
Abstract
Effects of Fe ↔ Al substitution on triclinic SFCA-I-type compounds with general formula A40O56 (A: Ca, Al, Fe3+, Fe2+) have been studied using single-crystal X-ray diffraction. Crystals of sufficient quality and size were synthesized in the temperature range between 1200 and 1300 °C. Six samples with Al/FeTot ratios of 0.127, 0.173, 0.216, 0.310, 0.349 and 0.459 have been structurally characterized. SFCA-I can be described with a modular approach involving the stacking sequence < PSS > of “P” and “S” modules that can be imagined as being cut from the well-known pyroxene (P) and spinel (S) structure types. Furthermore, SFCA-I is related to the sapphirine supergroup of minerals. Within the present solid-solution series, the contents in calcium show only minor variations (≈ 6.7 a.p.f.u.). The twenty crystallographically independent tetrahedrally (T) and octahedrally (M) coordinated cation sites exhibit considerable differences concerning the Al uptake. Indeed, Al is preferentially incorporated into the tetrahedra belonging to the single-chains located in the pyroxene modules. Ferrous iron, on the other hand, is restricted to one of the T-positions within the spinel blocks. Most structural aspects from unit-cell parameters and cell volumes to site occupancies, tetrahedral chain kinking as well as polyhedral distortions are defined by linear or nearly linear trends when plotted against the Al/FeTot ratio. Analysis of the < T–O > and < M–O > distances showed a complex interplay between the different coordination polyhedra resulting in a contrasting behavior of these values with positive or negative change rates as a function of composition. Evaluation of the average chemical strain tensor derived from the sets of lattice parameters for the two samples of the abovementioned series showing the highest and lowest Al/FeTot ratios indicated, that the major contraction with increasing Al content is perpendicular to the pyroxene- and spinel modules. Furthermore, the pyroxene module seems to be more affected when compared with the spinel block. There is evidence that the SFCA-I-type solid-solution series is limited on both the Al- and Fe-rich sides. The present investigation provides—for the first time—a detailed crystallographic analysis on the impact of chemical variations on a compound that is of relevance to the field of applied mineralogy related to the technologically important process of iron-ore sintering.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Steel—an alloy of iron and carbon—is a strategic product for the global economy. Its unique properties in combination with low costs make steel an ideal material for a plethora of industrial and engineering applications (Holmes and Lu, 2015). Data available from the Organization for Economic Co-operation and Development suggest that the global steel-producing capacity in 2019 was about 2362.5 million metric tons (OECD 2019).
At the heart of the steel-making process is the blast furnace, where iron ore is reduced to metallic iron. Currently, the group of iron formation-hosted ore deposits including the famous banded iron formations (BIFs) account for the majority of world production (Clout and Manuel, 2015). After a first mechanical processing of the ores, two particle fractions can be distinguished: lump (31.5 to 6.3 mm) and fine (< 6.3 mm) products. Because of blockage of the gas permeability of the furnace, however, the fine fraction of the iron ores cannot directly be used as a feedstock (Kalenga and Garbers-Craig 2010). The fines first need to undergo an agglomeration process that is frequently accomplished via sintering. In the European Union alone, about 130 million tons of iron ores have recently been sintered per annum (Fernandez-Gonzalez et al. 2017), making this material one of the most produced inorganic commodities.
During sintering, the loose fine particles are bonded together at temperatures between 1250 and 1350 °C into larger centimeter-sized particles by partial melting of the solids, which is followed by re-crystallization. The resulting porous aggregates consist of ore particles (hematite and magnetite, constituting 40 to 70% vol.) cemented by a matrix of Ca-rich ferrites (20 to 50% vol.), up to 10% vol. quenched melt (glass) and about 10% vol. calcium silicates such as larnite (β-Ca2SiO4) (Patrick and Lovel 2001). Minor phases may include quartz, sulphides, pyroxenes, serpentine and lime, but the total of these rarely accounts for more than a few percent of the sinter volume. The calcium ferrites usually contain some silica and alumina and are thus referred to as Silico Ferrites of Calcium and Aluminum, abbreviated to SFCA (Hamilton et al. 1989; Mumme et al. 1998). Actually, these compounds form the main bonding phase in the sinter (Scarlett et al. 2004).
To date, two representatives have been reported from industrial sinters in larger quantities: SFCA and SFCA-I. According to Scarlett et al. (2004), SFCA has a more prismatic habitus and can be described as columnar or lath-shaped. SFCA-I, on the other hand, should display a more platy morphology and in cross section appears needle-like or acicular. Apart from apparent morphological differences, both compounds are definitely distinct crystallographic phases and powder X-ray diffraction (PXRD) can be used to tell them apart. Their complex crystal structures exhibit low triclinic symmetry, comparatively large unit-cell volumes, and there is a clear structural relationship between them. Indeed, SFCA and SFCA-I belong to a so-called polysomatic series with general composition A14+6nO20+8n (A: Ca, Al, Mg, Si, Fe3+, Fe2+) where they represent the members with n = 0 and 1, respectively (see Kahlenberg et al. 2019 and references cited therein). The crystal structures of the series allow for a couple of cation substitution mechanisms, which can be summarized according to the following exchange reaction: 2(Fe3+,Al3+) = (Ca2+,Fe2+) + Si4+ (Patrick and Lovel 2001; Patrick and Pownceby 2002). Mg2+ can also be incorporated by replacing Fe2+ and/or Ca2+ (Sugiyama et al. 2005; Zöll et al. 2017).
SFCA (A14O20) found in plant sinters shows a highly flexible chemistry, typically containing about 60–76% wt. Fe2O3, 13–16% wt. CaO, 3–10% wt. SiO2, 4–10% wt. Al2O3 and 0.7–1.5% wt. MgO (Nicol et al. 2018). In the past, many studies have been performed on its compositional range, stability and structural characteristics (Hancart et al. 1967; Inoue et al. 1982; Dawson et al. 1985; Hsieh and Whiteman 1989; Patrick and Lovel 2001; Patrick and Pownceby 2002; Kim et al. 2002; Sugiyama et al. 2005; Lisles et al. 2016).
By contrast, for SFCA-I (A20O28 or A40O56) no comparably rigorous studies on the solid-solution limits, thermal stability and cation substitutions have been conducted. According to Mumme et al. (1998), SFCA-I in sinters contains about 84% wt. Fe2O3, 13% wt. CaO, 1% wt. SiO2 and 2% wt. Al2O3. However, Si-free representatives have also been synthesized under laboratory conditions (Mumme et al. 1998; Arakcheeva et al. 1991; Zöll et al. 2017). There are indications that the SFCA-I solid-solution series (a) is more limited in comparison with SFCA but (b) can probably accommodate more Fe2+ (Mumme et al. 1998; Webster et al. 2013).
