
Abstract
Introduction
The purpose of the present investigation is to report on detailed complications among a much larger group of 2372 orthopaedic patients treated with stem cell injections who were followed in a treatment registry for up to nine years.
Methods
All patients underwent an MSC-based, percutaneous injection treatment of an orthopaedic condition between December 2005 and September 2014 at one of 18 clinical facilities. Treated areas of the body included the knee, hip, ankle/foot, hand/wrist, elbow, shoulder, and spine. The patients were followed prospectively via enrollment in a treatment registry. Patients were followed prospectively at one, three, six and 12 months, and annually thereafter, using an electronic system, ClinCapture software.
Results
A total of 3012 procedures were performed on 2372 patients with follow-up period of 2.2 years. A total of 325 adverse events were reported. The majority were pain post-procedure (n = 93, 3.9 % of the study population) and pain due to progressive degenerative joint disease (n = 90, 3.8 % of the study population). Seven cases reported neoplasms, a lower rate than in the general population. The lowest rate of adverse events was observed among patients injected with BMC alone.
Conclusion
Lowest rate of adverse events was among those patients receiving BMC injections alone, but the higher rate of AEs for BMC plus adipose and cultured cells was readily explained by the nature of the therapy or the longer follow-up. There was no clinical evidence to suggest that treatment with MSCs of any type in this study increased the risk of neoplasm.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autologous mesenchymal stem cells (MSCs) have been utilized to treat degenerative and post-traumatic orthopedic conditions for more than two decades [1]. Because MSCs can differentiate into bone, cartilage, muscle, tendon, and ligament tissue and can use paracrine and other effects to elicit significant changes in injured tissues, their use for treating orthopaedic conditions holds significant promise [2–5]. In a clinical setting MSCs are typically harvested from bone marrow, then isolated and either re-injected or implanted in the same surgical procedure or culture expanded and then used clinically.
The same surgical procedure use of bone marrow aspirate is known as bone marrow concentrate (BMC). This is a fraction of the whole marrow which is isolated via centrifugation and subsequently injected into joints and surrounding tissue [3]. BMC contains MSCs and other nucleated cells, including hematopoietic stem cells, endothelial progenitor cells, macrophages, and platelets [6]. MSCs can also be isolated from marrow aspirate and then expanded in culture as a means of increasing the MSC dose [7]. In contrast with therapy utilizing BMC in which the entire procedure is performed in the same procedure, in vitro culture-expansion of MSCs requires a one to two week period of preparation and incubation.
A number of studies published over an 18-year span have described the safe use of autologous bone marrow derived MSCs to treat orthopaedic conditions [1, 5, 8–12]. The results of these studies, bolstered by the results of in vitro and animal studies, indicate that bone marrow derived MSCs carry little to no risk of malignant transformation, and that they are likely safe for use in human orthopaedic applications [4, 7, 13–15]. However, no large scale investigations exist with long-term patient follow-up where all complications have been reported, adjudicated, and classified.
We have previously published the results of two treatment registry studies that followed reported complications among 227 (in 2010) and 339 (in 2011) orthopaedic patients treated with culture-expanded MSCs [9, 14]. The purpose of the present investigation is to report on detailed complications among a much larger group of 2372 orthopaedic patients treated with stem cell injections who were followed in a treatment registry for up to nine years. The patients in the present analysis fall into one of the following treatment groups: 1) those who were treated with BMC only; 2) those who were treated with BMC along with an adipose graft, and 3) those who were treated with culture-expanded MSCs.
Methods
Participants and settings
Subjects included in the present study are all patients who underwent an MSC-based, percutaneous injection treatment of an orthopaedic condition between December 2005 and September 2014 at one of 18 clinical facilities located in the United States or Australia and who had attained at least a three month follow-up period. Treated conditions included those resulting from degenerative joint changes (i.e., osteoarthritis, degenerative disc disease, degenerative disc disease) as well as trauma (e.g., anterior cruciate ligament injuries, rotator cuff tears, etc.). Treated areas of the body included the knee, hip, ankle/foot, hand/wrist, elbow, shoulder, and spine. Knee, hip, and shoulder patients constituted approximately 87 % of the population.
