mini Kit” (QIAGEN GmbH, Hilden, Germany). Amplification was performed with K-ras and N-ras specific primers containing 5′ universal M13 primer sequence attached, which were then used for genomic sequencing (ABI PRISM® BigDye Terminator Cycle Sequencing Kit and ABI310 Genetic Analyzer-System, Applied Biosystems, Foster City, USA)
Results
-
1.
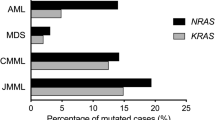
Frequency of N-ras and K-ras mutations in 23 patients with several hematological disorders and in one healthy control
Table 2 shows results of Ras activation in peripheral blood and bone marrow WBCs from a total of 23 patients: seven with AML, four with ALL, two with CML, four with JMML and one with lymphoma. No diagnosis could be given to five of the patients.
Mutation analysis confirmed mutations in the positive controls, and showed mutations in two of the seven AML samples (29%), one in codon 12 (exon1) and one in codon 61 (exon2). The latter data are consistent with the mutation frequencies reported in the literature. Our results are also in accordance with recent data indicating that the frequency of Ras mutations in AML and MDS has been overestimated in the past (M. Lübbert, personal communication).
In the samples from patients with MPS again no mutation of Ras could be detected, a finding which has been previously explained by constitutive Ras activation via the uncontrolled Bcr-Abl signal upstream of Ras [31]. None of the four ALL samples showed a mutation. This is compatible with the lower frequency of ras mutations in this entity. Ras mutations were also found in two of the JMML samples. A N-Ras mutation has been found in codon 12 (exon 1) of a sample from a patient with a plasma cell leukemia.
-
2.
Frequency of Ras activation in 23 patients with several hematological disorders and in one healthy control (Fig. 2) and in six cell lines (Fig. 1)

Ras Activation in 12 different determinations (dots) of Ras activation [GTP/(GDT+GTP)%] of control cells lines according to their mutational status. Samples with known WT sequences are shown separate from samples with known activating mutation. The 95% CI are shown by bars. Average ras activation in WT versus mutation samples is 4.75% (0.8–8.7%) vs. 23.06% (12.7–38%) respectively. This difference is significant (p = 0.006; t-test, ungrouped)
Recent mathematical models of the Ras signaling pathway supported by experimental observations have underlined the function of Ras as a ‘molecular switch’ in signal transduction. Given four hypothetical disruptions in Ras signaling—GAP insensitivity, reduced GTPase activity, increased effector affinity and competitive inhibition of Ras-GAP by Ras RasVal12—it was hypothesized that cells harboring a heterozygous ras mutation resulting in a GAP-insensitive RasVal12 show an hypothetical activation (total Ras bound GTP) of approximately 28%. By contrast, wildtype (WT cells) should show an activation of approximately 5% [27].
In Fig. 1, Ras activation from 12 different determinations [GTP/(GDT+GTP)%] were grouped according to their mutational status. The 95% CIs are represented by bars. Average Ras activation in WT was 4.75% (0.8–8.7%) vs. 23.06% (12.7–38%) respectively. This difference proved significant (p = 0.006; t-test, ungrouped).
In Fig. 2, Ras activation of patient samples is shown according to their mutational status. Patient samples (n = 23) are separated by their hematological entity. A patient sample was considered to contain activated Ras if the activation level was within the 95% CI of the positive controls, and negative if it was within the 95% CI of the negative controls. Both 95% CIs of negative and positive control cells are indicated by bars.

Ras activation of patient samples according to their mutational status. Numbers in bold represent average levels of Ras activation. A patient sample was considered to have activated Ras if activation level was within the 95% confidence interval (CI) of the positive controls, and negative for Ras activation if it was within the 95% CI of the negative controls. The 95% CIs of the positive and negative controls are represented by vertical bars
The average activation of patient samples with proven ras mutations (n = 5) was 10.8% (1.1–19.8%; 95% CI, 0.333–23.3). The average activation of patient samples with ras WT sequence was 7.41% (0.6–38.6%; 95% CI, 1.32–13.44). Since the 95% CIs overlap in this analysis, the difference is not significant (p = 0.565, t-test ungrouped).
-
3.
Discrepancy between Ras activation and ras activating mutations
One of the two samples with activating Ras from patients with AML, the a N-ras mutation of codon 61 in exon 2 resulting in a CAA-AAA sequence, did not show a higher percentage of GTP-Ras which would be indicative of an activation of the protein, whereas the N-ras mutation in codon 12 of Exon resulting in a GGT-TGT sequence was correlated with a GTP/GTP+GDP ratio of 19.8%. In the samples from patients with JMML, one sample had no increased Ras-GTP in spite of an activating mutation. One wildtype sample showed a Ras-GTP percentage of 38.6% without an activating Ras-mutation.
Discussion
The frequencies of mutations found in our study for different forms of hematological malignancies do not reach significance levels due to the low number of samples, but in general they reflect the tendencies found in previous reports [35].