The aim of the present paper was to investigate the cation distributions in crystalline SFCA-I-type compounds using single-crystal structure analysis. For this study, we focused on solid-solutions within the simplified chemical system CaO–Fe2O3(+ FeO)–Al2O3. More detailed knowledge about the cation substitution patterns of Al3+, Fe3+ and Fe2+ among the large number of tetrahedral and octahedral sites and their influence on the structural features is definitely of great interest for a better understanding of the SFCA-I-type mixed crystals. As has been demonstrated recently for SFCA, crystallographic information about site preferences of the involved cation species has been of fundamental importance for the successful thermodynamic modeling of this solid-solution series (Murao et al. 2018, 2020).
Experimental details
Starting materials for the synthesis experiments were Fe2O3 (99.9%, Alfa Aesar), γ-Al2O3 (99.997%, Alfa Aesar), and CaCO3 (99.995% Merck). Phase purity of the educts was checked by PXRD before they were dried at 300 °C for 24 h. After storing in an evacuated desiccator stoichiometric amounts of the reagents for a total of 2 g were weighed on an analytical balance. Seven different mixtures were prepared for a hypothetical SFCA-I solid-solution series with nominal composition Ca8AlxFe32-xO56 and x = 0, 0.8, 2.0, 4.0, 5.0, 6.0 and 8.0. For easy reference, we will denote these samples SFCA-I-00 to SFCA-I-06. Homogenization for 45 min was performed in a planetary mill operated at 600 rpm under ethanol. The resulting slurry was dried for 12 h at 50 °C to remove the alcohol completely and manually re-homogenized for 15 min in an agate mortar. Subsequently, 0.5 g of the powder was pressed into pellets with a diameter of about 12 mm and a thickness of about 2 mm. In the next step, two pellets of each mixture were transferred into an open Pt-crucible. Sintering was performed in air using a resistance heated chamber furnace. Samples were fired from 20 °C (with a ramp of 120 °C/h) to the maximum temperature of 1300 °C followed by cooling with a rate of 5 ℃/h to 1200 ℃ where the crucible was immediately quenched in air. Weight losses were determined from weight differences before and after heating.
Powder diffraction patterns have been collected with a Stoe-MP diffractometer system (STOE & Cie GmbH, Darmstadt, Germany) configured in Bragg–Brentano geometry using a Mythen 1 K detector (Dectris Ltd., Baden-Daettwil, Switzerland). The device is equipped with a primary-beam Ge(111) monochromator yielding Kα1 radiation only. The diffraction studies were performed with a cobalt tube operated at 40 kV and 30 mA. Data were acquired at ambient temperature over a 2θ range between 5° and 120° using a 2θ step size of 0.015°. The total measurement time per scan was about 48 h. Qualitative phase analysis was performed with the 2018 release of the PDF-4+ Powder Diffraction File database of the International Centre for Diffraction Data (Gates-Rector and Blanton 2019).
Chemical composition of the compounds obtained from the sinter experiments was determined using electron microprobe analysis (EMPA) on a JEOL JXA SUPERPROBE 8100 in wavelength-dispersive mode. For sample preparation, fragments of the pellets were embedded in epoxy resin and polished with diamond paste (10, 5 and 1 μm grain size). After sputter coating with carbon, measurements were performed with an acceleration voltage of 15 kV, a beam current of 10 nA and counting times of 20 s (on peaks) and 10 s (for background on each side of the peaks). Depending on the size, at least five data points per crystallite were analyzed. Diopside (Ca-Kα), corundum (Al-Kα) and magnetite (Fe-Kα) were used as standard reference materials. Furthermore, a so-called PRZ matrix correction was applied. Finally, the amount of Fe2+ was calculated from the crystal-chemical formula obtained by the microprobe measurements (normalized to the expected number of oxygen atoms) and charge balance considerations (Droop 1987).
From each synthesis run between 5 and 8 crystals with diameters up to 200 μm have been retrieved. The single crystals were mounted on the top of glass fibers with nail polish and screened on a Rigaku Oxford Diffraction Gemini R Ultra diffractometer equipped with a four-circle kappa-goniometer and a Ruby CCD detector. Measurements were conducted with Mo-Kα radiation at ambient conditions. Pre-experiments aiming at the determination of unit-cell parameters indicated the presence of a so-called α-CFF phase with composition Ca3.43Fe14.39O25 (for SFCA-I-00) as well as SFCA-I-type compounds (in all other cases). For each starting composition, the crystal showing the best diffraction quality was selected for full structure analysis. Details of the data collections covering at least a half-sphere of reciprocal space are summarized in Table 1. The CrysAlisPRO software package (Rigaku 2015) was employed to process the data. After indexing, the diffraction patterns were integrated. Intensities were corrected for Lorentz-polarization and absorption effects. For the SFCA-I-type samples, the structure model in space group P \(\bar{1}\) published by Mumme et al. (1998) was used as a starting point for the full-matrix least-squares refinements. These calculations were performed with the program SHELXL (Sheldrick 2008) within the WinGX environment (Farrugia 1999). X-ray scattering factors including anomalous dispersion terms were taken from the International Tables for Crystallography, Vol. C (Wilson 1995). The refinements based on fractional atomic coordinates, anisotropic displacement parameters and Ca/Fe or Fe/Al populations on the relevant sites converged to residual values for R1 between 3.09 and 4.37%. The largest shifts/esd in the last cycles of each refinement were < 0.001. As will be outlined further below, the final assignment of different chemical species (Fe3+, Fe2+, Al, Ca) to each of the 20 cation positions was performed using the program OccQP (Wright et al. 2000). Coordinates, site occupancies and equivalent isotropic displacement parameters of the atoms as well as selected inter‐atomic distances are summarized in supplementary Tables S1 and S2. Anisotropic displacement parameters are listed in Table S3. Figures showing structural features were prepared with the program VESTA 3 (Momma and Izumi 2011). For the calculation of the difference mean-square displacement parameters, the program XtalDraw (Downs and Hall-Wallace 2003) was employed. Crystallographic Information Files (CIF) for all six compounds have been uploaded as supplementary information.
Results
Synthesis and electron microprobe analysis
For all pellets, a significant reduction in volume was noticed after the heat treatment. On the surface, as well as in the interior of the sintered compacts, blackish crystals with maximum diameters of about 250 μm and a lath-shaped to platy habitus were observed. Notably, a distinct decrease in size with increasing Al-concentration was evident. The partially inter-grown crystals were embedded in a polycrystalline matrix of gray color from which they could be separated mechanically. Subsequent EMP analysis in combination with PXRD proved the multiphase character of most samples. The Al-free specimen (SFCA-I-00) consisted of a phase adopting the so-called α-CFF structure type (Karpinskii and Arakcheeva 1987) as the main constituent. For all other compositions, the platy crystals corresponded to SFCA-I-type compounds with varying Al/Fetot ratios. As expected, the analyzed Al-concentrations of the SFCA-I crystals increased with the alumina content of the starting mixtures. Notably, the amounts of calcium and divalent iron remained virtually constant. Table 2 summarizes the average compositions of the spot analyses for the different phases that could be identified.
