
Abstract
We synthesized six samples in the compositional field NaMgAl(SO4)3–KMgAl(SO4)3 in 20 mol% increments from pure Na to pure K compounds. We investigated them by Powder X-Ray diffraction, 23Na, and 27Al Nuclear Magnetic Resonance spectroscopy. The results confirm NaMgAl(SO4)3 as a unique phase identical to a presumed new mineral found in the fumaroles of Eldfell and Hekla volcanoes in Iceland. It tolerates less than 10 mol% K substitution for Na. There exists a compositional gap to approximately Na0.65K0.35MgAl(SO4)3 from where a solid solution extends to KMgAl(SO4)3. The mineral koryakite [NaKMg2Al2(SO4)6] is a member of the latter solid solution series. The crystal structures of all (Na,K)MgAl(SO4)3 phases are akin to NASICON (NA Super Ionic CONductor). NaMgAl(SO4)3 has \(R\overline{3}c\) symmetry and a disordered distribution of Mg and Al among the octahedral sites with only one unique site for the alkali atom. The members of the solid solution have \(R\overline{3}\) symmetry with ordered Mg–Al distribution and two unique alkali sites with different preferences for Na and K. In the crystal structure, the coordination of Na and/or K is trigonal antiprismatic, and these share bases with two octahedral Mg (Na) or Al (K) coordinations. These polyhedra are arranged in columns parallel to [001] and interconnected by SO4 tetrahedral groups. The alkali atoms from a column lie in the same (001) layers as the octahedrally coordinated atoms from the three neighboring rows. On the same level, parallel to (001), there are gaps in the other three neighboring columns forming channels containing Na+ or K+ ions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In July 1991 Sveinn Peter Jakobsson collected a sample with a presumed new mineral later labelled EA (Jakobsson et al. 2008) on the Hekla volcano, Iceland, which erupted earlier that year (January 18–19). He found the same presumed new mineral four years later in three samples collected in the fumaroles on the Eldfell volcano, Iceland, still active after the eruption of 1973. The presumed new mineral was identified by Erik Leonardsen using Powder X-Ray diffraction (PXRD) as a hitherto unknown species. The long-lived fumaroles of Eldfell yielded in 2009 a new sample containing EA among the material collected during a new expedition where one of the authors of this article (TBZ) took part. This sample was investigated by Scanning Electron Microscopy with Energy-Dispersive Spectrometer. The results showed it to contain another presumed new mineral labelled EN by Jakobsson et al. (2008), a description of which is presently in preparation, in a matrix of needles with composition (Na,K)MgAl(SO4)3 and a Na:K ratio of approximately 3:1. PXRD showed EN as the main phase, with EA and a second minor phase. A closer investigation suggested that EA and the new phase must have very similar structures, just with different sizes of the unit cells. We supposed it was due to different amounts of K replacing Na in the structure and decided to synthesize (Na,K)MgAl(SO4)3 compounds with different Na:K proportions to verify this assumption. Subsequently, an article was published describing the new fumarolic mineral, koryakite, discovered at Tolbachick volcano (Siidra et al. 2020) with composition reported as NaKMg2Al2(SO4)6, thus fitting into our research. We here explain its place among the (Na,K)MgAl(SO4)3 compounds.
Experimental
For PXRD measurements, we used a Bruker-AXS Advance 8 diffractometer equipped with a primary Ge111 monochromator giving practically monochromatic radiation (1.54059 Å wavelength) and a LynxEye silicon-strip detector with an active surface covering 3.3°. The technique used for room temperature measurements was the reflection Bragg–Brentano technique with a fixed beam divergence of 0.25°, between 5° and 90° 2θ in steps of 0.02°.
High-temperature (HT) PXRD measurements we performed using a cylindrical MRI TC oven centered close to the focus of the primary beam. We mounted samples in capillaries with a diameter of 0.5 mm. For temperature control, we used a 2404 Eurotherm temperature controller with a NiCr/NiAl thermocouple calibrated with the MgO standard. Measurements were carried out between 5° and 70° 2θ in steps of 0.01°.