The patients were followed prospectively via enrollment in a treatment registry. Patients were grouped by type of MSC treatment (see below). The choice of treatment type was left to the treating physician and while there were no exclusion criteria for MSC-treated patients to enter the registry, patients were naturally excluded from treatment if they were found not to be a candidate for the treatment by the attending physician. Reasons for exclusion from treatment included conditions for which the only therapeutic alternative was deemed to be surgery as well as medical conditions that would make MSC therapy difficult. Examples include a completely torn and retracted tendon or ligament, a severely osteoarthritic knee with deformity, severe spinal stenosis with neurologic compromise, and severe rheumatologic conditions like rheumatoid arthritis or systemic lupus erythematosus. Institutional Review Board oversight for the registry protocol was provided by a U.S. Office of Human Research Protections registered organization (#IRB00002637). Outcomes and efficacy of each procedure have been reported previously [2, 8, 13, 16]. Prior to each procedure, physicians discussed risks, benefits, and alternatives to the procedure. Each subject gave both oral and written informed consent for procedure.
Baseline information collected in the registry included primary diagnosis, patient demographics, medical history, and physical examination. Patients were followed prospectively at 1, 3, 6, and 12 months post treatment, and annually thereafter, using an electronic system, ClinCapture software (Clinovo Clinical Data Solutions, Sunnyvale, California). Patients were sent automated e-mails that asked them to respond to a number of questions regarding outcomes, function, and general health. Three e-mails were sent once a week and if the patient failed to respond after three e-mails, the registry staff initiated two phone calls. If the patient failed to respond to these additional two queries, then the time point was considered lost to follow-up and the process began again at the next time point. Attending physicians participating in the registry were also encouraged to report any complications.
In addition to outcome information, patients were also asked the following two questions regarding possible treatment-related adverse events (AEs): “Did you experience any complications you believe may be due to the procedure (i.e., infection, illness, etc.)? If yes, please explain;” and “Have you been diagnosed with any new illness since the procedure? If yes, please explain.” The complications questions were intentionally broad in order to capture any change in the patient’s health status that could possibly be related to the MSC procedure.
Treatment groups
The patients were grouped based on type of MSC treatment, as follows: SD (same day aspiration, isolation, and re-injection procedure with BMC), AD (same day aspiration, isolation, and re-injection procedure with BMC plus adipose graft), and CE (culture expanded MSCs re-implanted weeks or months after bone marrow aspiration) (see Supplement 1). All physicians were trained to use the same protocol for bone marrow aspiration, adipose graft, and re-injection procedures.
Two weeks prior to the bone marrow harvest procedure, patients in all groups were restricted from using steroidal and non-steroidal anti-inflammatory drugs in order to avoid possible cytotoxic effects on MSCs [17]. All injections in this study were confirmed with ultrasound or fluoroscopic imaging to ensure accurate placement. Two to five days prior to the administration of the MSCs to the treatment area, the patient’s joint, ligament, or tendon was pre-injected with 12.5 % hypertonic dextrose to promote an inflammatory response and begin the process of tissue repair. The decision to use this protocol was based on promising earlier observations in animal models that this protocol aided tendon healing and improved function in knee osteoarthritis patients and confirmed more recently through stabilization of cartilage volume on MRI in patients receiving only this treatment [18]. A detailed description of the procedures performed for the SD, AD, and CE groups are provided in our earlier publications [7, 9, 16]. Briefly, bone marrow harvest was completed via the collection of approximately 10–15 cc of bone marrow aspirate from the six to ten total sites from the bilateral posterior iliac crests. For the BMC injections (SD and AD groups), the aspirate was centrifuged to separate the buffy coat, resulting in 1–3 ml of BMC generally containing 0.2-1.5 × 108 nucleated cells. Platelet rich plasma (PRP) and platelet lysate (PL) was concurrently prepared and injected along with the BMC into the target region on the same day as the bone marrow aspiration. In the AD group, an additional component of minimally processed lipo-aspirate which had been separated from the aqueous and oil components was co-injected along with the BMC (3–7 cc) and PRP and PL solution [7]. All isolation techniques for PRP, PL, SD, AD, and CE were standardized using a standard operating procedure (SOP) protocol that has been described in previous publications [7, 9, 16]. Specifically, purpose built kits were not used, but all sites used the same off the shelf disposable lab supplies and the same or similar equipment such as centrifuges, pipettes, and microscopy. Staff at each site were trained in these SOP protocols. Based off the PLRA classification, the type of PRP produced is 1 cc of 14x/−/−/NO [19] but baseline platelet counts were not obtained. In the CE group, MSCs isolated from the bone marrow aspirate were expanded in an autologous based culture media for 12–16 days prior to injection into the joint space (1–3 cc in PL with dose ranges generally from 0.1-6 × 107 MSCs) or musculoskeletal structure (see Supplement 1 which elaborates on treatment differences between groups) [14]. Injectate volumes and dose were recorded, but not controlled and were determined by the treating physician.