We showed that cell lines with known constitutively activating ras mutations had significantly higher levels of Ras-GTP (mean activation 23.1%, p = 0.006) than did cell lines with WT-Ras (mean activation 4.8%). These data show the high degree of selectivity of the assay in distinguishing between cells with activated and non-activated Ras and are consistent with hypothetical and observed activations [28]. Determination of a threshold for positive Ras activation in patient samples is difficult. Given the aspect of Ras functioning as ‘molecular switch’ and taking into account the expected heterogenity of patients’ primary cells (Ras isoforms, clonality, etc.) we established two ranges (95% CIs) of cell populations with either WT ras or mutated ras in order to be able to classify patients’ values as activated or not activated in the setting of our assay. A patient sample was considered to have an activated Ras if the activation level was within the 95% CI of the positive controls. Three of the five patient samples (60%) with proven ras mutations belong in this category. On the other hand, 12 of 14 patient samples (approximately 86%) with wildtype sequence are within the 95% CI of the negative controls.
The lack of intermediate values reflects the function of Ras as a ‘molecular switch’. Therefore, patient samples with increased Ras activation lacking a mutational activation are of interest for further analysis of the Ras signal transduction cascade, since various up- and downstream effectors contribute to Ras activation.
Ras mutations were found in hematological neoplasia known to have mutated Ras: two of six AML cases, one of two plasma cell leukemias (PCL), and two of four JMML cases. In two of the five patients with Ras mutations, Ras showed increased activation. However, one AML and one JMML sample showed noticeably low levels of activation though harboring a ras mutation.
In addition to technical difficulties with the applied highly sensitive enzyme-based method, heterogeneity of the underlying patients’ samples has to be taken into account. In further studies, the issue of clonality of samples could be overcome by pre-sorting cells, e.g., by flourescence activated cell sorter (FACS). Another explanation might be additional compensatory factors bypassing Ras such as, for example, by GAP [29] regulation or compensatory up-regulation of WT-Ras.
In the patient sample with a Ras activation of 32.7% but no underlying mutation, the diagnosis was pre-B-ALL. In this context, CD40-stimulated Ras activation in the early developmental stages of B-cells has been described [30].
Assessing Ras quantitatively in hematopoietic neoplasia may provide a basis for understanding the role of Ras in various signal transduction pathways and may provide new routes toward targeted treatment strategies. The most promising category influencing Ras signalling is farnesyltransferase inhibitors (FTIs), which inhibit the attachment of the farnesyl moiety to the Ras protein [32, 33]. In addition, drugs which interfere with the interaction of the farnesyl group with the cell membrane, such as farnesyl thiosalicylic acid, also influence the activity of Ras on a protein level [34].
References
Fasano O, Aldrich T et al (1984) Analysis of the transforming potential of the human H-ras gene by random mutagenesis. Proc Natl Acad Sci U S A 81(13):4008–4012. doi:10.1073/pnas.81.13.4008
Guo W, Tang B et al (1998) N-ras mutations in 43 Chinese cases of acute myeloid leukemia. Chin Med J (Engl) 111:343–345
Farr C, Gill R et al (1991) Analysis of ras gene mutations in childhood myeloid leukaemia. Br J Haematol 77:323–327. doi:10.1111/j.1365-2141.1991.tb08578.x
Shih LY, Huang CF et al (2004) Acquisition of FLT3 or N-ras mutations is frequently associated with progression of myelodysplastic syndrome to acute myeloid leukemia. Leukemia 18:466–475. doi:10.1038/sj.leu.2403274
Padua RA, Guinn BA et al (1998) RAS, FMS and p53 mutations and poor clinical outcome in myelodysplasias: a 10-year follow-up. Leukemia 12:887–892. doi:10.1038/sj.leu.2401044
Hunter T (1997) Oncoprotein networks. Cell 88:333–346. doi:10.1016/S0092-8674(00)81872-3
Tobal K, Pagliuca A et al (1990) Mutation of the human FMS gene (M-CSF receptor) in myelodysplastic syndromes and acute myeloid leukemia. Leukemia 4:486–489
Kiyoi H, Naoe T et al (1999) Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood 93:3074–3080
Rohrschneider LR, Bourette RP et al (1997) Growth and differentiation signals regulated by the M-CSF receptor. Mol Reprod Dev 46:96–103. doi:10.1002/(SICI)1098-2795(199701)46:1<96::AID-MRD15>3.0.CO;2-1
Skorski T, Kanakaraj P et al (1994) Negative regulation of pl20GAP GTPase promoting activity by p210bcr/abl: implication for RAS-dependent Philadelphia chromosome positive cell growth. J Exp Med 179:1855–1865. doi:10.1084/jem.179.6.1855
Sawyers CL, McLaughlin J et al (1995) Genetic requirement for Ras in the transformation of fibroblasts and hematopoietic cells by the Bcr-Abl oncogene. J Exp Med 181:307–313. doi:10.1084/jem.181.1.307