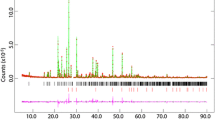
For Rietveld refinements, we used Topas, version 6 (a Bruker AXS product) applying a fundamental parameters approach for the description of profile shapes. For an external calibration of emission lines contribution, we used a CeO2 standard (NIST 674a) with an average crystallite size of 595 nm (Balzar et al. 2004).
Solid-state 23Na and 27Al Magic Angle Spinning (MAS) Nuclear Magnetic Resonance (NMR) spectra were recorded on an Agilent 600 MHz NMR spectrometer (14.1 T) equipped with a 3.2 mm triple resonance MAS NMR probe with 15 kHz spinning speed. The 23Na and 27Al NMR spectra were recorded with a short (≈10°) excitation pulse to ensure uniform excitation and are referenced to 1 M NaCl (δiso(23Na) = 0 ppm) and 1 M AlCl3 (δiso(27Al) = 0 ppm), respectively, and analyzed using ssNake (van Meerten et al. 2019).
The TGA was performed on a Mettler Toledo TGA using Nitrogen as the purge gas. Approximately 50 mg of the samples were placed in an alumina crucible on the sample stage. The samples were ramped to 100 °C and kept there until the temperature of the sample was uniform and stable. Hereafter it was heated at a rate of 10 °C/min to 800 °C.
The samples were obtained by solid-state synthesis through mixing powders of Na2SO4, MgSO4(H2O)6, Al2(SO4)3(H2O)17, and K2SO4 in stoichiometric amounts and heating the mixtures in an oven in covered ceramic crucibles to obtain the six compositions listed in Table 1. The initial treatment was five days at 230 °C. A check of products by PXRD showed those rich in K to be largely amorphous, so the temperature was increased to 300 °C and heating continued for ten days. After that, PXRD showed in all cases similar diagrams with a crystalline phase resembling the pattern of NaMgFe(SO4)3 (Slater and Greaves 1994) just with different sizes of unit cells. Therefore, we concluded at that stage that all were isostructural with the Fe3+ compound, which was supported by satisfactory Rietveld refinements using this structure’s crystallographic data to build a starting model.
Results
We performed TGA and HT PXRD analysis of samples (Figs. S2, S3, Table S2). TGA showed all samples to be stable up to around 650 °C (Fig. S2). Sample EA0 showed the smallest volume thermal expansion but the largest anisotropy (strong expansion along [001] but very small along [100]). The other samples had a relatively small anisotropy of expansion, EA40, and EA60 larger along [001], EA80 and EA100 along [100]. EA20 was later shown to be a mixture of two phases and, therefore, irrelevant for comparison (Fig. S1 and Table S1).
We continued the heat treatment of the same samples for one month further at 300 °C and at 550 °C for an additional 200 h to obtain better crystallinity. We made the final Rietveld analyses on samples after this treatment and the main Rietveld results, we presented in Table 1, and in Fig. 1 one can see the development of unit cell parameters with the content of K. The atomic displacement factors were constrained to have the same value for Na and K sites, Mg and Al sites and for all O sites, to diminish the number of free parameters and their influence on occupancy factors. The details of Rietveld refinements are in the supplementary information (Table S1) as well as the full lists of bond lengths, angles, and coordination distortion parameters calculated according to Balic-Zunic and Vickovic (1996) and Makovicky and Balic-Zunic (1998) (Table S3). Calculations are carried out using the program IVTON (Balic-Zunic and Vickovic, 1996).

Unit cell parameters at various K contents (X) normalized by the unit cell parameters at K content of 0% (X0) (for absolute values see Table S1). Red: \(R\overline{3}c\) phases, black: \(R\overline{3}\) phases. The data for two phases in EA20 (at K content 7 and 34 mol%) are probably biased by diffraction overlap (see text). The data points are connected to guide the eye. 3 × ESD are smaller than the symbols for all data points
All samples contained major phases with crystal structures of the same general type. As an example, the crystal structure of EA60 is shown in Fig. 2. The crystal structures of all synthesized EA phases are built upon the same structural principles as a material group called NASICON (NA Super Ionic CONductor) (Boilot et al. 1988), more specifically they have the same architecture as the pure phosphorus end-member of this group, NaZr2(PO4)3 (Hong 1976). This structure type contains columns of Na coordinations (in our case also K) sandwiched between two Zr (in our case Mg or Al) octahedral coordinations with which they share bases. The columns are connected through tetrahedral groups (SiO4 and PO4 in the original NASICON and SO4 in the EA phases).

