
Abstract
Background
Systematic, well-controlled clinical trials of botulinum toxin type A (BoNTA) in diverse patient populations are needed. The aim of this study was to characterize the safety and efficacy of 10-U and 20-U BoNTA doses versus placebo for treating glabellar lines in Japanese subjects.
Methods
A 16-week, multicenter, double-blind, randomized, placebo-controlled trial comparing 10 or 20 U of BoNTA versus placebo in 142 Japanese subjects with glabellar lines of at least moderate severity at maximal contraction. The primary efficacy endpoint was physician-rated line severity at maximal contraction 4 weeks after treatment. Secondary efficacy endpoints included physician/subject ratings and estimates of the effect’s duration.
Results
Response rates by physician-rated line severity at maximal contraction (week 4) were 86.4% (10 U), 88.6% (20 U), and 0% (placebo, p < 0.001). Line severity at maximal contraction in each BoNTA group (p < 0.001) improved significantly from baseline at each visit. BoNTA and placebo differed significantly on all other efficacy measures. Mean duration of effect was 9.4 weeks in the 20-U group and 7.9 weeks in the 10-U BoNTA group. No serious adverse events occurred.
Conclusion
Doses of BoNTA of 10 and 20 U are effective and safe for treating glabellar lines in Japanese subjects, and the 20-U dose provides greater efficacy and longer duration of effect.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Botulinum toxin type A (BoNTA) is a cornerstone in the rapidly expanding field of minimally invasive aesthetic medicine. BoNTA’s safety and efficacy, as well as its recommendations for use in treating glabellar and other facial lines, have been well documented in randomized controlled trials, numerous clinical studies, and consensus recommendations based on experts’ clinical experience [1–4]. Nevertheless, the preponderance of this research has been based on clinical trials involving female Caucasian subjects. It is well recognized that ethnicity and skin color are associated with variations in properties of facial skin and musculature; therefore, it was deemed important to establish the safety and efficacy of BoNTA in Japanese subjects in a systematic, well-controlled clinical trial.
Randomized controlled trials in predominantly Caucasian populations have shown that 20-U doses of BoNTA are effective and well tolerated in treating glabellar lines [1–4]. A pilot study conducted in 125 Japanese subjects showed similar results (unpublished data). The objectives of this study were to confirm and validate the effective dose of this formulation of BoNTA (BOTOX®; Allergan, Inc., Irvine, CA) and characterize the efficacy and safety profile in this patient population. This study included physician assessments and subject-reported outcomes, which have become increasingly recognized as key outcome measures in aesthetic trials and clinical practice [5–8].
Methods
Study Design
This multicenter, 16-week, double-blind, randomized, placebo-controlled trial compared the efficacy and safety of two doses (10 and 20 U) of BoNTA versus placebo in Japanese subjects with glabellar lines of at least moderate severity at maximal contraction. This study was conducted in compliance with the Declaration of Helsinki and the Standards for the Implementation of Clinical Trials on Pharmaceutical Products, and with the approval of the institutional review boards of the participating centers. The planned number of enrolled subjects was 135 (45 for each of the three treatment groups).
Subjects
BoNTA-naïve patients aged 20–64 years with glabellar lines of at least moderate severity at maximal contraction, based on standardized photography, were eligible for the study. Key exclusion criteria included any condition (such as myasthenia gravis, Lambert-Eaton syndrome, amyotrophic lateral sclerosis, or systemic neuromuscular junction disorder) that could influence the effect of treatment (e.g., deterioration in atonia); pregnancy; history of hypersensitivity to any component of the treatment product; any condition that could impair the safety of the subject (e.g., severe heart, kidney, liver, or respiratory disease); infection or skin disease at the injection site(s); use of a peripheral muscle relaxant within 2 weeks of the start of the study; history of surgery at the treatment site(s); or previous aesthetic procedures within 6 months of the beginning of the study. Subjects had to provide written informed consent after receiving an explanation of the study. Discontinuation criteria included voluntary retraction of consent, serious clinical abnormality, unsuitability for evaluation, inability to present for evaluation, protocol violation, or investigator’s decision due to medical or nonmedical reasons.