Adverse events adjudication
AEs accessed from the treatment registry were initially sorted into one of 20 categories: allergic, bone, cardiac, endocrine, gastrointestinal, immune, infection, lab work, neoplasm, neurologic, pain-post procedure, pain due to progressive DJD, pain-other areas, pain-other, pulmonary, renal, rheumatological, skin, vascular, and other.
AEs were further categorized by the attending physician as: (1) serious adverse events (SAEs) or non-SAEs (2) expected or unexpected, and, as appropriate (3) related to the implantation procedure or related to stem cells (not mutually exclusive). AEs related to the implantation procedure or the stem cells were further defined as “definite,” “possible,” “unlikely,” or “not related.” SAEs were defined using guidelines developed by the United States Department of Health and Human Services [20]. This is defined as any untoward event that results in death, is life-threatening, requires inpatient hospitalization or causes prolongation of existing hospitalization, results in persistent or significant disability/incapacity, or requires intervention to prevent permanent impairment or damage. All “possible” SAEs were tabulated by one author (CJC) and then provided to five independent physician reviewers who were blinded to any initial or subsequent adjudication by another reviewer. The tabulating author (CJC) also remained blinded as to the identity of the physician who performed any specific independent adjudication. The independent reviewers were unrelated to the treating physicians in the study. Independent reviewers were recruited via an electronic discussion board for physicians if they: (1) had experience in using platelet rich plasma or stem cells for orthopaedic conditions (2) were a practicing physician in private or academic practice (Mishra, Feb 2009). In order to estimate the AE incidence, a person-time metric was calculated based on the number of patients and the amount of time they were followed from the time of treatment. The follow-up period was calculated from the date of the procedure to the date of data access or study exit.
Statistical analysis
The treatment groups were described by age, body-mass index (BMI), follow-up time, gender, and the joint/area treated. Frequency, proportion, and the rate of AEs by category were reported for each treatment group. AE rates were compared between treatment groups using a chi-square test. Frequency, proportion, and rate were also reported for SAEs, expected AEs, procedure-related AEs, and stem cell-related AEs. Categorical differences in proportions and rates between groups were analyzed using a chi-square test. Post hoc pair-wise comparisons between groups were made using chi-square or Fisher’s exact test, as appropriate. AE incidence rates were calculated by dividing the frequency of a specific AE by the total person-year (PY) denominator, with the results reported per 100 PY. Logistic regression analysis for binary outcomes was used to quantify the risk of reporting an AE, SAE, and treatment-related AE by treatment group, and adjusted for potential predictive or confounding factors (i.e., length of follow-up, age, gender, and body area treated). All statistical analyses were performed using SAS software version 9.4 [21].
Results
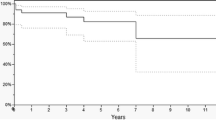
There were 2372 patients in the registry who were treated with any one of the three autologous MSC protocols in the period between December 2005 and September 2014. The follow-up period ranged from 1 month to 8.8 years, with 2.2 years mean follow-up time. In the SD group 1590 patients were treated (1949 BMC injections), 247 patients were treated in the AD group (364 BMC injections with adipose graft), and 535 patients were treated in the CE group (699 culture-expanded MSCs procedures). The higher number of procedures than patients indicates both serial procedures that occurred at different times and/or bilateral or multiple joint procedures that occurred in the same treatment session. The CE group was followed for an average of 4.4 years (3 months to 9 years), and the SD and AD groups were followed for an average of 1.1 (3 months-5 years) and 1.8 years (3 months to 4 years), respectively (see Fig. 1). Other baseline characteristics are reported in Table 1.

