
Abstract
Anti-CTLA-4 antibodies faced challenges due to frequent adverse events and limited efficacy, which spurred the exploration of next-generation CTLA-4 therapeutics to balance regulatory T cells (Tregs) depletion and CD8 T cells activation. CCR8, identified primarily on tumor-infiltrating Tregs, has become a target of interest due to the anti-tumor effects demonstrated by CCR8 antibody-mediated Tregs depletion. Single-cell RNA sequencing analysis reveals that CCR8-positive Tregs constitute a small subset, with concurrent expression of CCR8 and CTLA-4. Consequently, we proposed a novel bispecific antibody targeting CCR8 and CTLA-4 that had the potential to enhance T cell activation while selectively depleting intratumor Tregs. The candidate molecule 2MW4691 was developed in a tetravalent symmetric format, maintaining a strong binding affinity for CCR8 while exhibiting relatively weaker CTLA-4 binding. This selective binding ability allowed 2MW4691 to target and deplete tumor-infiltrating Tregs with higher specificity. In vitro assays verified the antibody’s capacity for antibody-dependent cellular cytotoxicity (ADCC) to Tregs with high level of CTLA-4 expression, but not CD8 T cells with relatively low level of CTLA-4 on cell surface. Also, 2MW4691 inhibited the CTLA-4 pathway and enhanced T cell activation. The in vivo therapeutic efficacy of 2MW4691 was further demonstrated using hCCR8 or hCTLA-4 humanized mouse models and hCCR8/hCTLA-4 double knock-in mouse models. In cynomolgus monkeys, 2MW4691 was well-tolerated, exhibited the anticipated pharmacokinetic profile, and had a minimal impact on the peripheral T cell population. The promising preclinical results supported the further evaluation of 2MW4691 as a next-generation Treg-based therapeutics in clinical trials.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Immunotherapy has revolutionized cancer treatment. Ipilimumab, the first FDA-approved immunotherapeutics in 2011, commenced a new epoch of cancer immunotherapy [1, 2]. Immune checkpoint inhibitors (ICB), such as CTLA-4 and PD-1/PD-L1, serve as crucial negative modulators of T cell function. Inhibition of such ICBs brought unprecedented success across various cancers. In head-to-head comparison between anti-CTLA-4 and anti-PD-1 therapies, the former yields lower response rates and a higher incidence of immunotherapy-related adverse effects (irAEs) than the latter [3,4,5]. That is why anti-PD-1/PD-L1 rapidly expanded indications but anti-CTLA-4 was approved only in melanoma as a monotherapy or as an agent combined with anti-PD1/PD-L1 in clinical [6]. Despite this, long-term benefits have been observed in patients treated with anti-CTLA-4 therapies [7]. A landmark analysis involving 1861 patients treated with ipilimumab indicated a survival plateau commencing around the 3rd year, extending up to a decade [8]. This underscores the enduring value of anti-CTLA-4 as a component of cancer immunotherapy, urging the development of safer and more efficacious CTLA-4-targeted therapies.
Two mechanisms of action of anti-CTLA-4 were proposed, one of which is the blockade of CTLA-4/B7 interaction to release inhibited conventional T cells [9]. CD28, a costimulatory molecule essential for T cell activation, competes with CTLA-4 for the same ligands, B7.1 and B7.2. Given that CTLA-4 has a higher affinity for these ligands, it outcompetes CD28, inhibiting the activation signal of T cells [10, 11]. The second mechanism involves the selective depletion of Tregs, which constitutively express CTLA-4, in contrast to the transient expression on activated conventional T cells. While debates persist regarding the precise mechanisms of anti-CTLA-4, there is a consensus that the maximal therapeutic efficacy of anti-CTLA-4 antibodies is achieved by preserving both the antagonist activity and the Treg depletion function [12,13,14].
Treg cells play a vital role in maintaining immune homeostasis and self-tolerance [15]. Treg cells are also abundantly present in tumors and exert immune suppressive function. Systemic removal of Tregs can evoke robust tumor immunity but also severe unwanted autoimmunity and immunopathology in normal tissue [16]. Another significant challenge in targeting Tregs is that many markers present on Tregs are expressed in activated conventional cells as well, such as CD25. Recently CCR8 was identified as a specific marker of intratumor Tregs [17,18,19]. CCR8 is selectively expressed in the tumor-infiltrating Tregs, but not in conventional T cells, and is almost undetectable in periphery Tregs. CCR8 was upregulated on Tregs in multiple cancers, including breast, lung, colorectal cancers, and so on [20]. The expression pattern positions it as an ideal target for Treg depletion of anti-tumor immunotherapy [21, 22].
It is reported that CCR8 was expressed in a highly active and strongly suppressive subgroup of intratumor Tregs [18]. Research conducted by Helena Van Damme et al. demonstrated that CCR8 is notably present on activated and suppressive tumor-infiltrating Tregs in both human and mouse tumors, with minimal expression in peripheral Tregs [20]. Dawei Sun et al. also observed pronounced expression of CCR8 in tumor-infiltrating Tregs compared to peripheral Tregs [23]. However, the prevalence of CCR8 + Tregs varies across different tumor types, with single-cell RNA sequencing (scRNA-seq) showing a range from 6.8% to 38% [17]. This positive ratio indicates the potential limited efficacy of the CCR8-targeting antibody. Several CCR8 antibodies have entered clinical trials, such as BMS-986340 (NCT05537740), and GS-1811 (NCT05007782). Patient selection for these treatments is challenging due to the dramatic variation in CCR8 + Treg levels among individuals and the minimal expression of CCR8 on peripheral cells.
In this research, we analyzed the subpopulation of Treg in the tumor using scRNA-seq and identified that the CCR8 + subset represents highly active Tregs along with elevated CTLA-4 expression. Consequently, we designed a bispecific antibody, 2MW4691, simultaneously targeting CCR8 and CTLA-4 with differentiated binding affinity aiming to deplete intratumor Tregs with high potency while activating T cells effectively. Our results demonstrated that 2MW4691 had superior Treg depletion ability without adverse impact on CD8 T cells in the periphery. 2MW4691 also could effectively inhibit tumor proliferation in vivo. Furthermore, 2MW4691 exhibited favorable stability and safety in non-human primates (NHPs). Our work provides a comprehensive insight into the Treg subpopulation and introduces a novel approach to Treg depletion therapy.
Materials and methods
Cells
HEK293-6E cells were obtained from the American Type Culture Collection (ATCC) and fed with FreeStyle™ F17 medium (Invitrogen, USA). HEK293-6E (FUT8-/-) cells were generated following the instructions of a FUT8 Knock Out Kit (Genloci Biotechnologies Inc. China) which based on CRISPR/Cas9 technology. The guide RNA sequence was as follows: GAATGAGCATAATCCAACGCCAGG. A monoclonal HEK293-6E (FUT8-/-) cell line was subsequently established through limiting dilution. Jurkat cells with stable NFAT-luc/hFcγRIIIa or NFAT-luc/mFcγRIV expression were purchased from Genomeditech Co., Ltd (Shanghai, China). Jurkat-NFAT-luc/hFcγRIIIa cells were cultured in RPMI 1640 medium (Gbico, USA) supplemented with 10% FBS (Gibco), 0.4 mg/mL G418 (Gibco), and 0.75 μg/mL puromycin (Gibco). Jurkat-NFAT-luc/mFcγRIV cells were cultured in RMPI 1640 containing 10% FBS, 3.5 μg/mL Blasticidin (Gbico), and 0.75 μg/mL puromycin. CHOK1-hCCR8 and CHOK1-hCTLA-4 were maintained in Ham’s F-12 K medium (Gibco) with 10% FBS and 4 mg/mL puromycin. Jurkat-cynoCCR8 cells were fed with RPMI 1640 containing 10% FBS, and HEK293T-hCCR8 cells were cultured in DMEM containing 10% FBS and 1 μg/mL puromycin. PBMCs were purchased from Schbio Biotechnology Co, Ltd. (Shanghai, China). The culture condition of all cells was 37 °C with 5% CO2. All cell lines were routinely tested using a Mycoplasmacontamination kit (R&D Systems, USA).