Crystal structure of EA60. A Projected on (001). B Projected on (110). Cyan = O atoms, light violet = AI (Na/K), dark violet = AII (K), blue = MI (Mg and its octahedral coordination), red = MII (Al and its octahedral coordination), yellow = S and its tetrahedral coordination. The trace of a unit cell is indicated
Among EA phases, NaMgAl(SO4)3 exhibits \(R\overline{3}c\) symmetry, which implies it only has one type of alkali-atom site and a disordered distribution of Mg and Al among the octahedrally coordinated sites that “sandwich” it. EA phases with K replacing Na partially (more than 35 mol%) or completely have \(R\overline{3}\) symmetry, which implies two symmetrically independent alkali-atom sites (AI at 3a; 0,0,0 and AII at 3b: 1/3,2/3,1/6) and two symmetrically independent octahedral sites (MI and MII both at 6c: 2/3,1/3, z). The refinement of the NaMgAl(SO4)3 was also attempted in the lower symmetry group, but R factors were very similar and, even more importantly, NaMgAl(SO4)3 showed the systematic extinctions consistent with the \(R\overline{3}\)c space group and the atomic coordinates were practically the same, suggesting that all octahedral coordinations are of the same size due to the high disorder of Mg and Al. The alkali sites have a coordination number (CN) of six and form a much-distorted octahedron that can be more accurately described as a trigonal antiprism. It was possible to refine directly the occupancies of AI and AII by Na and K due to a large difference in atomic numbers. The results can be seen in Fig. 3. K exhibits a large preference for AII which is also supported by the significantly higher cation-oxygen bond lengths in AII compared to AI observed for all EA phases (see Table S3). For compositions with K ≥ 50 mol%, this site is completely occupied by K. This is confirmed by solid-state NMR spectroscopy (see below).

Distribution of K and Na between the AI (Na1 and K1) and the AII site (Na2 and K2). Error bars represent 3 × ESD. The data points are connected to guide the eye
The occupancy of Mg and Al at the M sites cannot be directly refined due to their similar atomic numbers but can be inferred from coordination polyhedra volumes. The results can be seen in Fig. 4. The polyhedra volumes suggest a large preference for Mg for the MI site (sandwiching AI) and Al for the MII site (sandwiching AII) in all \(R\overline{3}\) phases. For comparison, the volume of MgO6 octahedron in periclase is 12.499 Å3 and of AlO6 octahedron in corundum has a volume of 9.08 Å3. The conclusions from the polyhedra volumes are further supported by bond valence calculations. Bond valence sums for the M1 site occupied by Mg range between 2.4 and 2.5, whereas for the MII site occupied by Al, it ranges from 3.0 to 3.2.

Coordination polyhedra volumes for the MI and MII sites containing Mg and Al. Error bars represent three times the standard deviation. The data points are connected to guide the eye
In Figs. 3 and 4 the data for the \(R\overline{3}\) phase in EA20 are not included because the diffraction overlaps of the two EA phases present in this sample do not allow the same accuracy of results as in the other samples.
To gain further insight into the local structure around Al and Na, 27Al and 23Na MAS NMR spectra were recorded for all samples. The 23Na MAS NMR spectra (Fig. 5) show a gradual evolution. EA0 (nominally one crystallographic alkali site) is very complex and contains several 23Na NMR resonances, which reflects a high degree of local disorder. This most likely reflects the displacement of some of the Na atoms from the 6b position to the 18e position, which is occupied in NASICON phases with “excess” Na coupled with Si for P substitution. As the 23Na content decreases, the spectra become less complex. For example, the 23Na MAS NMR spectrum of EA80 and EA60 can be simulated convincingly with a single site (δiso(23Na) = − 16.7(8) ppm, CQ = 2.78(5) MHz and ηQ = 0.00(5)) confirming the preference of Na for one of the two crystallographic positions. A small shoulder is observed on the right side, which reflects the presence of a small amount at the second site (AII). The 23Na MAS NMR spectrum of EA40 contains two distinct 23Na resonances with well-defined second-order quadrupolar line shapes implying a high degree of local order and the presence of 23Na on two crystallographic sites in different concentrations. The major site can be simulated with the parameters above, whereas the second site is characterized by (δiso(23Na) = − 28.3(8) ppm, CQ = 2.10(5) MHz, and ηQ = 0.01(5)). The 23Na MAS NMR spectra of EA20 and EA0 are too complex for detailed analysis but clearly contain several more resonances. From the intensity, there is a strong preference for Na to be located on one of these sites, which supports the observations based on PXRD (Fig. 3).