Golub TR, Barker GF et al (1994) Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell 77:307–316. doi:10.1016/0092-8674(94)90322-0
Golub TR, Goga A et al (1996) Oligomerization of the ABL tyrosine kinase by the Ets protein TEL in human leukemia. Mol Cell Biol 16:4107–4116
Elmberger PG, Lozano MD et al (1995) Transcripts of the npm-alk fusion gene in anaplastic large cell lymphoma, Hodgkin’s disease, and reactive lymphoid lesions. Blood 86:3517–3521
Waggott W, Lo YM et al (1995) Detection of NPM-ALK DNA rearrangement in CD30 positive anaplastic large cell lymphoma. Br J Haematol 89:905–907
Xu GF, O’Connell P et al (1990) The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 62:599–608. doi:10.1016/0092-8674(90)90024-9
DeClue JE, Papageorge AG et al (1992) Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell 69:265–273. doi:10.1016/0092-8674(92)90407-4
Side L, Taylor B et al (1997) Homozygous inactivation of the NF1 gene in bone marrow cells from children with neurofibromatosis type 1 and malignant myeloid disorders. N Engl J Med 336:1713–1720. doi:10.1056/NEJM199706123362404
Miyauchi J, Asada M et al (1994) Mutations of the N-ras gene in juvenile chronic myelogenous leukemia. Blood 83:2248–2254
Kalra R, Paderanga DC et al (1994) Genetic analysis is consistent with the hypothesis that NF1 limits myeloid cell growth through p21ras. Blood 84:3435–3439
Tartaglia M, Niemeyer CM et al (2003) Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 34:148–150. doi:10.1038/ng1156
Kratz CP, Niemeyer CM et al (2005) The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood 106:2183–2185. doi:10.1182/blood-2005-02-0531
Pandit B, Sarkozy A et al (2007) Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet 39(8):1007–1012. doi:10.1038/ng2073
Razzaque MA, Nishizawa T et al (2007) Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet 39(8):1013–1017. doi:10.1038/ng2078
Scheele JS, Rhee JM, Boss GR (1995) Determination of absolute amounts of GDP and GTP bound to Ras in mammalian cells: Comparison of parental and ras-overproducing NIH 3T3 fibroblasts. Proc Natl Acad Sci U S A 92:1097–1100. doi:10.1073/pnas.92.4.1097
Von Lintig FC, Huvar I et al (2000) Ras activation in normal white blood cells and childhood acute lymphoblastic leukemia. Clin Cancer Res 6:1804–1810
Stites EC, Trampont PC et al (2007) Network analysis of oncogenic Ras activation in cancer. Science 318(5849):463–467. doi:10.1126/science.1144642
Shen WP, Aldrich TH et al (1987) Expression of normal and mutant ras proteins in human acute leukemia. Oncogene 1:157–165
Bernards A, Settleman J (2005) GAPs in growth factor signalling. Growth Factors 23(2):143–149. doi:10.1080/08977190500130480
Gulbins E, Brenner B et al (1996) Activation of the Ras signaling pathway by the CD40 receptor. J Immunol 157:2844–2850
Garicochea B, Giorgi R, Odone VF, Dorlhiac-Llacer PE, Bendit I (1998) Mutational analysis of N-RAS and GAP-related domain of the neurofibromatosis type 1 gene in chronic myelogenous leukemia. Leuk Res 22:1003–1007. doi:10.1016/S0145-2126(98)00076-9
Cortes J, Quintás-Cardama A, Garcia-Manero G, O’Brien S, Jones D, Faderl S et al (2007) Phase 1 study of tipifarnib in combination with imatinib for patients with chronic myelogenous leukemia in chronic phase after imatinib failure. Cancer 110(9):2000–2006. doi:10.1002/cncr.23006
Reichert A, Heisterkamp N, Daley GQ, Groffen J (2001) Treatment of Bcr/Abl-positive acute lymphoblastic leukemia in P190 transgenic mice with the farnesyl transferase inhibitor SCH66336. Blood 97:1399–1403. doi:10.1182/blood.V97.5.1399
Halaschek-Wiener J, Wacheck V, Schlagbauer-Wadl H, Wolff K, Kloog Y, Jansen B (2000) A novel Ras antagonist regulates both oncogenic Ras and the tumor suppressor p53 in colon cancer cells. Mol Med 6:693–704
Scheele JS, Raepple D, Luebbert M (2000) The role of Ras and other low-molecular weight guanine nucleotide (GTP)-binding proteins during hematopoietic cell differentiation. Cell Mol Life Sci 57:1950–1963. doi:10.1007/PL00000675
Acknowledgment
This work was supported by the Deutsche Krebshilfe Grant 10-728 sche2. We thank Prof. Dr. C. Niemeyer, Department of Pediatric Hematology and Oncology, Centre for Pediatric and Adolescent Medicine Freiburg, for providing the samples of JMML patients.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplement 1
Primers used for mutation analysis (DOC 24.5 KB)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Raepple, D., von Lintig, F., Zemojtel, T. et al. Determination of Ras-GTP and Ras-GDP in patients with acute myelogenous leukemia (AML), myeloproliferative syndrome (MPS), juvenile myelomonocytic leukemia (JMML), acute lymphocytic leukemia (ALL), and malignant lymphoma: assessment of mutational and indirect activation. Ann Hematol 88, 319–324 (2009). https://doi.org/10.1007/s00277-008-0593-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-008-0593-6