Crystal structure
The refinement calculations confirmed an isotypic relationship between the members of our series and Mumme’s phase. To group the octahedral (M) and tetrahedral (T) sites, we decided to use a slightly different nomenclature and numbering for the cations as the one suggested in the previous investigation.
The crystal structure of SFCA-I contains an alternating sequence of two different types of fundamental layers (I and II) oriented parallel to (2 0\(\bar{1}\)). Within layer-type I, bands of edge-sharing [MO6]-octahedra (M: Ca, Fe3+, Al) running parallel to [010] can be identified (see Fig. 1a). The bands are not planar but exhibit a distinct curvature, i.e. the rims are thicker than the central parts (see Fig. 2a). It is noteworthy, that not all of the potentially available octahedral sites inside the bands are occupied resulting in the formation of ordered vacancies. Actually, central cavities surrounded by the octahedra around M11, M8, M5, M10, M9 and M6 as well as lateral gaps between M1 and M3 can be distinguished. Layer-type II, on the other hand, can be built from [MO6]-octahedra as well as [TO4]-tetrahedra (T: Al, Fe3+, Fe2+) (see Fig. 1b). The [MO6]-groups around M7 and M12 each share common vertices with two neighboring tetrahedra (T1/T3 and T2/T5, respectively) forming [MT2O12]-clusters or “winged-octahedra”. Linkage between adjacent strips of these moieties is provided by tetrahedral single chains built from the polyhedra about T4, T6, T7 and T8. T1 and T2 are directly connected to these chains by sharing common corners. In the classification of Liebau (1985), the whole assemblage could also be described as an open-branched vierer single chain. Concerning the connectedness of the six [TO4]-groups, which is defined as the number of other tetrahedra with which they are connected by sharing common oxygen atoms (Liebau 1985), two primary (Q1: T1, T2), two secondary (Q2: T4, T6) and two tertiary (Q3: T7, T8) tetrahedra can be distinguished inside the chains. The remaining two tetrahedra around T3 and T5 are not linked to the chains. They reside above and below the central octahedral vacancies of layer-type I (see Fig. 2b). The tetrahedra about T1 and T2 in turn “cover” the lateral gaps of the bands of layer-type I from both sides. Notably, the base triangles of the “up” and “down” pointing pairs of tetrahedra (T3 and T5 as well as T1 and T2) are rotated by about 60° relative to each other, i.e. they are in an almost staggered conformation (see Fig. 2b). One basal plane of each of the four tetrahedra forming the backbone of the abovementioned vierer chain connects adjacent bands from layer-type I by sharing common corners with the three octahedra around M1 and M3.

Principal layers observed in SFCA-I. a Band-like units of octahedra (layer-type I) and b so-called winged-octahedra as well as vierer single chains of tetrahedra (layer-type II). Layers are shown in projections perpendicular to the sheets. Dark-gray octahedra represent pure [CaO6] units. The Ca-dominated polyhedron around M2 is presented in medium gray. The octahedra about the remaining M-sites are given in light-gray. Blue and red tetrahedra indicate the presence of Fe3+ and Fe2+, respectively. The O–O–O angles ϕ1–ϕ4 can be used to characterize the degree of stretching of the chains (see text). (Color online)

The whole crystal structure of SFCA-I in projections a parallel and b perpendicular to the principal layers. The sub-division into spinel (S) and pyroxene (P) modules is indicated. A < PSS > stacking sequence is obvious. (Color online)
Alternatively, SFCA-I can be described using a modular approach involving stacking of sequences of “P” and “S” modules that can be imagined as being cut from the well-known pyroxene (P) and spinel (S) structure types. Indeed, these modules are more or less perpendicular to the sheets of type I and II (see Fig. 2a). This modular approach has been already outlined in detail using concepts of OD-theory in several previous publications for the different members of the SFCA family (Merlino & Zvyagin, 1998; Zvyagin & Merlino 2003; Kahlenberg et al. 2019). As can be seen from Fig. 2a, the whole crystal structure of SFCA-I can be understood using the stacking sequence < PSS > .
Unit-cell parameters
The substitution of a smaller cation like Al for the larger Fe decreases, as expected, the size of the unit-cell volume. Indeed, the volume reduction between SFCA-I-01 and SFCA-I-06 corresponds to – 20.3 Å3 or − 1.82%. As can be seen from Fig. 3, the volume data do not exhibit a significant deviation from a linear behavior when plotted against the Al/Fetot ratio. The volume changes are also reflected in a decrease of the lattice parameters a, b and c (see Fig. 4), which are reduced by about − 0.52, − 0.51 and − 0.82%, respectively. This indicates that the c direction is more affected by the aluminum incorporation when compared with a and b, showing almost identical changes. For the angles α, β and γ, two different trends have been observed. While α and β open with increasing Al content, the angle γ narrows at a roughly doubled rate. The evolution of the unit-cell volume and the individual lattice parameters as a function of the Al/Fetot ratio X can be described by the following linear expressions:
R corresponds to the correlation coefficient. A more detailed investigation of the compositionally induced chemical strains will be presented in the discussion section.

Evolution of the unit-cell volumes V of the six members of the SFCA-I solid-solution series with the total Al/FeTot ratios

Evolution of the six lattice parameters a, b, c, α, β and γ of the SFCA-I solid-solution series as a function of the total Al/FeTot ratios
Cation distributions
For the assignment of the cation species (Ca, Al, Fe3+, Fe2+) to the different M- and T-positions, the following procedure has been applied: The total scattering power for each site was obtained by allowing for potential Fe3+ ↔ Al or Fe3+ ↔ Ca substitutions under the assumption of the absence of vacancies. A detailed investigation of all site populations, including an analysis of ferric and ferrous iron contents was performed a posteriori with the program OccQP (Wright et al. 2000). This software uses the full information from chemical analysis (as determined from EMP), refined total scattering powers for the positions (as obtained from single-crystal structure analysis), bond valences and bond distances to find an optimized distribution of the cations. The mathematical algorithm is based on a least-squares method and a weighted linear system. The strength of this approach is the minimization of the residual between formal and apparent valences for an acceptable crystal-chemically solution concerning the site assignments (Wright et al. 2000). However, the program does not provide any standard uncertainties for the optimized site populations.
In the present case, we used the default weights of unity for site scattering, bond-valence, and bond-length terms. Because EMPA results indicated only minor compositional variations from point to point in the SFCA-I-type crystals, for the chemistry term an infinite weight was selected to assure that the resulting bulk chemistry matches the one obtained by the independently performed chemical analysis. Furthermore, Ca-avoidance on the tetrahedral sites has been used as an additional constraint. For the six samples, the resulting cation distributions obtained by OccQP for the individual M- and T-sites are listed in supplementary Table S1. The most important points can be summarized as follows.