Animals
Six-to-eight-week female hCCR8 knock-in C57BL/6 mice, hCTLA-4 knock-in C57BL/6 mice, and hCCR8/hCTLA-4 double knock-in C57BL/6 mice were procured from Biocytogen Pharmaceuticals Co., Ltd (Beijing, China).
Single-cell RNA-seq data
The data for non-small cell lung adenocarcinoma and colorectal adenocarcinoma patients were sourced from GSE139555, wherein the author sequenced 330 million mRNA transcripts from immune cells. For this study, tumor samples excised during surgery were selected for analysis. The detailed statistics information and patient information were recorded in Wu TD, et al.’s paper [24]. Colorectal cancer patients data from GSE146771 was generated from Smart-seq2 platform, including clinical characteristics of 10 CRC patients involved in this study [25]. Data from triple-negative breast cancer (TNBC) patients, available in GSE110686, were derived from single-cell suspensions prepared from two individual TNBC primary tumor samples.
The above scRNA-seq data underwent stringent quality control, with the exclusion of low-quality cells based on the number of genes expressed and the proportion of mitochondrial gene expression. Batch correction was performed across different patients by using harmony in Seurat (version 4.3.0).
Plasmids construction and antibody expression
Heavy (H) and Light (L) chain genes of antibodies were cloned into eukaryotic expression pTT5 vectors (Addgene, USA) and transfected into HEK293-6E cells with polyethyleneimine (PEI) (Polysciences, USA). Seven days later, supernatants containing target antibodies were centrifugated and purified by Protein A affinity chromatography. Ipilimumab and HBM4003 were also produced in-home using the above procedure. FUT8 gene knock-out HEK293-6E cells were adopted to manufacture antibodies without fucosylation. The transfection of antibody plasmids was carried out using the same protocols as for the transfection of plasmids into wild-type HEK293-6E cells.
Binding activity characterization by flow cytometry
Jurkat-hCCR8, Jurkat-cynoCCR8, CHOK1-hCCR8, or CHOK1-hCTLA-4 cells were collected and washed with fluorescence-activated cell sorting (FACS) buffer (1% FBS in PBS) and then incubated with serial dilutions of antibodies at 4 °C for 1 h. After three times washing, cells were stained with the secondary antibody APC goat anti-human IgG Fc antibody (Jackson ImmunoResearch, USA) for 1 h. Finally, the fluorescence intensity was detected by the iQUE® Flow Cytometer.
Blocking activity analysis by flow cytometry
CHOK1-hCTLA-4 cells were digested and washed twice with FACS buffer. The cells were then incubated with biotin-labeled human CD80 (ECD, Fc tag) or CD86 (ECD, his tag) in the presence of a range of concentrations of bispecific antibodies or the parental anti-CTLA-4 antibody ipilimumab. After a 1-h reaction at 4 °C, the cells were rinsed twice and stained by APC Streptavidin (BioLegend, USA). An hour later, cells were resuspended and analyzed by the corresponding channel of the flow cytometer.
ADCC reporter bioassay
ADCC reporter bioassay was briefly a bioluminescence assay catalyzed by luciferase to quantify the ADCC activity of antibodies. Jurkat-NFAT-luc/hFcγRIIIa or Jurkat-NFAT-luc/mFcγRIV cells were used as effector cells, while CHOK1-hCCR8 and CHOK1-hCTLA-4 cells were as target cells. The protocol was as follows: target cells were harvested and suspended in RPMI 1640 medium at a density of 0.5 × 106/mL, and 2 × 104 cells were seeded into a white opaque 96-well plate. Serial dilutions of antibody were added to the plate for 30-min incubation. Simultaneously, effector cells were collected and adjusted to 3.75 × 106/mL. 1.5 × 105 effector cells were mixed with target cells at a ratio of 7.5:1. After 6-h co-culture at 37 °C 5% CO2 incubator, 100 μL Bio-Lite detection substrate (Vazyme, China) was pipetted into the plate to react at least 3 min. Relative luminescence units (RLU) were read at SpectraMax M5e Microplate Reader (Molecular Devices, USA).
PBMC ADCC bioassay
To determine the ADCC activity of CCR8 monoclonal antibodies, HEK293T-hCCR8 and PBMCs were utilized as target cells and effector cells, respectively. In the presence of serially diluted CCR8 monoclonal antibodies, PBMCs were incubated with HEK293T-hCCR8 cells at a ratio of 15:1 at 37 °C for 5 h. Then, the LDH-Glo Cytotoxicity Assay Kit (Promega, USA) was adopted to measure the luminescence of the supernatants.
Tregs separated from PBMCs by Regulation T cell Isolation Kit (Stemcell Technologies, USA), and CD8 T cells isolated from PBMCs by Human CD8 T cell Isolation Kit (Stemcell Technologies) were also used as target cells. Tregs or CD8 T cells were cultured in RPMI 1640 medium containing 50 ng/mL IL-2 (Peprotech, USA) and CD3/CD28 activator (Gibco, USA) for three days. The day before the assay, frozen PBMCs were thawed and pre-treated with IL-2 overnight. The next day, Tregs or CD8 T cells were harvested and labeled with Calcein AM (Invitrogen, USA). The stained cells were incubated with 2MW4691 and control antibodies for 0.5 h at 37 °C. PBMCs were added to the plate at a ratio of 50:1 and cocultured for 5 h. The supernatants were taken out to measure Calcein AM release.
SEB stimulation assay
Frozen PBMCs from two donors were carefully thawed and adjusted with RPMI 1640 medium to a density of 2 × 106/mL. 1 × 105 PBMCs were pre-cocultured with 2MW4691 (50, 10, 2, 0.4 nM) in 96-well plate for 30 min. Then, 400 ng/mL SEB was added for an additional 4 days culture at 37 °C, 5% CO2. Supernatants were gathered to measure IL-2 concentration based on HTRF® technology (PerkinElmer, Inc., USA).