23Na MAS NMR spectra from EA0 to EA80 (Na-containing phases)
The 27Al NMR spectra show a similar trend. EA0 shows a single asymmetric line shape, which is characteristic of a 27Al site in a disordered local environment with a maximum intensity at δ(27Al) = − 27 ppm, whereas the major resonance is at δ(27Al) = − 21 to − 22 ppm for the members of the solid solution series from EA40 to EA100. Thus, for Al, the chemical shift significantly changes from EA0 through EA20 (mixture of phases) to EA40-EA100. This is in accordance with a change from a disordered to a largely ordered distribution in the crystal structure. Again, 27Al NMR confirms the miscibility gap. Moreover, a minor resonance is seen just below δ(27Al) ≈ − 20 ppm. This is either from a small impurity phase (KAl(SO4)2) or from a small amount of Al located on Mg sites (Fig. 6)

27Al MAS NMR spectra for EA0 to EA100
Discussion and conclusions
Slater and Greaves (1993) synthesized NaMgAl(SO4)3 among other sulfates/selenates of the NASICON type where it is described as a rhombohedral phase with unit cell parameters similar to those obtained in this work (see supplementary information), and reported its conductivity. Moreover, syntheses and measurements of the phases where Na is substituted by K and Ag are mentioned and further details were to be published elsewhere. However, we were not able to find such an article so, to the best of our knowledge, this work gives the first account of the relations among (Na,K)MgAl(SO4)3 phases with their full structural characterization.
Our results show that NaMgAl(SO4)3 is a unique phase in the compositional field NaMgAl(SO4)3–KMgAl(SO4)3 whereas a quenchable solid solution is formed from around Na0.65K0.35MgAl(SO4)3 to KMgAl(SO4)3 at 550 °C. NaMgAl(SO4)3 most probably can tolerate a small K for Na replacement up to around 10 mol%, so a compositional gap exists from around 10% to around 35% K in the formula. The results therefore confirm NaMgAl(SO4)3 as a new mineral species observed in Iceland. At the same time, koryakite is shown to have a composition and structure close to a low-K end-member of a solid solution series that extends to KMgAl(SO4)3. The appearance of the two minerals intimately mixed in the same fumarolic sample on Eldfell can be explained by a Na:K proportion of the source falling inside the compositional gap (around 20 mol% K) between NaMgAl(SO4)3 and koryakite.
The reason for a compositional gap between NaMgAl(SO4)3 and the koryakite solid solution series is an interesting question. The gap has been identified both in synthetic and natural samples, so the reason must be in the intrinsic properties of these closely related structures. We propose the following explanation: The \(R\overline{3}\)c structure of NaMgAl(SO4)4 with uniform size of the alkali site can tolerate only very small amounts of a much larger K atoms substituting for Na. On the other side, the mixed Na–K compositions of the koryakite solid solution series require a K amount larger than approximately 40 mol% to stabilize a structure with two alkali coordinations of distinctly different sizes followed by the ordering of Mg and Al among the octahedral sites.