Calcium cations can be found on all twelve octahedrally coordinated M-sites. Yet, their concentrations differ considerably. The positions M1 and M3 located at the edges of the octahedral bands are exclusively occupied with calcium. The M2-site in the central part of the band is strongly enriched in calcium (about 70%) but contains considerable amounts of Fe3+ and Al as well. Within the solid-solution series, the ratios between these three cation species sharing M2 are almost constant. This observation holds also for the M6-site exhibiting Ca-concentrations of about 22%. The remaining eight M-positions (M4, M5, M7–M12) located in the spinel-type modules show significantly lower percentages of calcium (2.7–8.5%). Furthermore, the variation in the calcium levels among individual sites is not pronounced. However, a clear trend concerning the Fe3+/Al ratios can be observed (see Fig. 5). With increasing alumina content in the bulk SFCA-I composition, the Al contents of these sites increase by a factor of 2.2–2.7.

Variation of the Al-concentrations within the M-sites of the spinel modules with the Al/FeTot ratio in the solid-solution series
For the tetrahedral positions, the following correlations were observed. The T-sites belonging to the central part of the vierer single chains (T4, T6, T7 and T8) located in the < P > modules are the primary sink for aluminum (see supplementary Table S1). Within the solid-solution series, these polyhedra show the highest increase rates in Al content. For the T8 site, for example, the Al-occupancy increases from 28% (for SFCA-I-01) to 91% (for SFCA-I-06). From SFCA-I-01 to SFCA-I-06, the tetrahedral centers T1 and T2 representing the branched part of the chains (located in the < S > modules) also incorporate an increasing amount of aluminum, but to a much lesser extent. The corresponding Al contents for the two “insular” T-positions vary between 0.09 and 0.14 (for T5) as well as 0.23 and 0.29 (for T3). The latter site, however, deserves special mention. According to the OccQP analysis divalent iron is solely restricted to this position. Notably, the Fe3+/Fe2+ ratio for this position is pretty constant: 0.11(2). Further proof for this assignment comes from bond-distance considerations which will be presented in the next section. A graphical representation of the evolution of the Al-concentrations for all tetrahedra can be found in Fig. 6.

Variation of the Al-concentrations within the eight T-sites as a function of the Al/FeTot ratio in the solid-solution series
Bond distances and angles
To an upper limit of 3.2 Å, the two pure calcium sites M1 and M3 are coordinated by 6 + 1 oxygen anions. In both cases, the six closest ligands define a strongly distorted octahedron with average bond distances of about 2.39 Å. The gap between the distances to these six next neighbors and the respective additional oxygens (O20, O22) is very pronounced, having a value of about 0.6 Å. Even though these two sites are not directly involved in the Fe ↔ Al substitution, two opposing trends can be recognized among their M–O bond lengths. With increasing overall Al-concentration of the mixed crystals the < M–O > values for the bonds defining the “inner” octahedrally coordination environments increase by 1.36% (for M1) and 1.43% (for M3), respectively, while the distance to the seventh ligand decreases from SFCA-I-01 to SFCA-I-06 by 0.18 Å or 5.92% (for M1) and 0.24 Å or 7.63% (for M3). Consequently, for the Fe-rich members, the abovementioned gap is much more obvious. The Ca-dominated M2 position located in the central regions of the octahedral bands exhibits only a minor shortening of the average M–O distances by 0.38% with increasing Al content.
Most of the remaining M-sites show slightly decreasing < M–O > values with increasing Al/Fetot ratios, directly reflecting the substitution of the smaller Al- by the larger Fe3+-cations (see Fig. 7). The corresponding value for M5, however, is almost constant. In accordance with the only minor reduction of the < M–O > values for the M-sites of layer-type I, the volume of the octahedral vacancy in this sheet decreases negligibly from 14.18 Å3 (for SFCA-I-01) to 13.96 Å3 (for SFCA-I-06).

Variation of the < M–O > distances within the M-sites of the spinel modules with the Al/FeTot ratio in the solid-solution series
For each of the two “insular” tetrahedra (T3 and T5) located above and below this vacancy one short and three long T–O distances can be distinguished. This pattern in the distributions of the individual bond lengths can be rationalized by considering the number of cations that are bonded to the corresponding O atoms. Indeed, the oxygen atoms involved in the short distances (O23, O15) are connected to only two additional M-sites, while each of the remaining six oxygens is linked to three other M-positions. For the six tetrahedra of the open-branched vierer single chains, the distances to the non-bridging or terminal oxygens are always significantly longer than the distances to the bridging oxygen ligands. The average T–O values of the latter group are lower than the values for < T5–O > and dramatically lower than those for < T3–O > . This observation compares well with the analysis of the site occupancies where the total amount of Fe2+ has been allocated to the T3-site.
Concerning the evolution of the < T–O > values as a function of the total Al-concentration in the solid-solution series, two opposing trends have been found. While the values for the tetrahedra around T3 and T5 increase with increasing total Al/FeTot ratios, the reverse relationship has been observed for the tetrahedra of the open-branched vierer chains (see Fig. 8). Moreover, for the central four tetrahedra of the chains (T4, T6, T7, T8)—which show the largest variation in individual Al-concentrations—the rate of decrease is by a factor of four larger than for the branched parts (T1, T2).

Variation of the < T–O > distances as a function of the Al/FeTot ratio in the solid-solution series
Another notable change in the solid-solution series is the linear variation of the four angles between the bridging oxygen atoms of the central backbone of the tetrahedral chains (see Fig. 9) which belong to the pyroxene modules (ϕ1: O9-O20-O22, ϕ2: O20-O22-O21, ϕ3: O22-O21-O9 and ϕ4: O21-O9-O20; see Fig. 1b). The increase of these angles by 6.0°, 6.8°, 6.9° and 5.9°, respectively, indicates that the chains are becoming more extended with increasing Al/FeTot ratio, or in other words, less kinked. As the angles are less than 180°, the chains could be classified as “O-rotated” (Papike et al. 1973). Following the results presented by Thompson & Downs (2001) in a systematic study on structural variations in model pyroxenes as a function of tetrahedral rotation, the < P > modules in SFCA-I can be considered distorted cubic closest packed (ccp), and because \(\partial\)(< O–O–O >)/\(\partial\)(Al/FeTot) > 0, they become less ccp with increasing Al content.