In vivo activity studies
To evaluate the efficacy of 2MW4691, 5 × 105 MC38 cells suspended in 100 μL PBS were subcutaneously transplanted at the right flank of C57BL/6-hCCR8 or C57BL/6- hCTLA-4 mice. The mice were randomized into two groups with seven mice in each group when the tumor size was from 80 to 100 mm3. C57BL/6-hCCR8 mice were treated with 5 mg/kg isotype_AF or 6.66 mg/kg 2MW4691, and C57BL/6-hCTLA-4 mice were treated with 1 mg/kg isotype_AF or 1.33 mg/kg 2MW4691. Antibodies were intraperitoneally administrated weekly for 3 weeks. Subsequently, hCCR8 and hCTLA-4 double transgenic C57BL/6 mice were applied to further assess the in vivo efficacy of 2MW4691. The treatment groups were as follows: 0.3 mg/kg isotype_AF, 0.4 mg/kg 2MW4691, 0.3 mg/kg B9B11_AF, 0.3 mg/kg ipilimumab, and 0.15 mg/kg B9B11_AF plus 0.15 mg/kg ipilimumab. The intraperitoneal administration was conducted three times on days 6, 13, and 16 after tumor inoculation. Finally, C57BL/6-hCCR8/hCTLA-4 mice were recruited to compare the in vivo efficacy of 2MW4691 to HBM4003. The mice were intraperitoneally treated with 0.3 mg/kg isotype_AF, 0.4 mg/kg 2MW4691, or 0.16 mg/kg HBM4003 twice a week for 2 weeks.
Tumor size and mouse body weight were measured and recorded three times weekly. Tumor volumes were figured out as the following fundamental equation: tumor volume (mm3) = (length) × (width)2 × 0.5. Tumor growth inhibition (TGI) was calculated according to the formula: TGI (%) = (1 – TVt/TVc) × 100%, where TVt and TVc were, respectively, the average tumor volume of the treated and control group. The mice were sacrificed when the average tumor size of that group was more than 2000 mm3.
Pharmacokinetic and toxicity study
One female and one male cynomolgus monkeys were recruited to conduct the single-dose pharmacokinetic study and toxicity in NHPs. The monkeys received a dosage of 3 mg/kg 2MW4691 through a 0.5-h intravenous infusion. For the pharmacokinetics study, serums were collected immediately after infusion, and at 2, 8, 24, 48, 72, 96, 120, 168, 336, and 504-h post-administration. For the toxicity study, blood samples were collected before treatment and at 24, 72, and 168 h after transfusion. PBMCs were isolated from peripheral blood and then stained with specific markers of CD4 T cells, CD8 T cells, and Tregs for population analysis by FACS. Besides, the routine clinical observation, ethology, weight, and food consumption were monitored regularly.
The concentration of the target antibody in serum was measured using two different ELISA methods. Method A involved coating an ELISA plate with anti-human Fab (Sigma-Aldrich, USA) to capture serially diluted positive serum. This was then detected using HRP-labeled goat anti-human Fc (Jackson ImmunoResearch, USA). Method B comprised coating ELISA plate with anti-human Fab, adding serial dilutions of positive serum to the plate, adding biotin-labeled hCTLA-4 (ECD, his tag), detecting the reaction with streptavidin HRP (Thermo Fisher, USA). Concurrently, a standard binding response-concentration curve was established to calculate the serum antibody concentration.
Statistical analysis
Statistical analysis was performed using GraphPad 8.0 software. Unpaired Student’s t-test was applied to determine the statistical significance between the two groups. One-way ANOVA with Dunnett’s multiple comparisons test was performed when comparing more than two groups. P < 0.05, P < 0.01, P < 0.001 was noted as *, **, *** respectively, and P < 0.05 was considered statistically significant.
Results
Analysis of CCR8 and CTLA-4 expression profiles
There is a significant heterogeneity within the Treg subset, as indicated by numerous studies [26,27,28]. Utilizing single-cell RNA sequencing data from the GEO database, this study examined the expression of CCR8 and CTLA-4 genes in tumor-infiltrating Tregs from NSCLC patients. The dataset GSE139555, comprising tumor samples from six NSCLC patients, was chosen for this purpose. Immune cells within the tumor were isolated using FACS, specifically targeting CD45 and CD3 markers (Fig. 1A). Following quality control and filtration processes, seven major cell clusters were identified (Fig. 1B), with the characteristics of each cluster detailed in a dot plot (Fig. S1A). Expression levels of FOXP3, CCR8, and CTLA-4 are depicted in Fig. 1C–D. FOXP3 was predominantly expressed in Tregs, while CCR8 was found in only a small subset of these cells. CTLA-4 showed a broader expression pattern, not only in Tregs but also in exhausted T cells and some NK cells. Notably, CCR8 expression closely overlapped with that of FOXP3 and CTLA-4. Further analysis of Treg cells led to the identification of six subpopulations, with CCR8 being expressed in highly suppressive subpopulations alongside elevated levels of Treg suppressive genes such as LAG3, TNFRSF9, and TNFRSF4 [29, 30]. Additionally, the analysis revealed a consistency between the expression of CCR8 and the cytokines IL-10 and TGFβ, both of which play essential roles in Treg-mediated immune suppression (Fig. S1B). The ratios of CCR8 + and CTLA-4 + Tregs were calculated (Table S1), revealing that 56% of Tregs were CTLA-4 positive, whereas only 15% were CCR8 positive.

CCR8 and CTLA-4 expression levels in NSCLC and CRC by scRNA-seq. (A) Schematic overview of GSE139555 study design. Six NSCLC patients were included in the single-cell sequencing analysis. (B) Uniform manifold approximation and projection (UMAP) plots of NSCLC patient cells showing seven clusters. (C-E) UMAP plots show FOXP3 (C), CCR8 (D), and CTLA-4 (E) expression in NSCLC. (F) Schematic overview of the CRC scRNA-seq design. The data sources are from GSE146771. (G) UMAP plots of 10 CRC patient cells showing nine clusters. (H-J) UMAP plots show FOXP3 (H), CCR8 (I), and CTLA-4 (J) expression in CRC
To further corroborate our findings, we analyzed a dataset comprising transcriptomes of tumor cells from 10 treatment-naïve CRC patients using scRNA-seq through the Smart-seq2 platform [25]. Unsupervised clustering revealed nine distinct cell clusters (Fig. 1F, G), with the characteristics of each cluster detailed in a dot plot (Fig. S2A). Single-cell data demonstrated the expression of FOXP3, CCR8, and CTLA-4. Notably, CCR8 exhibited surprisingly high expression in Tregs, showing overlap with FOXP3 and partial overlap with CTLA-4 (Fig. 1H–J). Tregs were further categorized into four subpopulations, and the gene expression dataset for each was examined. CCR8 expression was consistent with suppressive gene profiles like LAG3, TNFRSF9, TNFRSF4, as well as IL-10 and TGFβ (Fig. S2B). The ratio of CCR8-positive Tregs was 88%, with CCR8 + CTLA-4 + Tregs accounting for 86% of the population. This suggests a highly suppressive tumor microenvironment in CRC (Table S1).
These findings validate the co-expression of CCR8 and CTLA-4 at the mRNA level in tumor-infiltrating Tregs. The proportion of CCR8-positive Tregs shows significant variation, ranging from 15 to 88%, depending on the tumor type. Meanwhile, CTLA-4 was found to be broadly expressed. The gene expression dataset highlights the heterogeneity within Tregs, with CCR8 primarily expressed in a highly suppressive subgroup.