Study Treatments
Active treatment was a 10- or 20-U dose of BoNTA supplied in vials containing 100-U doses of BoNTA, 0.5 mg albumin (human), and 0.9 mg sodium chloride. The placebo contained 0.9 mg sodium chloride only. Vials were reconstituted with physiologic saline (Japanese Pharmacopoeia) to deliver 2 or 4 U per site in equal volumes (total of five injection sites of 2 or 4 U/0.1 ml/site) or an identical volume of placebo. Vials used for treatment administration were coded to maintain the blind.
Study Procedures and Clinical Outcome Measures
Subjects meeting enrollment criteria provided a medical history before enrollment and treatment, and females of childbearing age had to have a negative pregnancy test. Previous and concomitant therapies were documented. Using the random-number-generation function of SAS (Statistical Institute, Inc., Cary, NC), subjects were allocated to one of three treatment groups: 10-U BoNTA, 20-U BoNTA, or placebo. Vital signs and laboratory tests were evaluated at pretreatment, week 4, week 16, and on the day the subject discontinued.
Pretreatment line severity at rest and maximal contraction was assessed by a physician and photographed on the day of treatment. Each subject then received two injections in each corrugator supercilii muscle and one injection in the procerus muscle for a total of five injection sites (similar to other controlled trials of this formulation of BoNTA) [3, 4]. The volume of each injection was 0.1 ml for a total of 0.5 ml. Follow-up evaluations occurred at week 1 and then at 4, 8, 12, and 16 weeks after treatment. Each follow-up visit included a physician’s evaluation of line severity at rest and maximal contraction, photographic documentation, and the subject’s assessment of line improvement. The measurement scales for assessing line severity and improvement in line severity used the same scoring as the Facial Wrinkle Scale and subject’s global assessment of change in wrinkle appearance as the United States pivotal trials [3, 4]. In addition, subjects rated their degree of satisfaction with the effects of treatment at weeks 4 and 16 (Table 1). The week-16 satisfaction assessment requested subjects to consider their satisfaction over the period of the trial and whether they would like to be treated again. Adverse events (AEs) were monitored throughout the course of the study and assessed for severity and possible relationship to the treatment.
Efficacy Endpoints and Statistical Analyses
The primary efficacy endpoint was physician-rated line severity 4 weeks after treatment at maximal contraction (frown). Secondary endpoints included physician-assessed line severity at maximal contraction at all other posttreatment visits, line severity at rest at all visits, subject-assessed improvement ratings at each visit, and patient satisfaction ratings at weeks 4 and 16 and for the entire study period (rated at week 16).
All patients randomized to treatment, except those with severe protocol violations, comprised the full analysis set, which was used for efficacy analyses. The safety population was composed of all subjects randomized to treatment who received the trial substance at least once. Paired t tests were used to analyze continuous variables for change-from-baseline values. Analysis of variance was performed for between-group comparisons. Nonparametric statistical tests were conducted on categorical variables to analyze changes from baseline and between-group differences. For the primary efficacy endpoint, responders were defined as those with posttreatment scores of 0 or 1. For the secondary efficacy endpoint, responders for line severity were defined as those with a score of 0 or 1, and responders for subject-assessed improvement ratings were defined as those with a score of at least +2 (moderate improvement; definite improvement, about 50% improvement). Duration of efficacy by group also was estimated. Subgroup analyses were used to investigate the influence of baseline and subject demographic variables on outcomes. Results were deemed statistically significant for p ≤ 0.05.
Results
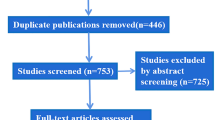
The full analysis data set comprised 140 patients (Fig. 1). There were no statistically significant differences among the groups. Groups did not differ in their pretreatment line severity either at rest or maximal contraction. The majority of subjects were female (90.0%), and the mean age was 45.7 (±9.1) years. All subjects had either moderate (50.7%) or severe (49.3%) glabellar lines at maximal contraction. The safety population was 139 subjects: 46 in the 10-U group, 44 in the 20-U group, and 49 in the placebo group. Six subjects in the full analysis data set discontinued: two in the 20-U group discontinued before treatment, two in the 10-U group moved away, one in the 20-U group retracted consent, and one in the placebo group became pregnant.

Study design and patient flow through study
Efficacy
Physician Assessments
Primary Endpoint Response rates in each active-treatment group differed significantly from placebo but not from each other. The response rates were 86.4% (38/44) in the 10-U group, 88.6% (39/44) in the 20-U group, and 0% (0/48) in the placebo group at week 4 posttreatment (Fig. 2).