Variation of the four kinking angles (ϕ1, ϕ2, ϕ3 and ϕ4) of the tetrahedral chains in the pyroxene modules with the Al/FeTot ratio in the solid-solution series. For more details, see text and Fig. 1b
Difference thermal displacement parameters
The anisotropic displacement parameters of the cations and anions are generally well behaved. We refrain from providing a detailed discussion of the influence of the total Al/FeTot ratio on these parameters but will rather focus on one specific aspect. As mentioned above, site occupancies studied using the OccQp software as well as inspection of the bond distances indicated the presence of ferrous iron on the T3-site. Further evidence for this assumption comes from an analysis of the differences between the anisotropic mean-square displacement parameters of the T and O atoms along the corresponding T–O bonds. According to Kunz and Armbruster (1990) these < ∆U > values can provide insight into static disorder when averaged over a complete tetrahedron. For the SFCA-I-03 data set, for example, the value for the tetrahedron around T3 (< ∆U > = 0.022(6) Å2) is 3.1 to 7.3 times larger than the corresponding values for the tetrahedra belonging to the branched tetrahedral chains and still 2.4 times larger than the one for the second “insular” tetrahedron (T5)—all of which have been assumed to contain ferric iron only. Consequently, the significantly increased < ∆U > value observed for T3 would reflect the disorder effects when a comparatively small ion (such as Al3+, r[4] = 0.39 Å) shares the same position with a much larger cation (such as Fe2+, r[4] = 0.63 Å) (Shannon 1976).
Discussion
In principle, our studies confirm the model for SFCA-I that was published by Mumme et al. (1998) for a synthetic Si-free sample with a comparable chemical composition (Ca6.36Al2.68Fe3+29.32Fe2+1.64O56). Yet, for several positions, we have obtained strikingly different results concerning the partitioning of the cations. For example, Mumme et al. described M2 to be fully occupied with calcium. This assignment, however, results in a strong overbonding with a bond valence sum as high as 2.6 v.u. Second, based on bond length considerations the content in ferrous iron was exclusively attributed to the octahedral site M6. Our own refinements corroborate that the average bond distances and polyhedral volumes of M6 are slightly bigger when compared to the other M-sites within layer-type I. However, Mumme’s assumption leaves us with a problem that there is no explanation for the dramatically increased size of the tetrahedron about T3—a structural feature that occurred in the former refinement as well, where this site was labelled Fe14 and supposed to contain Fe3+ (84%) and Al (16%). As outlined above, we believe that there are strong arguments for the incorporation of Fe2+ on this position. Our changes concerning the principal structural model of SFCA-I are minor modifications. Nevertheless, we consider them a valuable improvement and a better description of the crystallochemical features of the SFCA-I series.
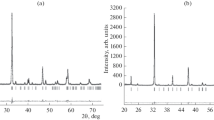
Powder X-ray diffraction is the method of choice for the characterization of polycrystalline materials. To study the influence of the cation substitutions, the diffraction patterns of all six members of the SFCA-I series have been calculated (see Fig. 10a–f). They can act as library-of-reference diffractograms for predicting how the Fe ↔ Al substitutions give rise to different signatures in the patterns. In general, the simulated powder diffraction data of the solid-solution series look quite similar. However, there are some features that are especially useful to tell the different members apart. For example, the first observable peak corresponding to the 001-reflection shows the most pronounced dependency of its intensity on the Al content, which increases significantly with increasing Al/FeTot ratio.

Simulated powder diffraction patterns for the different members of the SFCA-I solid-solution series based on the crystal structure analyses of this publication. For the calculations, the program FullProf.2 k (Rodriguez-Carjaval, 2011) has been employed (Co-Kα1 radiation, Bragg–Brentano geometry, TCH-pseudo Voigt functions) using typical peak widths and shapes for a Stoe-MP laboratory diffractometer. The 001-reflection is marked with a star (see text)
Figure 11 summarizes the evolution of the < T–O > values of the tetrahedra obtained for the SFCA-I series as a function of their Al-concentrations, xAl. A similar analysis has been recently performed in the course of a re-investigation of a SFCA-II sample (Kahlenberg et al. 2021) and the corresponding data have been included in the present study. The combined data sets now cover an almost complete range for xAl ranging from 0 to 1. Most of the values shown follow a trend that can be adequately described by the following linear relationship:

Evolution of the < T–O > values of the tetrahedra obtained for the present SFCA-I series and a recent analysis on SFCA-II (Kahlenberg et al. 2021) as a function of their Al-concentrations, xAl
Extrapolation of the linear trend to xAl = 0 and xAl = 1 gives values of 1.886 and 1.735 Å, respectively. Both numbers compare well with the mean bond lengths of 1.875 ± 0.022 (for < Fe3+-O >) and 1.746 ± 0.022 Å (for < Al–O >) obtained by Gagné and Hawthorne (2018, 2020) from a comprehensive statistical analysis of individual [Fe3+O4]- and [AlO4]-tetrahedra in inorganic compounds.
However, Fig. 11 also shows that there are some outliers. The average bond distances for T3 and two of the tetrahedra in SFCA-II, for example, deviate significantly from the trend line which predicts much lower values when compared with the actual results obtained from crystal structure analyses. It is exactly these polyhedra that have been demonstrated to contain large amounts of Fe2+.
The aforementioned equation could be useful in two ways. First, it may provide an estimation of the Al content of individual tetrahedra in other members of the SFCA family from purely geometrical parameters. Second, it could be employed for the identification of tetrahedra containing appreciable quantities of ferrous iron. With this respect, the data points for T5 are of interest. Actually, they are located slightly above the trend line. This finding could be taken as an indication for the incorporation of minor amounts of Fe2+ on this site. Yet, the OccQp analysis did not support this interpretation.
The data given in the results section indicate a complex interplay between chemical composition on the one and structural changes related to individual deformations of the coordination polyhedra on the other hand. To obtain a more holistic picture of the distortion patterns, the evaluation of the so-called compositional strain tensor can be a helpful tool. The derivation of the components of this second-rank tensor from two sets of lattice parameters corresponding to two different compositions has been described by Ohashi and Burnham (1973), for example. The necessary calculations based on the two limiting compositions of the present series (SFCA-I-01 and SFCA-I-06, respectively) have been performed with the program Win_Strain 4.11 (Angel 2011). Using a finite Eulerian strain formalism referred to an orthonormal coordinate system {x, y and z} with z // c, x // a* and y = z × x, the following 3 × 3 matrix for an average strain tensor εij was derived:
With respect to the Cartesian coordinate system of the principal axes {e1, e2 and e3}, the following three principal strains are obtained: \(\varepsilon\)1 = 0.0018(2), \(\varepsilon\)2 = 0.0067(1) and \(\varepsilon\)3 = 0.0098(2) with ε1:ε2:ε3 = 1:3.72:5.44, indicating a large anisotropy of the strain induced by the Fe ↔ Al substitution. As expected for the triclinic case, the principal axes are not related to the directions of the crystallographic coordinate system along a, b and c. Using Win_Strain, the following angles between the principal and the crystallographic axes have been derived. The values given in parentheses refer to the corresponding angles with a, b and c, respectively: e1: (124.3°; 125.2°; 64.9°); e2: (34.3°; 144.5°; 97.7°); e3: (90.9°; 85.9°; 26.4°).