Determination of the format of CCR8/CTLA-4 bispecific antibodies
The gene expression pattern of CCR8 and CTLA-4 within intratumoral Tregs suggests that targeting both CTLA-4 and CCR8 simultaneously could enhance the efficiency of Treg depletion. In light of this, we developed a bispecific antibody that targets CTLA-4 and CCR8, aiming to deplete Tregs that are either single positive for CCR8 or CTLA-4, or positive for both, through ADCC. We produced an in-house CCR8-specific antibody, B9B11, characterized by its high binding ability and ADCC activity. When compared with the 7B16 antibody, also known as GS-1811, which is under clinical trials by Gilead, B9B11 demonstrated comparable binding capability and ADCC activity (Fig. 2A, B). The specificity of B9B11 was confirmed through its nonbinding to unrelated cells, where it exhibited high specificity (Fig. S3A). Importantly, B9B11 was capable of interacting with cynomolgus CCR8, facilitating the extension of experiments to cynomolgus monkeys (Fig. 2C). B9B11 was also shown to effectively block the CCR8 signaling pathway, potentially enhancing anti-tumor efficacy (Fig. S3B). Moreover, B9B11 significantly reduced tumor growth in human CCR8 transgenic mice and exhibited normal pharmacokinetics in cynomolgus monkeys (Fig. S3C, D). These findings advocate for the selection of B9B11 in the construction of the bispecific antibody. Given the extensive research on CTLA-4 as a target, we chose ipilimumab, a well-established anti-CTLA-4 antibody, for the development of the bispecific antibody.

Bispecific antibody design, construction, and screening. (A) Binding ability characterization of CCR8 antibodies was performed using Jurkat-hCCR8 stable cells, which were incubated with serially diluted antibodies and detected by FACS using APC-labeled secondary antibody. (B) ADCC activity of B9B11 was evaluated by PBMC-mediated HEK293T-hCCR8 lysis. PBMCs were incubated with HEK293T-hCCR8 cells (E:T = 15: 1) for 5 h in the presence of CCR8 antibodies, and the reactions were detected by LDH detection reagents. (C) B9B11 binding to cynoCCR8 was evaluated using Jurkat-cynoCCR8 stable cells. Serial dilutions of CCR8 antibodies were incubated with Jurkat-cynoCCR8 and detected by FACS using APC-labeled goat anti-human IgG Fc antibody. (D) The format schematic diagram of CCR8/CTLA-4 bispecific antibodies. A total of eight distinguished bispecific antibody formats were designed and constructed. (E) Comparison of bispecific antibodies binding ability to CHOK1-hCCR8. Threefold dilutions of bispecific antibodies and parent antibody were incubated with CHOK1-hCCR8 cells and detected by FACS using APC-labeled secondary antibody. (F) The binding activity measurement of bispecific antibodies to CHOK1-hCTLA-4. Serially diluted antibodies starting from 100 nM were incubated with CHOK1-hCTLA-4 cells and detected by FACS using APC-labeled secondary antibody. (G) The blocking ability of bispecific antibodies to CHOK1-hCTLA-4 /CD80. 2 μg/mL biotin-labeled hCD80 was incubated with CHOK1-hCTLA-4 cells in the present of serial dilutions of antibodies and detected by FACS using APC streptavidin. (H) The blockade of association of CHOK1-hCTLA-4 with CD86 by bispecific antibodies. CHOK1-hCTLA-4 cells were incubated with 2 μg/mL biotin-labeled hCD86, followed by the addition of serially diluted antibodies and detection by FACS using APC streptavidin
Several strategies were employed to engineer eight distinct bispecific antibodies targeting CCR8 and CTLA-4, as depicted in Fig. 2D. Bispecific antibody (BsAb) formats 1 and 2 utilized CrossMab and knob-into-hole technologies simultaneously, to achieve a configuration with monovalent binding to CTLA-4 and bivalent binding to CCR8 [31]. BsAb format 3 was developed based on the Dual Variable Domain Immunoglobulin (DVD-Ig) platform [32]. Formats 4 and 5 applied CrossMab technology to create a tetravalent symmetric structure, incorporating two anti-CCR8 Fab arms and two anti-CTLA-4 Fab arms. For formats 6, 7, and 8, the N-terminal was the Fab of anti-CCR8, then indirectly or directly connected with the single-chain variable fragment (scFv) of ipilimumab in in-tandem manner. The scFv for anti-CTLA-4 consisted of the heavy and light chain variable domains of ipilimumab connected by a flexible (Gly4Ser)4 linker. To enhance stability and reduce aggregation of the bispecific antibody, an interchain disulfide bond was introduced between positions VH44 and VL100 [33]. All bispecific antibodies were anchored to a human IgG1 scaffold, providing a stable and functional framework for therapeutic application.
CHO-K1 cells overexpressing hCCR8 or hCTLA-4 were utilized to assess the binding and blocking activities of bispecific antibodies. Initially, the binding ability of these antibodies to CHOK1-hCCR8 was determined using flow cytometry. All formats, except format 2, maintained the binding ability of the parental antibody, indicated by the similar half-maximal concentration (EC50) to that of parental B9B11 (Fig. 2E). However, when the anti-CCR8 arms were positioned externally to the anti-CTLA-4 arms, as seen in formats 1, 3, 4, and 8, a more obvious binding response was observed. The binding ability to CHOK1-hCTLA-4 was also measured by flow cytometry, revealing a general reduction in binding potency of the bispecific antibodies compared to the parental ipilimumab (Fig. 2F). Among the eight formats, only formats 7 and 8 preliminarily met the CTLA-4 binding activity requirements for the bispecific antibody. The blocking ability was also characterized, showing that formats 7 and 8 could partially block the interaction between CTLA-4 and its ligands CD80 and CD86 (Fig. 2G, H), a mechanism reported to suppress tumor growth [34]. Consequently, blocking activity is a desirable feature for bispecific antibodies. After a comprehensive analysis of binding and blocking activities, format 7 was selected as the candidate bispecific antibody and named 2MW4691. The depletion of tumor-infiltrating Tregs through ADCC activity is a key mechanism of 2MW4691, which was engineered for enhanced ADCC effect through afucosylation to improve affinity for FcγRIIIa. Prior to in vitro and in vivo evaluation, 2MW4691 was characterized for purity, thermal stability, and affinity through SDS-PAGE, size-exclusion chromatography (SEC), and differential scanning fluorimetry (DSF), confirming its correct molecular weight and satisfactory purity and thermal stability (Fig. S4A–C). It was observed that 2MW4691 dissociated faster than the parental ipilimumab antibody, resulting in a weaker affinity (Fig. S4D). Given its head-to-tail structure, the CHOK1-hCCR8 overexpressing cell line and soluble CTLA-4 protein was employed to test simultaneous binding, with 2MW4691 demonstrating strong simultaneous binding to both targets (Fig. S4E).
2MW4691 was developed to specifically bind to CCR8 and CTLA-4, featuring enhanced ADCC activity while exhibiting reduced binding and blocking activity toward CTLA-4.
In vitro characterization of 2MW4691 by ADCC and SEB stimulation assays
The ADCC activity of 2MW4691 was evaluated using a reporter bioassay with FcγRIIIa-expressing Jurkat cells as effector cells and CHOK1-hCCR8 or CHOK1-hCTLA-4 cells as target cells. It demonstrated potent ADCC activity against CHOK1-hCCR8 cells, similar to the parental B9B11 antibody, with an EC50 value of 0.062 nM for 2MW4691 compared to 0.027 nM for B9B11 (Fig. 3A). The ADCC effect of CTLA-4 binding arms was attenuated due to their C-terminal configuration (data not shown). However, afucosylated Fc fragments with enhanced hFcγRIIIa binding ability could restore their function. As displayed, 2MW4691’s ADCC activity against CHOK1-hCTLA-4 was significantly stronger than that of the parental ipilimumab antibody, which used a normal human IgG1 Fc (Fig. 3B). This suggests that the structural design of 2MW4691 does not impair the interaction between afucosylated hIgG1Fc and hFcγRIIIa. Thus, 2MW4691, with its dual binding to CCR8 and CTLA-4 and enhanced ADCC activity, maximizes the depletion of intratumor Tregs.