Percentage of responders (line severity = none or mild) at maximal contraction (physician ratings). *p < 0.001, 10 U vs. placebo and 20 U vs. placebo; 10 U vs. 20 U not significant. Calculated in the full analysis set
Secondary Endpoints Each active-treatment group showed significant improvements in line severity at maximal contraction relative to baseline ratings (p < 0.001), in contrast to the placebo group, which did not improve. The percentage of responders (line severity none or mild at maximal contraction) at each visit is shown in Fig. 2. Each active-treatment group differed significantly from placebo at each visit (p < 0.001) but did not differ from each other, although the higher dose resulted in a somewhat higher response rate that was sustained over the course of the study, especially from week 8 onward. The degree of mean change from baseline in line severity at maximal contraction also differed significantly between the active-treatment groups and placebo (p < 0.001), but the active groups again did not differ from each other (Fig. 3).

Mean change from baseline in line severity at maximal contraction (physician ratings). * p < 0.001, 10 U vs. placebo and 20 U vs. placebo; 10 U vs. 20 U not significant
At rest, patterns of response were similar to those at maximal contraction. The 10-U and 20-U BoNTA groups differed significantly from placebo (p < 0.05) but not from each other. The peak response rates in the active-treatment groups occurred at week 4 for the 10-U group (84.1%) and at weeks 4 and 8 for the 20-U group (93.2%). The 20-U group differed from the placebo group at all visits (p < 0.05). The degree of change at rest was not as dramatic as at maximal contraction, in part because the majority of subjects in each group (62.2, 52.3, and 69.4% of 10-U, 20-U, and placebo groups, respectively) had baseline scores of 0 (lines are not noticeable) or 1 (lines are somewhat noticeable). The greatest proportion of subjects whose lines at rest improved by at least one level occurred at week 8 in the 20-U group (68.2%). The greatest proportion of subjects with a similar level of improvement in the 10-U group was 56.8% at week 4. The active-treatment groups differed significantly from placebo throughout the study period (p < 0.001) and from each other at week 8 (p = 0.021), at which time the 20-U group had a greater degree of change.
Because of the high proportion of subjects with baseline resting scores of ≤1, a subgroup analysis of subjects with scores of ≥2 was performed (Fig. 4). This comprised a total of 53 subjects: 17 in the 10-U group, 21 in the 20-U group, and 15 in the placebo group. In this analysis, the 20-U dose resulted in higher levels of improvement throughout the study. The 10-U and 20-U groups differed significantly from placebo (p < 0.019 and p < 0.001, respectively) and each other at week 8 (p = 0.037).

Subgroup analysis: percentage of responders among subjects with pretreatment line severity at rest of ≥2 (lines are noticeable or readily apparent; physician ratings). 10 U vs. placebo, all visits, p ≤ 0.019; 20 U vs. placebo, all visits, p < 0.001. *10 U vs. 20 U, week 8, p = 0.037; all other visits, not significant. Calculated in the full analysis set
Subject Assessments
Line Improvement and Responder Rates At each posttreatment visit subject assessment of line improvement was significantly greater (p < 0.001) than placebo for each of the two active-treatment groups. Moreover, the 20-U group differed significantly from the 10-U group at week 8 (p = 0.001). The percentage of responders (subject’s improvement assessment of ≥+2) at each visit demonstrated that both active treatments consistently resulted in a significantly (p < 0.001) greater proportion of responders than placebo (Fig. 5). In addition, the 20-U treatment resulted in a significantly (p = 0.035) greater proportion of responders at week 8 than did the 10-U treatment.