Number of patients categorized by length of follow-up, in number of years
There were a total of 325 AEs reported by 287 patients (12.1 % of the study population), with 36 reported SAEs, representing 1.5 % of the study population and incidence of 0.7/100 PY (see Fig. 2 and Table 2). SAE incidences were significantly different between groups, with the CE group reporting the highest incidence at 1.1/100 PY, versus 0.9/100 PY in the AD group and 0.4/100 PY in the SD group (P = 0.006). There were 38 AEs that were deemed to be definitely related to the procedures (1.6 % of the total population) and ten AEs definitely related to stem cells (0.4 % of the total population). Incidences of procedure- and stem cell-related AEs were not significantly different between treatment groups.

Flow chart demonstrating the distribution and number of serious adverse events, as they related to to procedure type or stem cells. AE = adverse event
The majority of AEs were post-procedure pain or attributed to degenerative joint disease (DJD) for which the treatment was sought (Fig. 3). There were 93 reports of post-procedure pain (3.9 % of the study population), and 90 reports of pain due to DJD (3.8 % of the study population) (Table 2). There were 27 AEs classified as “other” (i.e., that did not fit into any of the described categories) and “pain in other areas” was reported by 16 patients. This last category describes AEs where the patient reported pain in an area that was not treated (i.e., the knee was treated and the patient reported new onset shoulder pain). Frequencies of neurologic, vascular, and allergic AEs were 14 (0.6 %), 14 (0.6 %), and 11 (0.5 % of the study population), respectively (Table 2). Among SAEs the most frequent categories were neoplasm, neurologic, and vascular events (Table 3). There were seven neoplasm cases representing 0.3 % of the study population, with an incidence of 0.14/100 PY. The difference in neoplasm rates between groups was not statistically significant. Serious neurologic and vascular events were six and five cases, respectively, representing 0.25 % and 0.21 % of the total population.

Proportions of adverse event (AE) subcategories versus the total number of AEs. “Other systems” include endocrine, renal, gastrointestinal, pulmonary, bone, and rheumatological, with <1 % each. DJD = degenerative joint disease
Results of the SAE adjudication are reported in Table 4 and the Addendums. In Addendum 1, the adjudications of the six reviewers regarding the relatedness of the 36 SAEs are recorded. A majority opinion (as defined by >50 % agreement) was present in all but two SAEs (#15 and #30). Addendum 2 includes the results, by reviewer, of the relationship of the SAE to the procedure. In total, 19/36 (53 %) of the SAEs were considered as not related or unlikely to be related to the procedure. There were 13/36 cases or 36 % in which at least one reviewer indicated that the SAE was possibly related. Four of the 36 cases, or 11 %, of SAEs were adjudicated as definitely related to the procedure by a minority of reviewers (i.e., one or two of the six reviewers). These four cases were categorized as neoplasm, pain post procedure, rheumatological, and other. Addendum 3 contains adjudication information from the reviewers regarding the relationship of the SAE to the stem cells or other biologic agent used. Fourteen of the 16 cases (39 %) of the SAEs were categorized as not related or unlikely to be related, while 16/22 (61 %) were adjudicated by one or more reviewer as possibly related. None of the SAEs were considered to be likely or definitely related to the stem cells or other biologic agent.
Logistic regression modeling revealed that patients in both the AD and CE groups were more likely to report an AE than in the SD group; ORs = 1.64 (95 % CI; 1.03, 2.61) and 1.68 (95 % CI; 1.11, 2.54), respectively (Table 5). Further analysis showed that, compared to the SD group, the increase in AE rate was largely attributable to post-procedure pain in the AD group, and pain due to DJD in the CE group (Figs. 4 and 5). A longer follow-up period, older age, and female gender increased the risk of reporting an AE. SAEs were more common in patients with a longer follow-up period and of older age [OR = 1.51 (95 % CI; 1.37, 1.67) and 1.03 (95 % CI; 1, 1.06), respectively]. Patients treated for spinal conditions were more likely to report any AE in comparison with patients undergoing knee procedures [OR = 2.17 (95 % CI; 1.13, 4.15)].