In vitro efficacy evaluation of 2MW4691. (A) ADCC effect of 2MW4691 on CHOK1-hCCR8 was performed based on reporter-based bioassay. Jurkat-NFAT-luc/hFcγRIIIa cells were incubated with CHOK1-hCCR8 cells (E:T = 7.5:1) in the presence of serial dilutions of antibodies for 6 h before luminescence detection. (B) ADCC effects of 2MW4691 on CHOK1-hCTLA4. Jurkat-NFAT-luc/hFcγRIIIa cells were stimulated with serially diluted antibodies in the presence of CHOK1-hCTLA-4 cells for 6 h before measuring luminescence. (C) Analysis of CTLA-4 and CCR8 expression on Tregs and CD8 T cells. ScRNA-seq data from the TNBC dataset GSE110686 were collected to analyze the expression pattern of CTLA-4 and CCR8 in the two kinds of cells. (D) RNA sequencing data from the COAD dataset DSE139555 were gathered to analyze the expression of CTLA-4 and CCR8 in Tregs and CD8 T cells. (E) Tregs isolated from PBMCs were activated by CD3/CD28 beads for 72 h and the CTLA-4 and CCR8 expression on Tregs was analyzed by FACS using PE-labeled ipilimumab and B9B11. (F) CTLA-4 and CCR8 expression on CD3/CD28-activated CD8 T cells was analyzed by FACS using PE-labeled ipilimumab and B9B11. (G) Activated Tregs were applied to the PBMC-mediated ADCC assay to evaluate the function 2MW4691. Calcein AM-stained Tregs were cocultured with PBMCs at a ratio of 1:50 in the presence of antibodies for 5 h before measuring Calcein AM release by M5e Microplate Reader. (H) PBMCs were cocultured with activated CD8 T (E:T = 50:1) in the presence of antibodies for 5 h, and cell lysis was characterized by measuring Calcein AM release. (I) The measurements of secretion of IL-2 in SEB stimulation assay. PBMCs (1 × 105/well) from two different donors were stimulated by 400 ng/mL SEB in the presence of antibodies for 96 h. Released IL-2 in the supernatant was measured by HTRF® technology
CTLA-4 is highly expressed in intratumor Tregs but is also found on the surface of activated CD8 T cells. To explore CTLA-4 expression profiles across different immune cells, particularly in Tregs and CD8 T cells, scRNA-seq data from the GEO database were analyzed. Figure 3C, D revealed that CTLA-4 expression was significantly higher in Tregs than in CD8 T cells in both triple-negative breast cancer and colorectal adenocarcinoma, and similar results were obtained in the CCR8 expression analysis.
To validate CTLA-4 expression and assess the ADCC activity of 2MW4691 further, isolated Tregs and CD8 T cells were examined. Tregs, once isolated and activated, showed surface expression of CCR8 and CTLA-4, with CTLA-4 being more prominently expressed (Fig. 3E). CD8 T cells were also isolated and activated to mimic tumor-resident CD8 T cells, revealing minimal expression of CTLA-4 and CCR8 (Fig. 3F), aligning with tumor tissue expression profiles. Calcein AM stained Tregs were cocultured with PBMCs in the presence of 2MW4691 or control antibodies. Compared with the parental monoclonal antibodies, 2MW4691 could lysate more Tregs, which was indicated by more fluorescence intensity (Fig. 3G). This indicated that 2MW4691 significantly enhanced PBMC-mediated cytotoxicity. ADCC assays on CD8 T cells, across various antibody concentrations, showed a slight increase in cytotoxicity with ipilimumab and 2MW4691 compared to the isotype control, but no significant difference between ipilimumab and 2MW4691 (Fig. 3H). This lack of difference was likely due to the low expression of CTLA-4 on activated CD8 T cells, insufficient to trigger significant ADCC activity. CTLA-4 interaction with its ligands provides an inhibitory signal, reducing T cell activity. Blocking the CTLA-4/B7 pathway with an inhibitor can restore T cell effector function. SEB stimulation assay was used to evaluate the T cell activation potential of 2MW4691. PBMCs from various donors, cultured with 2MW4691 and SEB, showed a concentration-dependent increase in IL-2 cytokine secretion (Fig. 3I), highlighting 2MW4691’s ability to significantly enhance T cell activation.
2MW4691 demonstrated strong ADCC activity, suggesting a potent Treg depletion function, significant T cell activation capability, and no apparent cytotoxic effect on activated CD8 T cells.
In vivo efficacy evaluation of 2MW4691
To evaluate the in vivo efficacy of 2MW4691, humanized mice were employed. Theoretically, using CCR8 and CTLA-4 transgenic mice with humanized FcγRs would be more appropriate for characterizing the anti-tumor effects of 2MW4691, considering its mechanisms of action. However, such models are not currently available. Therefore, we performed alternative in vitro experiments to assess the interaction of afucosylated human IgG1Fc with mouse FcγRIV, which is generally considered to be an analog of human FcγRIIIa due to its similar affinities and expression pattern [35]. The results demonstrated that 2MW4691 could induce effective ADCC responses when using the Jurkat-mouse FcγRIV effector cells (Fig. S5). Additionally, afucosylation enhances the binding ability of hIgG1Fc to both mFcγRIV and hFcγRIIIa [36], leading us to believe that the current ADCC data from mice can largely reflect ADCC responses in humans.
For the in vivo efficacy evaluation of 2MW4691, MC38 syngeneic tumor models were employed. Initially, hCCR8 or hCTLA-4 transgenic mice were used to assess the anti-tumor efficacy of targeting CCR8 or CTLA-4 separately. MC38-inoculated hCCR8 knock-in mice were divided into two groups when the average tumor volume reached approximately 100 mm3. They were then treated with 2MW4691 at a dose of 6.66 mg/kg and an isotype control at 5 mg/kg through intraperitoneal injection. On day 17 post-MC38 inoculation, the tumor growth inhibition (TGI) rate for 2MW4691 was found to be 67% (Fig. 4A). This significant difference in anti-tumor efficacy between 2MW4691 and the isotype control highlighted the anti-tumor effect of targeting CCR8 (Student’s t-test, P value: 0.0045). Throughout the treatment period, the mice’s body weight increased steadily without any abnormalities (Fig. 4B). Mice from the isotype and 2MW4691 groups were sacrificed on days 17 and 21 after tumor inoculation, respectively. The tumor tissues were then isolated for photographic documentation (Fig. 4C).