Subject assessment of line improvement: Percentage of responders. All comparisons between 10 U vs. placebo and 20 U vs. placebo, p < 0.001; *10 U vs. 20 U, p = 0.035 at week 8; all other visits are not significant. Calculated in the full analysis set
Subject Satisfaction Ratings Subjects treated with BoNTA had significantly higher satisfaction scores than those treated with placebo at both evaluation time points as well as for the entire study period (p < 0.001). The satisfaction ratings did not differ significantly between the two active-treatment groups at any time point. The proportions of subjects who rated themselves as at least somewhat satisfied (scores of 1, 2, or 3) are shown in Fig. 6. In the two active-treatment groups, mean satisfaction rating scores peaked at 4 weeks at 2.2 (where 1 is very satisfied) for the 10-U group and at 2.1 for the 20-U group. At 16 weeks, the scores were 3.5 and 3.2, respectively, and for the entire observation period they were 2.5 and 2.1, respectively. Analysis of covariates indicated that at week 4, dose of BoNTA, line severity rating during maximal contraction and at resting position (both with baseline level subtracted), and subject-assessed line improvement were significantly (p < 0.001) related to subject satisfaction. Analyses of the week-16 data indicated that in addition to the preceding variables, the subjects’ gender and age were associated with satisfaction. Specifically, female sex and increasing age were associated with a lower probability of satisfaction. Analysis of the entire observation period resulted in similar findings; however, the subject’s gender did not have an effect on subject satisfaction. Age remained the only variable with an upper confidence limit of <1, indicating a decreasing probability of satisfaction with advancing age.

Subject satisfaction ratings (scores of 1, 2, 3). All comparisons between 10 U vs. placebo and 20 U vs. placebo, p < 0.001; no significant differences between 10 U and 20 U. Calculated in the full analysis set
Duration of Effect
The duration of BoNTA’s effect was based on the change in line severity at maximal contraction from week 1 to week 16 posttreatment. The treatment was determined to remain effective for the amount of time during which line severity at maximal contraction was 0 (none) or 1 (mild), not on return to baseline levels of severity. The mean duration of effect was 9.4 weeks for the 20-U treatment group and 7.9 weeks for the 10-U group. The analysis of duration for 10-U and 20-U groups by the Kaplan–Meier curve is shown in Fig. 7.

Duration of effect of BoNTA: Kaplan–Meier curve of treated subjects with scores of 0 (none) or 1 (mild)
Safety and Tolerability
No subject discontinued the study because of an adverse effect (AE). Neither the incidence of AEs nor that of adverse drug reactions (ADRs) differed significantly between groups. The incidence of AEs was 67.4% (31/46) in the 10-U group, 75.0% (33/44) in the 20-U group, and 59.2% (29/49) in the placebo group. The rate of ADRs was 32.6% in the 10-U group, 27.3% in the 20-U group, and 22.4% in the placebo group. There were no deaths, nor any serious AEs requiring emergency interventions. Of the 139 subjects, 2 developed blepharoptosis (1 in each active-treatment group), and 12 experienced heavy eyelids (5 in the 10-U group, 6 in the 20-U group, and 1 in the placebo group). Each of these events was mild in severity and resolved over the course of the study.
Discussion
This study confirms the utility of BoNTA for the treatment of glabellar lines and demonstrates its safety and efficacy in Japanese subjects. Doses of 10 U and 20 U were effective in treating glabellar lines based on both physician and subject ratings. BoNTA treatment resulted in significant improvement of line severity at both maximal contraction and rest, which is consistent with previous controlled clinical trials of this formulation [9]. Subjects rated their satisfaction with treatment as high at weeks 4 and 16, and for the entire study period. Overall duration of effect in this study also was similar to that of earlier studies.
The overall results of this study indicate that the two doses of BoNTA did not differ significantly on several of the endpoints but suggest that the 20-U dose may provide somewhat higher response levels, greater improvement from baseline, and a somewhat longer duration of effect, as measured by between-group differences at the various time points. This study did not assess the absolute duration of effect because relapse rates (return to pretreatment line severity) were not included. Also, as all subjects in this study were naïve to BoNTA treatment, repeated treatment without dosing changes could perhaps extend the duration of benefit or result in progressive improvement [1, 10, 11].
The findings of this study are consistent with those of other studies in which responses appear to be dose dependent but which were not always statistically significant [9, 12]. In the present study, only two doses were used, both of which were effective overall. These findings, coupled with the subgroup and covariate analyses, suggest that fruitful areas for additional research include the impact on efficacy and the interaction of variables such as age, gender, BoNTA dose, and pretreatment line severity.
Conclusions
Doses of 10 U and 20 U of BoNTA are effective and well tolerated in treating glabellar lines in Japanese subjects; however, the 20-U dose provides greater efficacy and a somewhat longer duration of effect than does the 10-U dose.