Incidence of the most common adverse event categories, per 100 person-years (PY). SD = same-day bone marrow concentrate; AD = bone marrow concentrate with adipose graft; CE = culture expanded stem cells; DJD = degenerative joint disease

Percentages of total patients reporting adverse events, for the most commonly reported categories. SD = same-day bone marrow concentrate; AD = bone marrow concentrate with adipose graft; CE = culture expanded stem cells; DJD = degenerative joint disease
Discussion
In the present study we generally observed low rates of reported AEs among patients treated with MSC procedures, and substantially lower rates of serious or treatment-related AEs. The finding that the majority of AEs were post-procedure pain or pain due to DJD that pre-existed the treatment was not surprising, and consistent with the progressive nature of the treated disorders.
While there have been several publications that have described the safety and efficacy of bone marrow derived stem cell therapies for orthopaedic applications [1, 7, 9–13, 15], to our knowledge the current investigation is the most comprehensive report of its kind, following the largest population for the longest time, and incorporating an analysis of the relative safety of several different approaches. Our findings are consistent with prior investigations demonstrating a favorable safety profile for the percutaneous use of BMC and MSC injections for the treatment of orthopaedic conditions of the peripheral and axial joints and surrounding tissues [7, 9, 13, 14]. The SAE rates observed in our study were substantially lower than those reported for more invasive orthopaedic surgical procedures [22]. As an example, the SAE rate for total knee arthroplasty among 260 patients at three months follow-up was 6 % [22]. In comparison, there were 13 possibly related SAEs in the present study among 2372 patients, approximately 0.55 %, and only four of these SAEs (0.17 %) were deemed definitely related to the procedure. While SAEs related to stem cell injections can and do occur, prior authors have indicated that the rate is not greater than that observed with other types of intra-articular injections, such as hyaluronic acid injections [23]. The findings in the present investigation reinforce this conclusion.
The differences observed in the AE rates between the treatment groups were not directly attributed to the treatment but rather to symptoms of progressive degenerative disease. Thus, the group that was tracked for the longest time (the culture expanded [CE] group) also had the highest incidence of AEs resulting from worsening of the treated condition over time. This observation is consistent with the natural history of painful degenerative joint disease [24, 25]. Further, the AEs reported in the first months of follow-up differ from those reported after several years of follow-up. For example, treatment-related AEs, including post-procedural pain, are more likely to be reported in the earliest few weeks after treatment; while unrelated or more serious AEs, such as neoplastic and cardiovascular events, are more likely to be reported after several years of follow-up (i.e., as patients age). The higher rate of AEs in the adipose graft (AD) BMC group versus the BMC only group (SD) was largely attributed to post-procedural pain. This difference may be explained by the pro-inflammatory effects of residual adipose oil in the injectate [26].
Of the seven reported cases of neoplasm among the registry patients, none occurred at the site of implantation despite all injections being confirmed with imaging guidance. Given the number and age of the patients followed in the registry, and the amount of time that the patients were followed, some cases of cancer were expected. According to the National Cancer Institute, the annual incidence of cancer in the U.S. population in 2011 was 0.44 % (438 cases per 100,000 individuals), and 0.78 % in adults 50–64 years [27]. In contrast, we observed a lower annual cancer rate (0.14 %) among our registry participants. These findings are consistent with previous reports indicating no increased risk of tumor formation following BMC injections or treatment with culture-expanded MSCs [9, 11, 13, 15].
Older age and longer follow-up times increased the risk of reporting of both AEs and SAEs. These findings are explained both by the fact that morbidity increases with age [28], and that older patients are more likely to report adverse events after orthopaedic procedures [29]. A gender effect was also observed, in that women were more likely than men to report AEs. While the nature of the registry data makes it difficult to determine the reason for this disparity, previous authors have noted that women are more likely to report post-operative pain after arthroscopic procedures [30]. Patients who underwent treatment for degenerative joint and disc changes in the spine also had a higher rate of AE reporting, including AEs related to the treatment. Most of the reports in this group were of pain due to degenerative joint disease and post-procedural pain. While the explanation for this observation is not readily apparent; it could be due to the nature of the treated condition or it could be entirely due to differences in treatment efficacy. Further study would be required to provide more meaningful insight.