In vivo efficacy determination of 2MW4691. (A) Two groups (n = 7/group) of C57BL/6-hCCR8 mice, bearing MC38 tumor cells, were treated with 6.66 mg/kg 2MW4691 or 5 mg/kg isotype at a frequency of once a week. Tumor size was measured three times a week and shown as mean ± SEM. Student’s t-test was applied and P value was labeled. (B) Mice’s body weight was measured during treatment. (C) The tumor tissue was isolated from mice of the isotype_AF group on day 17 after tumor inoculation and mice of the 2MW4691 group on day 24. (D) C57BL/6-hCTLA-4 mice (n = 6/group) were treated with 1.33 mg/kg 2MW4691 or 1 mg/kg isotype control on days 6, 13, and 20 after MC38 cells inoculation. The tumor volume change was recorded three times a week and shown as mean ± SEM. Student’s t-test was applied and P value was marked. (E) The average mice body weight change of each group was recorded during treatment. (F) Resected tumor tissues were photographed when isotype_AF treated mice were sacrificed on day 21 and 2MW4691 treated mice on day 28. (G) C57BL/6-hCCR8/hCTLA-4 mice (n = 7/group) bearing MC38 cells were intraperitoneally administrated with 2MW4691 (0.4 mg/kg), B9B11_AF (0.3 mg/kg), ipilimumab (0.3 mg/kg), B9B11_AF plus ipilimumab (0.15 + 0.15 mg/kg), and isotype_AF (0.3 mg/kg), respectively. The treatments occurred on days 9, 16, 19 post-tumor transplantation. Tumor volume was measured three times a week and shown as mean ± SEM. (H) Mice’s body weight was measured and recorded during treatment. (I) Statistical analysis of tumor volumes on day 21 after tumor inoculation was determined by one-way ANOVA with Dunnett’s multiple comparisons test. (J) Mice of isotype_AF and B9B11_AF groups were sacrificed on day 21 after tumor inoculation and mice of the other groups on day 24. The tumor tissue isolated from mice was photographed. (K) 0.3 mg/kg isotype_AF, 0.4 mg/kg 2MW4691, and 0.16 mg/kg HBM4033 was intraperitoneally administrated into C57BL/6-hCCR8/hCTLA-4 mice (n = 6/group) bearing MC38 tumors twice a week for 2 weeks. Tumor size was measured three times a week and displayed as mean ± SEM. Student’s t-test was applied and P value was marked. (L) The average body weight of mice of each was recorded during treatment. (M) Tumor tissues were removed from mice of 2MW4691 and HBM4003 groups when mice were sacrificed on day 26 post-tumor inoculation. The tumors were weighed and photographed. Statistical analysis of tumor weight was determined by Student’s t-test. *P < 0.05, ** P < 0.01, **P < 0.001
MC38-implanted hCTLA-4 knock-in C57BL/6 mice were utilized to evaluate the effect of targeting CTLA-4. When the average tumor size reached approximately 100 mm3, mice were treated intraperitoneally with 1.33 mg/kg 2MW4691 and 1 mg/kg isotype control once a week. By day 21 of the experiment, the TGI of 2MW4691 was 74%, indicating that 2MW4691 significantly suppressed tumor cell proliferation in vivo (Student’s t-test, P value: 0.031) (Fig. 4D). Importantly, there was no adverse effect on the body weight of the mice, suggesting that 2MW4691 was well-tolerated in hCTLA-4 transgenic mice (Fig. 4E). The tumors isolated from the isotype control and treated groups on days 21 and 28 after administration clearly demonstrated 2MW4691’s anti-tumor effect (Fig. 4F).
To further assess the in vivo efficacy of 2MW4691 compared to its parental antibodies and determine if 2MW4691 exhibits synergistic tumor suppression activity, hCCR8 and hCTLA-4 double humanized C57BL/6 mice were inoculated with MC38 cells. The mice were divided into five groups and received the following treatments: 0.4 mg/kg 2MW4691 (G1), 0.3 mg/kg B9B11 (G2), 0.3 mg/kg ipilimumab (G3), 0.15 mg/kg B9B11 plus 0.15 mg/kg ipilimumab (G4), and 0.3 mg/kg isotype control (G5). On day 21, the TGI for groups G1, G2, G3, and G4 was 75%, 45%, 68%, and 67%, respectively (Fig. 4G). 2MW4691 demonstrated a significant improvement in tumor inhibition compared to B9B11 at equivalent molarity, and it slightly outperformed ipilimumab and the combination therapy. The body weight of mice showed an increasing trend throughout the treatment period (Fig. 4H). Tumor volumes of the treated groups were significantly different from the isotype control group on day 21 post-transplantation, with statistical analysis indicating significant differences for all groups except G2 (One-way ANOVA with Dunnett’s multiple comparisons test, P values: 0.0018, 0.0965, 0.0054, 0.0055, respectively) (Fig. 4I). Mice from the isotype and anti-CCR8 groups were sacrificed on day 12 post-administration, while those in the remaining three groups were observed for an additional 3 days. The resected tumor tissues from all groups were documented to further validate the tumor suppression effects of 2MW4691 (Fig. 4J).
In an independent study, the efficacy of 2MW4691 was compared with the next-generation CTLA-4 antibody, HBM4003, currently under clinical trials and known for its efficacy and safety advantages [37]. Both 2MW4691 and HBM4003 demonstrated significant anti-tumor effects on day 20 post-tumor inoculation, with TGIs of 89% and 69%, respectively (One-way ANOVA with Dunnett’s multiple comparisons test, both P values: < 0.0001, verse isotype control). By day 26, 2MW4691 exhibited more potent efficacy than HBM4003 (Student’s t-test, P value: 0.0221) (Fig. 4K). The body weight of mice showed normal growth trends throughout the experiment (Fig. 4L). Mice from both the 2MW4691 and HBM4003 treatment groups were sacrificed on day 26, and the tumors were dissected, revealing a significant difference in tumor weight favoring 2MW4691 over HBM4003 (Fig. 4M). The individual tumor growth curves for all the in vivo efficacy studies are shown in Fig. S6.
Overall, 2MW4691 demonstrated high potency in vivo, primarily due to its ability to deplete tumor-infiltrating Tregs and block CTLA-4 signaling on CD8 T cells.
Preliminary pharmacokinetic and safety studies of 2MW4691
To preliminary evaluate the pharmacokinetics of 2MW4691, a single-dose pharmacokinetic study was carried out in rodents. Eighteen BALB/c mice were divided into three groups: receiving respective doses of 6.66 mg/kg 2MW4691, 5 mg/kg B9B11, and 5 mg/kg ipilimumab via tail intravenous injection. Blood samples were collected at various time points post-administration (0.5, 2, 8, 24, 48, 72, 96, 120, 168, and 240 h) for analysis (Fig. S7A). The serum concentration of the antibody was quantified using ELISA. The pharmacokinetic parameters of 2MW4691 showed no significant difference compared to those of its parental antibodies in rodents (Fig. S7B), indicating a comparable pharmacokinetic profile.
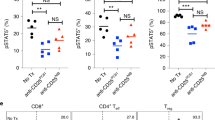
Following the rodent studies, a single-dose pharmacokinetic and safety evaluation of 2MW4691 was conducted in non-human primates (NHPs). The study included one male and one female cynomolgus monkey. The afucosylated 2MW4691 was administered at a dosage of 3 mg/kg through intravenous infusion. To serve various study objectives, serum and anticoagulated blood were collected at specific time points for analysis (Fig. 5A). Two ELISA methods were developed to accurately measure the blood concentration of 2MW4691. The resulting time-concentration curves revealed that 2MW4691 was detectable up to 168-h post-administration (Fig. 5B). The half-life of 2MW4691 was approximately 68 h, which was slightly shorter than that observed for traditional monoclonal antibodies. Additionally, peripheral blood was collected to assess the impact of 2MW4691 on the population of CD4 T cells, CD8 T cells, and Tregs. The results indicated a negligible impact on the peripheral T cell populations (Fig. 5C), suggesting that the treatment did not adversely affect these crucial immune cell types.