The crystal structures of NaMgAl(SO4)3 and the members of the koryakite solid solution series are built upon the same general principles. The main crystal structure distinction is Mg and Al disordering in NaMgAl(SO4)3, whereas they are highly ordered in the koryakite solid solution series. Likewise, K and Na are highly ordered in the solid solution structures. As regards K coordination, one can observe that there are additional six O atoms closer than 3.5 Å. There is, however, a large bond distance gap, which decreases somewhat with the increasing K content but even in KMgAl(SO4)3 the difference between nearest neighbors and the next neighbors remains too large (K1: 2.63, vs. 3.25; K2: 2.94 vs. 3.21 Å) to consider the next neighbors as an important part of the K coordination. More surprising is the short K–O bond distance in the K1 site, which is only 2.63 Å even in the fully K-occupied site, significantly different from 2.94 Å in K2. This, and the unusually low coordination number for K (6) testifies to a large flexibility of this structure type responsible for an unusually extensive solid solution between Na+ and K+, which are so different in size. As the changes of the unit cell parameters show (Fig. 1) the change in the size is mostly accommodated by increasing the c parameter or, in other words, increasing the extension of the [001] columns with increasing the content of K in the structure.
The structure of koryakite (Siidra et al. 2020) and all the EA structures are related to the crystal structures of millosevichite [Al2(SO4)3; Dahmen and Gruehn 1993] and isostructural mikasaite [Fe2(SO4)3 (Christidis and Rentzeperis 1976)]. The structures in the koryakite solid solution series can be regarded as stuffed derivatives of the millosevichite structure (Siidra et al. 2020). The deficit in the positive charge caused by the substitution of Mg2+ for Al3+ is compensated by the addition of alkali atoms in the voids of tetrahedral – octahedral framework. However, the alkali atoms do not occupy channels [001], as written in the article of Siidra et al. (2020). Such channels do not exist in millosevichite structure (Dahmen and Gruehn 1993; Fig. 2). The alkalis occupy the empty sites between the two Mg or two Al octahedra. The structural projections 3 (b) and (c) in the article by Siidra et al. (2020) give an impression of such channels, but one should consider that those projections represent only relatively thin (001) slices of the structure and not the full structure. The full structural projections of millosevichite, koryakite, NaMgAl(SO4)3 and NaZr2(PO4)3 are compared in Fig. 7. One can see the actual situation in it. The coordinations of alkali cations, and the empty sites that correspond to them in millosevichite, are “plugged” in [001] direction by two octahedral coordinations with which they share basal faces. In Na–K mixed compositions, K shows a preference to occupy the sites between two AlO6 octahedra and Na between two MgO6 octahedra. The channels containing Na+ and K+ do exist but they run parallel to the a-axis which can be seen in Fig. 7. It can also be seen that these channels in \(R\overline{3}\)c structures [NaMgAl(SO4)3 and NaZr2(PO4)3] have a uniform profile, whereas in \(R\overline{3}\) structures (milloshevicite and koryakite) the two profiles are obtained, differently occupied by Na and K in koryakite solid solution. However, the Na+ ions do not move along these channels in NASICON. The distances between Na “neighbor sites” in a channel are too long (over 8 Å) to exert a significant electrostatic influence. The migration of Na+ occurs between the Na1 (0,0,0) and a closer Na2 (x,0,1/4) site, in compositions with surplus Na where there are still enough vacant sites in both places and follows a zig-zag path (Boilot et al. 1988).

Comparison of the structures of Al2(SO4)3, millosevichite (a, c), K0.5Na0.5MgAl(SO4)3, koryakite (b, d), NaMgAl(SO4)3 (e, g) and NaZr2(PO4)3 (f, h) shown in projections along [100] (a, b, e, f) and [001] (c, d, g, h). The coloring is as follows: yellow = sulphate tetrahedra; green = AlO6 octahedra; dark blue = MgO6 octahedra; dark green = Mg/AlO6 mixed octahedra; light blue = phosphate tetrahedra; purple = ZrO6 octahedra; orange = Na atoms; violet = K atoms
Data availability
Any additional data is available upon request.