The results of the SAE adjudication by the attending physician and the panel of independent and blinded reviewers indicated good agreement on the categorization of pre-existing conditions, with majority agreement on 34 of 36 SAEs. One of the cases in which a minority of reviewers judged an SAE to be related concerned a neoplasm that a single reviewer opined was definitely related to the mechanics of the draw or re-implant injection procedure (the other five reviewers judged the relationship to be unlikely or not related). The SAE concerned a patient who was diagnosed with aggressive stomach cancer three weeks following a knee BMC injection, and who died from the disease at approximately two months following the injection. The protocol of the blinded adjudication process made impossible any follow up with the reviewer for an explanation as to why he or she believed that the stomach cancer, which likely pre-existed the procedure in nearly the same state as it was in three weeks following the procedure, was definitely related.
Another SAE, consisting of severe post-procedure swelling, was judged by two reviewers as definitely needle trauma related, and two reviewers judged the condition as definitely caused by the stem cells or other injectates. A rheumatologic condition was deemed to be definitely related to an injection by two reviewers. In that case, the patient presented with severe knee swelling after a pre-injection procedure with hypertonic dextrose. The joint was drained and found to be purulent, but gram stain and culture were negative. Ultimately synovial fluid crystalline structures were revealed and a diagnosis of gout was made. Because of the pre-injection complication the patient did not undergo the stem cell injection.
An SAE following treatment of a degenerated and painful intervertebral disc was judged to be to be definitely related to the trauma of the stem cell injection by two reviewers. In that case, at approximately eight months post-procedure, the patient sustained an acute disc herniation at the injected level. Three of the reviewers considered the SAE to be possibly related and one determined that it was unlikely to be related to the injection. It is certainly plausible that the needle trauma could have resulted in injury to the disk annulus, resulting in structural compromise and the latent herniation.
The strengths of the current study are its large patient population, the fact that data was collected from multiple centers, that SAEs were adjudicated by multiple independent and blinded reviewers, that AE/SAE rates of multiple treatment types are compared, and that unlike prior large studies all AEs were reported and classified. The main weaknesses of the current research are that it is based on data accessed from a treatment registry. Thus, there is no control group with which the frequency and type of observed illnesses could be compared. Further, the majority of AEs were patient reported. Despite the fact that repeated efforts were made to contact non-responders and all treating physicians were encouraged to report any possible complications while patients were under their care, it is possible that adverse events were under-reported to some degree.
Conclusion
To our knowledge, the present investigation is the first report to compare the clinical safety of different bone marrow derived stem cell therapies to treat orthopedic diseases and the first multi-site, large scale report of all AEs in stem cell treated orthopaedic patients. We found that the lowest rate of adverse events was among those patients receiving BMC injections alone, but the higher rate of AEs for BMC plus adipose and cultured cells was readily explained by the nature of the therapy or the longer follow-up. There was no clinical evidence to suggest that treatment with MSCs of any type in this study increased the risk of neoplasm. Although efficacy is best demonstrated with randomized controlled clinical trials, it is reasonable to conclude that the results of the present study add to the existing body of evidence showing the safety of MSC based therapies for orthopaedic conditions.
Change history
06 November 2017
The published online version contains mistake in Acknowledgment Section. The author name "Steve Gorin, M.D." should have been "Steven Gorin, D.O, M.S.Ed.".