In the next step, the components of the principal axes in the crystallographic coordinate system have been calculated and—after appropriate scaling for better visualization—the corresponding vectors have been plotted together with specific elements of the SFCA-I structure using the program VESTA 3 (see Fig. 12a–d). This approach facilitates a direct comparison of the macroscopic compositional strain and certain structural features of the SFCA-I-type compounds. As can be seen from Fig. 12a showing a single < PSS > unit, the direction ε3 of major expansion with increasing Fe-concentration (major compression with increasing Al content) is almost perpendicular to the spinel- and pyroxene units, and simultaneously almost parallel to c*. Further on we tried to separate the contributions of both entities. For this purpose, the thicknesses of the < P >- and < SS > modules have been approximated by the perpendicular distances between least-squares planes defined by the oxygen atoms located at the boundaries of the modules. Figure 13a, b presents the variations of these parameters as a function of the Al/FeTot ratio. While the width of the two spinel modules is virtually constant, the pyroxenes module narrows significantly by 3.36%. The narrowing (widening) of the pyroxene module seems to dominate the contraction (expansion) of the structure in this direction with increasing (decreasing) Al/FeTot ratio. Figure 12b, c demonstrates that the intermediate principal strain axis ε2 is close to the apical O–M–O bonds in both the pyroxene and spinel modules. ε1—the least deformable direction—is parallel to the projections of the remaining four equatorial bonds of the octahedra onto the ε1–ε2 plane. Furthermore, ε1 is in line with a system of continuous linear edge-sharing octahedral chains present in both spinel modules that are additionally “stiffened” by two lateral tetrahedra (see Fig. 12d). Finally, the directions of the principal axes can be also related to the underlying distorted ccp arrangements of oxygen atoms that are present in both modules. ε2 is almost parallel to one of the four < 100 > ccp-directions in the ideal packing, say [001]ccp. ε3 and ε1, thus, run along [110]ccp and [\(\bar{1}\) 10]ccp, respectively.

Orientation of the principal axes of the compositional strain tensor for the SFCA-I solid-solution series and their relations to structural building blocks. The size of the axes is proportional to the principal strains reported in the text. a Projection of a single < PSS > unit parallel to the distorted close-packed oxygen monolayers (the levels of three successive layers are indicated by stars); b and c show the orientation of a single < P >- and < SS > module, respectively, within the ε1-ε2-plane; d linear edge-sharing octahedral chains in a single < S > block. Oxygen atoms are shown as red spheres. Orange spheres represent pure Ca-sites. Gray spheres indicate mixed cation positions. (Color online)

Evolution of the thickness of (a) the < SS > module and (b) the < P > module as a function of the Al/FeTot ratio in the solid-solution series
Concluding remarks
Most structural aspects, from unit-cell volumes to site occupancies, tetrahedral chain kinking and polyhedral distortions, are defined by nearly linear trends when plotted against the Al/FeTot ratio. From a thermodynamic perspective, the linear relationships observed in this study imply that the present SFCA-I solid solution is likely close to be ideal.
When compared with the compositions of the starting mixtures, the retrieved SFCA-I single crystals of the series were consistently depleted in calcium and iron as well as enriched in aluminum. Moreover, all samples contained ferric and minor amounts of ferrous iron. The concentrations of Ca were almost constant. The last observation indicates that under the given experimental conditions the current Ca contents of about 6.6 to 6.7 a.p.f.u. may represent an upper threshold. The fact, that not all of the divalent cations required for electroneutrality are calcium ions is in accordance with earlier studies by Mumme et al. (1998) and Arakcheeva et al. (1991) who consistently reported less than 8 Ca a.p.f.u. to be present in their Si- and Mg-free SFCA-I samples: Ca6.36Al2.68Fe3+29.32Fe2+1.64O56 and Ca7.12Al8.18Fe3+23.82Fe2+0.88O56, respectively. The incorporation of more than about 7 a.p.f.u. of the large Ca cations into the M-positions located in the octahedral bands is probably not compatible with the short M–O distances implied by the comparatively rigid connectivity of the edge-sharing [MO6]-polyhedra. Furthermore, the necessity to match the tetrahedral chains with the octahedra within the pyroxene modules may also induce an upper limit for calcium incorporation.
The four tetrahedra T4, T6, T7 and T8 belonging to the chains in the < P > modules of SFCA-I are the preferred sites for the Al-uptake within the solid-solution series. This observation compares well with the outcome of structural studies on the related SFCA-type A14O20 compounds where a similar tendency was recognized (Sugiyama et al. 2005; Murao et al. 2018, 2020).
A further major feature found in the site occupancy patterns of the present SFCA-I solid-solution series concerns the mean Al-concentration < xAl > of the Q1-, Q2- and Q3-sites in the branched chains as well as the mean connectedness < Q > for Al in the chains. It is evident, that for all members of the series (a) < xAl > for the Q3-positions (T7, T8) is larger than for Q2 (T4, T6) and Q1 (T1, T2) and (b) < Q > for Al is > 2. The finding, that the minority species (Al) of the cations in the chains concentrates on the most polymerized sites has also been reported for structurally related compounds containing the same type of branched chains such as sapphirine ((Mg,Al)8(Al,Si)6O20), for example (Christy 2009 and references cited therein). For sapphirine, which is isostructural to SFCA, the most abundant species in the chains (Al) is the least polymerized and, conversely, Si as the substituting ion shows a high degree of polymerization. Using Monte Carlo simulations, Christy (2009) demonstrated that this effect is related to the exothermic nature of the Al–O–Si linkages and the same explanation may be applicable for the Fe–O–Al connections in SFCA-I. Studies on the interplay between short- and long-range order as well as configurational entropy in the chains may be an interesting topic for further research activities.
Moreover, one could ask whether there exists an upper limit for the Al content in SFCA-I. For the case that all T-sites within the < P > modules are completely filled with Al, one possibility to accommodate additional aluminum could be realized by increasing the 1:2 ratio between the < P >- and < S > modules, which naturally leads to a new structure type. Interestingly, only very recently we demonstrated that for the more alumina-rich starting composition Ca8Al16Fe16O56, a SFCA-II type phase with a < PSPSS > stacking sequence (< P > : < S > ratio 2:3) occurred in the same temperature region that was selected for the synthesis experiments of the present study (Kahlenberg et al. 2021). The Al/FeTot ratio in this phase is 0.6. On the other hand, our results indicate that there is probably also a lower threshold for Al-incorporation. Actually, a pure “Fe-SFCA-I” phase could not be obtained. Of course, further experimental work is needed to chart the boundaries between the different members of the SFCA family in the system CaO–Fe2O3–FeO–Al2O3 in more detail and to verify our hypotheses concerning the limits of solid-solution in SFCA-I.