References
Balić Žunić T, Makovicky E (1996) Determination of the centroid or “the best centre” of a coordination polyhedron. Acta Cryst B52:78–81. https://doi.org/10.1107/S0108768195008251
Balzar D, Audebrand N, Daymond MR, Fitch A, Hewat A, Langford JI, Le Bail A, Louër D, Masson O, McCowan CN, Popa NC, Stephens PW, Toby BH (2004) Size–strain line-broadening analysis of the ceria round-robin sample. J Appl Crystallogr 37(6):911–924. https://doi.org/10.1107/S0021889804022551
Boilot JP, Collin G, Colomban P (1988) Relation structure-fast ion conduction in the NASICON solid solution. J Solid State Chem 73(1):160–171
Balic-Zunic T, Vickovic I (1996) IVTON–a program for the calculation of geometrical aspects of crystal structures and some crystal chemical applications. J Appl Cryst 29(3):305–306. https://doi.org/10.1107/S0021889895015081
Christidis PC, Rentzeperis PJ (1976) The crystal structure of rhombohedral Fe2(SO4)3. Z Für Krist 144:341–352
Dahmen T, Gruehn R (1993) Beiträge zum thermischen Verhalten von Sulfaten: IX.* Einkristallstrukturverfeinerung der Metall (III)-sulfate Cr2(SO4)3 und Al2(SO4)3. Z Für Krist Cryst Mater 204(1):57–65. https://doi.org/10.1524/zkri.1993.204.Part-1.57
Hong HY-P (1976) Crystal structures and crystal chemistry in the system Na1+xZr2SixP3−xO12. Mater Res Bull 11:173–182
Jakobsson SP, Leonardsen ES, Balic-Zunic T, Jónsson SS (2008) Encrustations from three recent volcanic eruptions in Iceland: the 1963–1967 Surtsey, the 1973 Eldfell and the 1991 Hekla eruptions. Fjölrit Náttúrufraedistofnunar 52:65
Makovicky E, Balić-Žunić T (1998) New measure of distortion for coordination polyhedra. Acta Cryst B54:766–773. https://doi.org/10.1107/S0108768198003905
Siidra OI, Nazarchuk EV, Zaitsev AN, Vlasenko NS (2020) Koryakite, NaKMg2Al2(SO4)6, a new NASICON-related anhydrous sulfate mineral from Tolbachik volcano, Kamchatka, Russia. Min Mag 84:283–287. https://doi.org/10.1180/mgm.2019.69
Slater PR, Greaves C (1993) Synthesis and conductivities of sulfate/selenite phases related to Nasicon: NaxM’(II)xM’’(III)2–x(SO4)3-y(SeO4)y. J Solid State Chem 107:12–18. https://doi.org/10.1006/jssc.1993.1317
Slater PR, Greaves C (1994) Powder neutron diffraction study of the nasicon-related phases NaxMIIxMIII2–x(SO4)3–y(SeO4)y: MII = Mg, MIII = Fe, In. J Mater Chem 4:1469–1473. https://doi.org/10.1039/JM9940401463
van Meerten SGJ, Franssen WMJ, Kentgens APM (2019) sNake: a cross-platform open-source NMR data processing and fitting application. J Magn Reason 301:56–66. https://doi.org/10.1016/j.jmr.2019.02.006
Acknowledgements
UGN acknowledges funding from the Danish Council for Independent Research Science and Universe (Grant DFF-7014-00198). We acknowledge and thank Professor Woźniak and the Crystallochemistry Laboratory at the University of Warsaw for the possibility of using their thermogravimetric analysis (TGA) equipment for measurements. We acknowledge the open-access funding provided by the Royal Danish Library. The authors thank the three anonymous referees for their comments, which improved the text.
Funding
Open access funding provided by Royal Library, Copenhagen University Library.
Author information
Authors and Affiliations
Contributions
P.G.J. carried out the bulk of Rietveld refinement and data analysis, analysis of HT-PXRD and prepared the final manuscript and most of the figures. T.B.Z. prepared the initial draft, conducted PXRD-measurements, prepared samples, carried out Rietveld refinement, general supervision, prepared figure 2, and PXRD-figures in the supplementary information. U.G.N. carried out NMR measurement and analysis and prepared figures 5 and 6. P.M.K. prepared samples, carried out HT-PXRD measurements and TGA. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jensen, P.G., Balic-Zunic, T., Nielsen, U.G. et al. The solid solution in the system NaMgAl(SO4)3–KMgAl(SO4)3. Phys Chem Minerals 50, 35 (2023). https://doi.org/10.1007/s00269-023-01259-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00269-023-01259-0