References
Hernigou P, Beaujean F (1997) Pseudarthrosis treated by percutaneous autologous bone marrow graft. Rev Chir Orthop Reparatrice Appar Mot 83(6):495
Centeno CJSJ, Cheever M, Freeman M, Robinson B et al (2011) A case series of percutaneous treatment of non-union fractures with autologous, culture expanded, bone marrow derived, mesenchymal stem cells and platelet lysate. J Bioengineer & Biomedical Sci S2:007. doi:10.4172/2155-9538.S2-007
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284(5411):143–147. doi:10.1126/science.284.5411.143
Cvetkovic VJ, Najdanovic JG, Vukelic-Nikolic MD, Stojanovic S, Najman SJ (2015) Osteogenic potential of in vitro osteo-induced adipose-derived mesenchymal stem cells combined with platelet-rich plasma in an ectopic model. Int Orthop 39(11):2173–2180. doi:10.1007/s00264-015-2929-x
Flouzat-Lachaniette CH, Heyberger C, Bouthors C, Roubineau F, Chevallier N, Rouard H, Hernigou P (2015) Osteogenic progenitors in bone marrow aspirates have clinical potential for tibial non-unions healing in diabetic patients. Int Orthop. doi:10.1007/s00264-015-3046-6
Lee GR, Wintrobe MM (1993) Wintrobe's clinical hematology, vol 9. Lea & Febiger, Philadelphia
Centeno CJ, Freeman MD (2014) Percutaneous injection of autologous, culture-expanded mesenchymal stem cells into carpometacarpal hand joints: a case series with an untreated comparison group. Wien Med Wochenschr 164(5):83–87. doi:10.1007/s10354-013-0222-4
Centeno CJ, Pitts J, Al-Sayegh H, Freeman MD (2015) Anterior cruciate ligament tears treated with percutaneous injection of autologous bone marrow nucleated cells: a case series. J Pain Res 8:437–447. doi:10.2147/JPR.S86244
Centeno CJ, Schultz JR, Cheever M, Freeman M, Faulkner S, Robinson B, Hanson R (2011) Safety and complications reporting update on the re-implantation of culture-expanded mesenchymal stem cells using autologous platelet lysate technique. Curr Stem Cell Res Ther 6(4):368–378
Hernigou P, Beaujean F (2002) Treatment of osteonecrosis with autologous bone marrow grafting. Clin Orthop Relat Res 405(405):14–23. doi:10.1097/00003086-200212000-00003
Hernigou P, Poignard A, Beaujean F, Rouard H (2005) Percutaneous autologous bone-marrow grafting for nonunions. Influence of the number and concentration of progenitor cells. J Bone Joint Surg (Am Vol) 87(7):1430. doi:10.2106/JBJS.D.02215
Hernigou P, Poignard A, Zilber S, Rouard H (2009) Cell therapy of hip osteonecrosis with autologous bone marrow grafting. Ind J Orthop 43(1):40–45. doi:10.4103/0019-5413.45322
Centeno C, Pitts J, Al-Sayegh H, Freeman M (2014) Efficacy of autologous bone marrow concentrate for knee osteoarthritis with and without adipose graft. BioMed Res Int 2014:370621–370629. doi:10.1155/2014/370621
Centeno CJ, Schultz JR, Cheever M, Robinson B, Freeman M, Marasco W (2010) Safety and complications reporting on the re-implantation of culture-expanded mesenchymal stem cells using autologous platelet lysate technique. Curr Stem Cell Res Ther 5(1):81
Hernigou P, Homma Y, Flouzat-Lachaniette C-H, Poignard A, Chevallier N, Rouard H (2013) Cancer risk is not increased in patients treated for orthopaedic diseases with autologous bone marrow cell concentrate. J Bone Joint Surg (Am Vol) 95(24):2215. doi:10.2106/JBJS.M.00261
Centeno CJ, Busse D, Kisiday J, Keohan C, Freeman M, Karli D (2008) Regeneration of meniscus cartilage in a knee treated with percutaneously implanted autologous mesenchymal stem cells. Med Hypotheses 71(6):900–908. doi:10.1016/j.mehy.2008.06.042
Wyles CC, Houdek MT, Wyles SP, Wagner ER, Behfar A, Sierra RJ (2015) Differential cytotoxicity of corticosteroids on human mesenchymal stem cells. Clin Orthop Relat Res 473(3):1155–1164. doi:10.1007/s11999-014-3925-y