Finally, our investigations on the SFCA-I series may have also implications for mineralogists and petrologists working on new minerals. Natural counterparts to SFCA have been already described from pyrometamorphic rocks all over the world including famous localities such as the Hatrurim complex (Negev Desert, Israel) or the Eifel region (Germany). The compositions of these minerals such as khesinite (Ca2MgFe10SiO20, Galuskina et al. 2017), dorrite (Ca2MgFe5Al5SiO20, Shchipalkina et al. 2019) or devilliersite (Ca3Fe10SiO20, Krüger et al. 2021) are related to those of the synthetic phases studied in the present investigation. Furthermore, addibischoffite (Ca2Al12O20)—the natural analog to the pure aluminum end-member of SFCA—has been found in a Ca–Al-rich inclusion of a chondrite meteorite. The compound probably formed under rather exotic conditions from a Ca- and Al-rich melt at high temperature (≈ 1575 °C) and low pressure (≈ 10–4 to 10–5 bar) in a solar-like gaseous reservoir near the protosun (Ma et al. 2017). Minerals with equivalent crystal structures to SCFA-I have not yet been discovered, but it is more than likely that their first observation in Nature is only a question of time and of the availability of powerful X-ray sources at synchrotron beamlines dedicated to the study of small crystalline samples. Our crystallographic investigations on synthetic compounds can aid in the identification of these potentially new minerals. Information concerning the synthesis conditions could also be useful to gain a better insight into the formation conditions of the relevant host rocks.
Availability of data and material
All primary structural information of the six SFCA-I phases is included in the corresponding CIF-files that have been uploaded as well.
References
Angel RJ (2011) Win_Strain 4.11, http://www.rossangel.com/home.htm, accessed November 3rd. 2020.
Arakcheeva AV, Karpinskii OG, Lyadova VY (1991) Crystal structure of CaFe3AlO7 aluminum-calcium ferrite of variable composition. Sov Phys Crystallogr 36:332–336
Christy AG (2009) A Monte Carlo study of short- and long range order of tetrahedral cations in sapphirine and khmaralite. Am Mineral 94:270–282
Clout JMF, Manuel JR (2015) Mineralogical, chemical, and physical characteristics of iron ore. In: Lu L (ed) Iron ore: mineralogy, processing and environmental sustainability. Woodhead Publishing, Cambridge UK, pp 45–84
Dawson PR, Ostwald J, Hayes KM (1985) Influence of alumina on development of complex calcium ferrites in iron-ore sinters. Trans Inst Min Metall C 94:C71–C78
Downs RT, Hall-Wallace M (2003) The American mineralogist crystal structure database. Am Mineral 88:247–250
Droop GTR (1987) A general equation for estimating Fe3+ concentrations in ferromagnesian silicates and oxides from microprobe analyses, using stoichiometric criteria. Mineral Mag 51:431–435
Farrugia LJ (1999) WinGX suite for small-molecule single-crystal crystallography. J Appl Crystallogr 32:837–838
Fernandez-Gonzalez D, Ruiz-Bustinza I, Mochon J, Gonzalez-Gasca C, Verdeja L (2017) Iron ore sintering: process. Miner Process Extr Metall Rev 38:215–227
Gagné OC, Hawthorne FC (2018) Bond-length distributions for ions bonded to oxygen: metalloids and post transition metals. Acta Cryst B 74:63–78
Gagné OC, Hawthorne FC (2020) Bond-length distributions for ions bonded to oxygen: results for the transition metals and quantification of the factors underlying bond-length variation in inorganic solids. IUCrJ 7:581–629
Galuskina I, Galuskin E, Pakhomova A, Widmer R, Armbruster T, Krüger B, Grew E, Vapnik Y, Dzierazanowski P, Murashko M (2017) Khesinite, Ca4Mg2Fe103+O4[Fe103+Si2O36], a new rhönite-group (sapphirine supergroup) mineral from the Negev Desert, Israel - natural analogue of the SFCA phase. Eur J Mineral 29:101–116
Gates-Rector SD, Blanton TN (2019) The powder diffraction file: a quality materials characterization database. Powder Diffr 34:352–360
Hamilton J, Hoskins B, Mumme W, Borbridge W (1989) The crystal-structure and crystal-chemistry of Ca2.3Mg0.8Al1.5Si1.1Fe8.3O20 (SFCA) - solid-solution limits and selected phase-relationships of SFCA in the SiO2-Fe2O3-CaO(-Al2O3) system. N Jb Mineral Abh 161:1–26
Hancart J, Leroy V, Bragard A (1967) A study of the phases present in blast furnace sinter. Some considerations on the mechanism of their formation. CNRM Metall Rep DS 24(67):3–7
Holmes RJ, Lu L (2015) Introduction: overview of the global iron ore industry. In: Lu L (ed) Iron ore: mineralogy, processing and environmental sustainability. Woodhead Publishing, Cambridge UK, pp 1–42
Hsieh L, Whiteman J (1989) Sintering conditions for simulating the formation of mineral phases in industrial iron-ore sinter. ISIJ Int 29:24–32
Inoue K, Ikeda T (1982) The solid solution state and the crystal structure of calcium ferrite formed in lime-fluxed iron ores. Tetsu to Hagané 68:2190–2199
Kahlenberg V, Krüger H, Goettgens VS (2019) Structural elucidation of triclinic and monoclinic SFCA-III – killing two birds with one stone. Acta Cryst B 75:1126–1136
Kahlenberg V, Krüger H, Tribus M, Anwander B (2021) SFCA-II type Ca2.46Fe3+8.57 Fe2+0.52Al5.45O24 – an improved structural model for an iron-ore sinter phase. Mineral Petrol 115:137–147
Kalenga M, Garbers-Craig A (2010) Investigation into how the magnesia, silica, and alumina contents of iron ore sinter influence its mineralogy and properties. J South Afr Inst Min Metall 110:447–456
Karpinskii OG, Arakcheeva AV (1987) Crystal structure of triple Ca-ferrite Ca3.0Fe14.82O25 (α-CFF-phase). Sov Phys Crystallogr 32:28–30
Kim HS, Park JH, Cho YC (2002) Crystal structure of calcium and aluminium silicoferrite in iron ore sinter. Ironmak Steelmak 29:266–270
Krüger B, Krüger H, Galuskina IO, Galuskin EV, Vapnik Y (2021) Devilliersite, IMA 2020-073. In: CNMNC Newsletter 59, Eur J Mineral 33. https://doi.org/10.5194/ejm-33-139-2021
Kunz M, Armbruster T (1990) Difference displacement parameters in alkali feldspars: effects of (Si, Al) order-disorder. Am Mineral 75:141–149
Liebau F (1985) Structural chemistry of silicates. Springer Verlag, Berlin
Lisles DC, deVilliers JPR, Kahlenberg V (2016) Refinement of iron ore sinter phases: a silico-ferrite of calcium and aluminium (SFCA) and an Al-free SFC, and the effect on phase quantification by X-ray diffraction. Mineral Petrol 110:141–147
Ma C, Krot AN, Nagashima K (2017) Addibischoffite, Ca2Al6Al6O20, a new calcium aluminate mineral from the Acfer 214 CH carbonaceous chondrite: a new refractory phase from the solar nebula. Am Mineral 102:1556–1560
Merlino S, Zvyagin B (1998) Modular features of sapphirine-type structures. Z Kristallogr 213:513–521
Momma K, Izumi F (2011) VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J Appl Crystallogr 44:1272–1276
Mumme W, Clout J, Gable R (1998) The crystal structure of SFCA-I, Ca3.18Fe14.663+Al1.34Fe0.822+O28, a homologue of the aenigmatite structure type, and Mg-free SFCA, Ca2.45Fe9.043+Al1.74Fe0.162+Si0.6O20. N Jb Mineral Abh 173:93–117
Murao R, Harano T, Kimura M, Jung IH (2018) Thermodynamic modeling of the SFCA phase Ca2(Fe, Ca)6(Fe, Al, Si)6O20. ISIJ Int 58:259–266
Murao R, Harano T, Kimura M, Jung IH (2020) Thermodynamic modeling of multi-component calcium ferrite for sintering reaction analysis. Nippon Steel Tech Rep 123:67–74
Nicol S, Chen J, Pownceby M, Webster N (2018) A review of the chemistry, structure and formation conditions of silico-ferrite of calcium and aluminum (‘SFCA’) phases. ISIJ Int 58:2157–2172
OECD (2019) Steelmaking capacity http://www.oecd.org/sti/ind/steelcapacity.htm, accessed November 1st. 2020.