PK and toxicity evaluation of 2MW4691. (A) Schematic illustration of the workflow of a single-dose pharmacokinetics and toxicity of 2MW4691 in NHPs. One male and one female cynomolgus monkeys were intravenously administrated 3 mg/kg 2MW4691 via intravenous infusion. Serums were collected 0, 2, 8, 24, 48, 72, 96, 120, 168, 336, and 504-h post-administration. Blood samples with anticoagulation were collected before treatment and at 24, 72, and 168 h after infusion. (B) Pharmacokinetics of 2MW4691 in NPHs after single intravenous administration. The concentration of 2MW4691 in serum was quantitated by two ELISA methods, one of which was detected using a general secondary antibody and the other using the biotin-labeled antigen. (C) Population analysis of CD4 T cells, CD8 T cells, and Tregs in peripheral blood of cynomolgus monkeys by FACS using CD45, CD3, CD4, CD8, CD25, and FOXP3 markers. The proportion change of such three kinds of cells was measured
Discussion
Ipilimumab stands as a landmark antibody in the realm of tumor immunotherapy, approved for various cancers including melanoma, renal cell carcinoma, and hepatocellular carcinoma, among others [38]. Yet, despite the significant successes of PD-1/PD-L1 antibodies, the development of CTLA-4-targeted therapies has encountered obstacles. The effectiveness of ipilimumab is limited to a subset of patients, and its use can result in severe adverse events due to over-activation of the immune system, which restricts its broader application [39]. In response to these challenges, considerable research efforts have been dedicated to exploring CTLA-4 as a therapeutic target. While still subjected to debate, a novel mechanism of action has been proposed that differs from the previously dominant theory of blocking activity. This new mechanism focuses on ADCC-mediated depletion of intratumor Tregs [40]. Several studies have underscored the necessity of incorporating Fc function to achieve anti-CTLA-4 efficacy, suggesting a promising direction for future therapeutic strategies that might overcome the limitations of current treatments and enhance the therapeutic landscape of tumor immunotherapy [12].
The foundational aspect of our research was established by analyzing the expression profiles and correlation between CCR8 and CTLA-4. We observed heterogeneity within intratumor Tregs, with CCR8 being expressed in a small subset of highly activated Tregs, whereas CTLA-4 exhibited broader expression across Treg cells. The overlapping expression pattern of CCR8 and CTLA-4 inspired the development of bispecific antibodies targeting both markers.
We have developed a bispecific antibody, 2MW4691, with a head-to-tail configuration targeting both CCR8 and CTLA-4. The CCR8 arms exhibit high affinity, while the CTLA-4 arms possess attenuated binding activity. The bispecific antibody 2MW4691 operates through dual anti-tumor mechanisms: disrupting the CTLA-4/B7 interaction to activate naïve T cells in peripheral organs and mediating the clearance of tumor-infiltrating Tregs via ADCC. According to our single-cell analysis data on CTLA-4 expression (Fig. 3C, D), CTLA-4 is more abundantly expressed on tumor-infiltrating Treg cells compared to peripheral CD8 T cells. Subsequent in vitro functional assays confirmed that 2MW4691 can effectively activate CD8 T cells, as the lower abundance of CTLA-4 on CD8 T cells prevents their depletion (Fig. 3H, I). Additionally, our single-cell data and in vitro activated Treg analysis reveal that CCR8 is highly expressed on tumor-infiltrating Treg cells but not on peripheral Treg cells (Fig. 1, 3E). Therefore, the high expression of both CTLA-4 and CCR8 on tumor-infiltrating Treg cells enables 2MW4691 to exert ADCC effects within the tumor microenvironment, efficiently eliminating these Treg cells, as demonstrated in our in vitro killing assays (Fig. 3G). To optimize ADCC activity, the hIgG1 Fc subclass was chosen, and afucosylation was employed to enhance the ADCC capability of 2MW4691. A previous study reported that the co-engagement of CD8 T cells with antigen-presenting cells (APC) by anti-CTLA-4 antibodies could enhance T cell activation [41]. We believe that 2MW4691 may similarly enhance T cell activation, in addition to depleting Tregs in the tumor microenvironment. Our previous SEB stimulation assay also showed that afucosylated ipilimumab had stronger T cell activation ability (data not shown). Hence, the afucosylated Fc engagement of 2MW4691 with FcγRIIIa might help T cell activation.
In our study, we conducted a comprehensive evaluation of 2MW4691, covering its key characteristics such as size, purity, binding and blocking capabilities, and the ability for simultaneous binding, employing various analytical methods. The mediated cytotoxicity and T cell activation potential of 2MW4691 were substantiated through in vitro experiments. Furthermore, its in vivo anti-tumor efficacy was validated using different mouse models, demonstrating significant anti-tumor effects in both hCCR8 and hCTLA-4 transgenic mice. An attempt was made to compare the in vivo efficacy of 2MW4691 with that of combination therapy. However, challenges in administering equal treatment to two groups and the sensitivity of the MC38 tumor model to these antibodies meant that the results did not fully meet our expectations. Consequently, the exploration of 2MW4691’s anti-tumor effects necessitates the use of additional tumor models to provide a more comprehensive assessment.
A preliminary safety evaluation conducted through a single-dose administration in cynomolgus monkeys has indicated the safety of 2MW4691. Plans for a repeat high-dose toxicity study are underway to further assess and confirm the tolerability of 2MW4691.
Overall, the preclinical data for 2MW4691 lay a robust foundation for its continued development and highlight its potential as a compelling candidate for cancer treatment. The results underscore the necessity for further investigation to fully realize the therapeutic promise of 2MW4691 in the realm of cancer immunotherapy.
Data availability
The data and materials used in this study are available upon reasonable requestion from the corresponding author (xun.gui@mabwell.com).