Rabago D, Kijowski R, Woods M, Patterson JJ, Mundt M, Zgierska A, Grettie J, Lyftogt J, Fortney L (2013) Association between disease-specific quality of life and magnetic resonance imaging outcomes in a clinical trial of prolotherapy for knee osteoarthritis. Arch Phys Med Rehabil 94(11):2075. doi:10.1016/j.apmr.2013.06.025
Mautner K, Malanga GA, Smith J, Shiple B, Ibrahim V, Sampson S, Bowen JE (2015) A call for a standard classification system for future biologic research: the rationale for new PRP nomenclature. Pm r 7(4 Suppl):S53–S59. doi:10.1016/j.pmrj.2015.02.005
Mashaw R (2007) OHRP issues new guidance on unanticipated problems, adverse events. Adverse Event Reporting News 4(2):1
SAS Institute (2014) SII (issued 2014) SAS software version 9.4. SAS Institute, Cary, NC
Kirschner S, Lützner J, Günther K-P, Eberlein-Gonska M, Krummenauer F (2010) Adverse events in total knee arthroplasty: results of a physician independent survey in 260 patients. Patient safety in surgery 4(1):12. doi:10.1186/1754-9493-4-12
Vangsness JCT, Farr NJ, Boyd J, Dellaero DT, Mills CR, LeRoux-Williams M (2014) Adult human mesenchymal stem cells delivered via intra-articular injection to the knee following partial medial meniscectomy: a randomized, double-blind, controlled study. J Bone Joint Surg (Am Vol) 96(2):90. doi:10.2106/JBJS.M.00058
Cicuttini FM, Jones G, Forbes A, Wluka AE (2004) Rate of cartilage loss at two years predicts subsequent total knee arthroplasty: a prospective study. Ann Rheum Dis 63(9):1124–1127. doi:10.1136/ard.2004.021253
Woollard JD, Gil AB, Sparto P, Kwoh CK, Piva SR, Farrokhi S, Powers CM, Fitzgerald GK (2011) Change in knee cartilage volume in individuals completing a therapeutic exercise program for knee osteoarthritis. J Orthop Sports Phys Ther 41(10):708–722. doi:10.2519/jospt.2011.3633
Tynan GA, Hearnden CH, Oleszycka E, Lyons CL, Coutts G, O'Connell J, Corrigan MA, Lynch L, Campbell M, Callanan JJ, Mok KH, Geoghegan J, O'Farrelly C, Allan SM, Roche HM, O'Shea DB, Lavelle EC (2014) Endogenous oils derived from human adipocytes are potent adjuvants that promote IL-1α-dependent inflammation. Diabetes 63(6):2037
SEER (2014) Fast stats: compare statistics by data type. http://seer.cancer.gov/faststats/
Piccirillo JF, Vlahiotis A, Barrett LB, Flood KL, Spitznagel EL, Steyerberg EW (2008) The changing prevalence of comorbidity across the age spectrum. Crit Rev Oncol Hematol 67(2):124–132. doi:10.1016/j.critrevonc.2008.01.013
Koenig K, Huddleston RJI, Huddleston H, Maloney WJ, Goodman SB (2012) Advanced age and comorbidity increase the risk for adverse events after revision total hip arthroplasty. J Arthroplast 27(7):1402–1407. doi:10.1016/j.arth.2011.11.013
Rosseland LA, Stubhaug A (2004) Gender is a confounding factor in pain trials: women report more pain than men after arthroscopic surgery. Pain 112(3):248–253. doi:10.1016/j.pain.2004.08.028
Acknowledgments
Steve Gorin, M.D. and R. Amadeus Mason M.D. for acting as independent adjudicators for SAEs.
Author information
Authors and Affiliations
Corresponding author
Additional information
A correction to this article is available online at https://doi.org/10.1007/s00264-017-3680-2.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplement 1
(DOCX 14 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Centeno, C.J., Al-Sayegh, H., Freeman, M.D. et al. A multi-center analysis of adverse events among two thousand, three hundred and seventy two adult patients undergoing adult autologous stem cell therapy for orthopaedic conditions. International Orthopaedics (SICOT) 40, 1755–1765 (2016). https://doi.org/10.1007/s00264-016-3162-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00264-016-3162-y