Ohashi Y, Burnham CW (1973) Clinopyroxene lattice deformations. The roles of chemical substitution and temperature. Am Mineral 58:843–849
Papike JJ, Prewitt CT, Sueno S, Cameron M (1973) Pyroxenes: comparisons of real and ideal structural topologies. Z Kristallogr 138:254–273
Patrick T, Lovel R (2001) Leaching dicalcium silicates from iron ore sinter to remove phosphorus and other contaminants. ISIJ Int 41:128–135
Patrick TRC, Pownceby MI (2002) Stability of silico-ferrite of calcium and aluminum (SFCA) in air-solid solution limits between 1240 °C and 1390 °C and phase relationships within the Fe2O3-CaO-Al2O3-SiO2 (FCAS) system. Metall Mater Trans B 33:79–89
Rigaku Oxford Diffraction (2015) CrysAlisPRO, version 1.171.38.43. Oxford: Rigaku Oxford Diffraction.
Rodriguez-Carjaval J (2011) FullProf.2k, Version 5.00, Jan2011-ILL JRC. Institut Laue-Langevin, Grenoble, France.
Scarlett NVY, Pownceby MI, Madsen IC, Christensen AN (2004) Reaction sequences in the formation of silico-ferrites of calcium and aluminum in iron ore sinters. Metall Mater Trans B 35:929–936
Shannon RD (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst A 32:751–767
Shchipalkina N, Pekov I, Chukanov N, Koshlyakova N, Ternes B, Schuller W (2019) Crystal chemistry of dorrite from the Eifel volcanic region, Germany, and chemical variations in the khesinite-dorrite-rhonite-kuratite solid-solution system. Mineral Petrol 113:249–259
Sheldrick GM (2008) A short history of ShelX. Acta Cryst A 64:112–122
Sugiyama K, Monkawa A, Sugiyama T (2005) Crystal structure of the SFCAM phase Ca2(Ca, Fe, Mg, Al)6(Fe, Al, Si)6O20. ISIJ Int 45:560–568
Thompson RM, Downs RT (2001) Model pyroxenes II: structural variation as a function of tetrahedral rotation. Am Mineral 89:614–628
Webster N, Pownceby M, Madsen I (2013) In situ X-ray diffraction investigation of the formation mechanisms of silico-ferrite of calcium and aluminium-I-type (SFCA-I-type) complex calcium ferrites. ISIJ Int 53:1334–1340
Wilson AJC (ed) (1995) International tables for crystallography, volume C mathematical, physical and chemical tables, 1st edn. Kluwer Academic Publishers, Dordrecht, Boston, London
Wright SE, Foley JA, Hughes JM (2000) Optimization of site occupancies in minerals using quadratic programming. Am Mineral 85:524–531
Zöll K, Manninger T, Kahlenberg V, Krüger H, Tropper P (2017) Investigations on the crystal structure and stability field of FCAM-I (Ca3MgAl6Fe10O28), an iso-structure to SFCA-I. Metall Mater Trans B 48:2207–2221
Zvyagin B, Merlino S (2003) The pyroxene-spinel polysomatic system. Z. Kristallogr 218:210–220
Acknowledgements
The authors thank Amelie Schickhoff for the preparation of the samples for EMP analysis and Ross John Angel (IGG-CNR, Padova) for providing a development version of the program CrystalPalace that eased the generation of the comprehensive tables from the CIF-files considerably. The manuscript benefited from the helpful comments of two anonymous reviewers.
Funding
Open access funding provided by University of Innsbruck and Medical University of Innsbruck.
Author information
Authors and Affiliations
Contributions
VK: conceived and planned the investigation. He performed the crystal growth experiments, single-crystal structure analyses and wrote the manuscript with the help of HK. HK: carried out the powder diffraction studies. MT: performed the EMP analyses.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
269_2021_1148_MOESM1_ESM.docx
Atomic coordinates, site populations and equivalent isotropic displacement parameters (Å2) for the members of the SFCA-I solid solution series. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor. All atoms in the asymmetric unit are located on general positions. Numbers given in column 1 refer to the Al/FeTot ratios. (DOCX 28 KB)
269_2021_1148_MOESM2_ESM.docx
Individual and average M-O and T–O bond distances (up to 3.15 Å) as well as polyhedral volumes (Å3) for the different members of the SFCA-I solid-solution series. The numbers in the first column refer to the Al/FeTot-ratios of the investigated compositions. (DOCX 27 KB)
269_2021_1148_MOESM3_ESM.docx
Anisotropic displacement parameters for the six members of the solid-solution series. The anisotropic displacement factor exponent takes the form: − 2π2 (h2a*2U11…+ 2 h k a* b* U12). (DOCX 25 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kahlenberg, V., Krüger, H. & Tribus, M. Structural systematics of SFCA-I type solid-solutions in the system CaO–Fe2O3–FeO–Al2O3. Phys Chem Minerals 48, 25 (2021). https://doi.org/10.1007/s00269-021-01148-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00269-021-01148-4