Note These results do not apply to any formulation of BoNTA other than that used in the present study, due to well-documented differences in formulations and lack of interchangeability [13, 14]. It should be noted that the results reported in this study refer to the Allergan (Irvine, CA, USA) formulation of BoNTA (BOTOX®, BOTOX® Cosmetic, Vistabel®) and cannot be generalized to other formulations or serotypes of botulinum toxin.
References
Carruthers A, Carruthers J, Lowe NJ, Menter A, Gibson J, Nordquist M, Mordaunt J, for the BOTOX® Glabellar Lines I & II Study Groups (2004) One-year, randomised, multicenter, two-period study of the safety and efficacy of repeated treatments with botulinum toxin type A in patients with glabellar lines. J Clin Res 7:1–20
Carruthers J, Fagien S, Matarasso SL, the Botox Consensus Group (2004) Consensus recommendations on the use of botulinum toxin type A in facial aesthetics. Plast Reconstr Surg 114(Suppl 6):1S–22S
Carruthers JA, Lowe NJ, Menter MA, Gibson J, Nordquist M, Mordaunt J, Walker P, Eadie N, for the BOTOX Glabellar Lines I Study Group (2002) A multicenter, double-blind, randomized, placebo-controlled study of the efficacy and safety of botulinum toxin type A in the treatment of glabellar lines. J Am Acad Dermatol 46:840–849
Carruthers JD, Lowe NJ, Menter MA, Gibson J, Eadie N, for the Botox Glabellar Lines II Study Group (2003) Double-blind, placebo-controlled study of the safety and efficacy of botulinum toxin type A for patients with glabellar lines. Plast Reconstr Surg 112:1089–1098
Carruthers A, Carruthers J (2007) Patient-reported outcomes with botulinum neurotoxin type A. J Cosmet Laser Ther 9(Suppl 1):32–37
Carruthers J, Carruthers A (2007) Botulinum toxin type A treatment of multiple upper facial sites: patient-reported outcomes. Dermatol Surg 33:S10–S17
Fagien S, Cox SE, Finn JC, Werschler WP, Kowalski JW (2007) Patient-reported outcomes with botulinum toxin type A treatment of glabellar rhytids: a double-blind, randomized, placebo-controlled study. Dermatol Surg 33:S2–S9
Stotland MA, Kowalski JW, Ray BB (2007) Patient-reported benefit and satisfaction with botulinum toxin type A treatment of moderate to severe glabellar rhytides: results from a prospective open-label study. Plast Reconstr Surg 120:1386–1393
Carruthers A, Carruthers J, Said S (2005) Dose-ranging study of botulinum toxin type A in the treatment of glabellar rhytids in females. Dermatol Surg 31:414–422
Carruthers JDA, Carruthers JA (1992) Treatment of glabellar frown lines with C. botulinum-A exotoxin. J Dermatol Surg Oncol 18:17–21
Gordon MF, Barron R (2006) Effectiveness of repeated treatment with botulinum toxin type A across different conditions. South Med J 99:853–861
Carruthers A, Carruthers J (2005) Prospective, double-blind, randomized, parallel-group, dose-ranging study of botulinum toxin type A in men with glabellar rhytids. Dermatol Surg 31:1297–1303
Allergan, Inc. (2005) Botox Cosmetic package insert. Irvine, CA: Allergan, Inc.
De Boulle KLV (2007) Botulinum neurotoxin type A in facial aesthetics. Expert Opin Pharmacother 8:1059–1072
Acknowledgments
This study was funded by Allergan, Inc., Irvine, California. The authors would like to thank the investigators: Ryuichi Utsugi, M.D., Head of Plastic Surgery, Kitasato Institute Hospital; Hidemi Nakagawa, M.D., Professor of Dermatology, Jikei University, School of Medicine; Ryuzaburo Tanino, M.D., Professor of Surgery, Director of Department of Plastic Surgery, Tokai University, School of Medicine; Yoshiaki Hosaka, M.D., Professor & Chairman, Department of Plastic & Reconstructive Surgery, Showa University, School of Medicine; Kitaro Ohmori, M.D., Head of Plastic Surgery, Tokyo Metropolitan Police Hospital.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Harii, K., Kawashima, M. A Double-Blind, Randomized, Placebo-Controlled, Two-Dose Comparative Study of Botulinum Toxin Type A for Treating Glabellar Lines in Japanese Subjects. Aesth Plast Surg 32, 724–730 (2008). https://doi.org/10.1007/s00266-008-9199-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00266-008-9199-6