References
Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, Davidson N, Richards J, Maio M, Hauschild A, Miller WH Jr, Gascon P, Lotem M, Harmankaya K, Ibrahim R, Francis S, Chen TT, Humphrey R, Hoos A, Wolchok JD (2011) Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 364(26):2517–2526. https://doi.org/10.1056/NEJMoa1104621
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbé C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363(8):711–723. https://doi.org/10.1056/NEJMoa1003466
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD (2015) Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 373(1):23–34. https://doi.org/10.1056/NEJMoa1504030
Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, Shaheen M, Ernstoff MS, Minor D, Salama AK, Taylor M, Ott PA, Rollin LM, Horak C, Gagnier P, Wolchok JD, Hodi FS (2015) Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 372(21):2006–2017. https://doi.org/10.1056/NEJMoa1414428
Weber J, Mandala M, Del Vecchio M, Gogas HJ, Arance AM, Cowey CL, Dalle S, Schenker M, Chiarion-Sileni V, Marquez-Rodas I, Grob J-J, Butler MO, Middleton MR, Maio M, Atkinson V, Queirolo P, Gonzalez R, Kudchadkar RR, Smylie M, Meyer N, Mortier L, Atkins MB, Long GV, Bhatia S, Lebbé C, Rutkowski P, Yokota K, Yamazaki N, Kim TM, de Pril V, Sabater J, Qureshi A, Larkin J, Ascierto PA (2017) Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med 377(19):1824–1835. https://doi.org/10.1056/NEJMoa1709030
Sanmamed MF, Chen L (2018) A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell 175(2):313–326. https://doi.org/10.1016/j.cell.2018.09.035
Maio M, Grob J-J, Aamdal S, Bondarenko I, Robert C, Thomas L, Garbe C, Chiarion-Sileni V, Testori A, Chen T-T, Tschaika M, Wolchok JD (2015) five-year survival rates for treatment-naive patients with advanced melanoma who received ipilimumab plus dacarbazine in a phase III trial. J Clin Oncol 33(10):1191–1196. https://doi.org/10.1200/jco.2014.56.6018
Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen T-T, Berman DM, Wolchok JD (2015) Pooled analysis of long-term survival data From phase II and phase III Trials of Ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 33(17):1889–1894. https://doi.org/10.1200/jco.2014.56.2736
Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA (1994) CTLA-4 can function as a negative regulator of T cell activation. Immunity 1(5):405–413. https://doi.org/10.1016/1074-7613(94)90071-x
Krummel MF, Allison JP (1995) CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 182(2):459–465. https://doi.org/10.1084/jem.182.2.459
Leach DR, Krummel MF, Allison JP (1996) Enhancement of antitumor immunity by CTLA-4 blockade. Science 271(5256):1734–1736. https://doi.org/10.1126/science.271.5256.1734
Ingram JR, Blomberg OS, Rashidian M, Ali L, Garforth S, Fedorov E, Fedorov AA, Bonanno JB, Le Gall C, Crowley S, Espinosa C, Biary T, Keliher EJ, Weissleder R, Almo SC, Dougan SK, Ploegh HL, Dougan M (2018) Anti–CTLA-4 therapy requires an Fc domain for efficacy. Proc Natl Acad Sci 115(15):3912–3917. https://doi.org/10.1073/pnas.1801524115
Lute KD, May KF, Lu P, Zhang H, Kocak E, Mosinger B, Wolford C, Phillips G, Caligiuri MA, Zheng P, Liu Y (2005) Human CTLA4 knock-in mice unravel the quantitative link between tumor immunity and autoimmunity induced by anti–CTLA-4 antibodies. Blood 106(9):3127–3133. https://doi.org/10.1182/blood-2005-06-2298
Liu Y, Zheng P (2020) Preserving the CTLA-4 checkpoint for safer and more effective cancer immunotherapy. Trends Pharmacol Sci 41(1):4–12. https://doi.org/10.1016/j.tips.2019.11.003
Sakaguchi S, Yamaguchi T, Nomura T, Ono M (2008) Regulatory T Cells and immune tolerance. Cell 133(5):775–787. https://doi.org/10.1016/j.cell.2008.05.009
Tanaka A, Sakaguchi S (2016) Regulatory T cells in cancer immunotherapy. Cell Res 27(1):109–118. https://doi.org/10.1038/cr.2016.151
Kidani Y, Nogami W, Yasumizu Y, Kawashima A, Tanaka A, Sonoda Y, Tona Y, Nashiki K, Matsumoto R, Hagiwara M, Osaki M, Dohi K, Kanazawa T, Ueyama A, Yoshikawa M, Yoshida T, Matsumoto M, Hojo K, Shinonome S, Yoshida H, Hirata M, Haruna M, Nakamura Y, Motooka D, Okuzaki D, Sugiyama Y, Kinoshita M, Okuno T, Kato T, Hatano K, Uemura M, Imamura R, Yokoi K, Tanemura A, Shintani Y, Kimura T, Nonomura N, Wada H, Mori M, Doki Y, Ohkura N, Sakaguchi S (2022) CCR8-targeted specific depletion of clonally expanded Treg cells in tumor tissues evokes potent tumor immunity with long-lasting memory. Proc Natl Acad Sci 119(7):e2114282119. https://doi.org/10.1073/pnas.2114282119
Whiteside SK, Grant FM, Gyori DS, Conti AG, Imianowski CJ, Kuo P, Nasrallah R, Sadiyah F, Lira SA, Tacke F, Eil RL, Burton OT, Dooley J, Liston A, Okkenhaug K, Yang J, Roychoudhuri R (2021) CCR8 marks highly suppressive Treg cells within tumours but is dispensable for their accumulation and suppressive function. Immunology 163(4):512–520. https://doi.org/10.1111/imm.13337
Villarreal DO, L’Huillier A, Armington S, Mottershead C, Filippova EV, Coder BD, Petit RG, Princiotta MF (2018) Targeting CCR8 induces protective antitumor Immunity and enhances vaccine-induced responses in colon cancer. Can Res 78(18):5340–5348. https://doi.org/10.1158/0008-5472.Can-18-1119
Van Damme H, Dombrecht B, Kiss M, Roose H, Allen E, Van Overmeire E, Kancheva D, Martens L, Murgaski A, Bardet PMR, Blancke G, Jans M, Bolli E, Martins MS, Elkrim Y, Dooley J, Boon L, Schwarze JK, Tacke F, Movahedi K, Vandamme N, Neyns B, Ocak S, Scheyltjens I, Vereecke L, Nana FA, Merchiers P, Laoui D, Van Ginderachter JA (2021) Therapeutic depletion of CCR8+tumor-infiltrating regulatory T cells elicits antitumor immunity and synergizes with anti-PD-1 therapy. J ImmunoTherapy Cancer. https://doi.org/10.1136/jitc-2020-001749
De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, Bonnal RJP, Provasi E, Sarnicola ML, Panzeri I, Moro M, Crosti M, Mazzara S, Vaira V, Bosari S, Palleschi A, Santambrogio L, Bovo G, Zucchini N, Totis M, Gianotti L, Cesana G, Perego RA, Maroni N, Pisani Ceretti A, Opocher E, De Francesco R, Geginat J, Stunnenberg HG, Abrignani S, Pagani M (2016) Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity 45(5):1135–1147. https://doi.org/10.1016/j.immuni.2016.10.021
Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, Chudakov DM, Rudensky AY (2016) Regulatory T cells exhibit distinct features in human breast cancer. Immunity 45(5):1122–1134. https://doi.org/10.1016/j.immuni.2016.10.032
Sun D, Sun Y, Janezic E, Zhou T, Johnson M, Azumaya C, Noreng S, Chiu C, Seki A, Arenzana TL, Nicoludis JM, Shi Y, Wang B, Ho H, Joshi P, Tam C, Payandeh J, Comps-Agrar L, Wang J, Rutz S, Koerber JT, Masureel M (2023) Structural basis of antibody inhibition and chemokine activation of the human CC chemokine receptor 8. Nat Commun. 14(1):7940. https://doi.org/10.1038/s41467-023-43601-8
Wu TD, Madireddi S, de Almeida PE, Banchereau R, Chen Y-JJ, Chitre AS, Chiang EY, Iftikhar H, O’Gorman WE, Au-Yeung A, Takahashi C, Goldstein LD, Poon C, Keerthivasan S, de Almeida Nagata DE, Du X, Lee H-M, Banta KL, Mariathasan S, Das Thakur M, Huseni MA, Ballinger M, Estay I, Caplazi P, Modrusan Z, Delamarre L, Mellman I, Bourgon R, Grogan JL (2020) Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 579(7798):274–278. https://doi.org/10.1038/s41586-020-2056-8
Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, He Y, Wang L, Zhang Q, Kim A, Gao R, Orf J, Wang T, Sawant D, Kang J, Bhatt D, Lu D, Li C-M, Rapaport AS, Perez K, Ye Y, Wang S, Hu X, Ren X, Ouyang W, Shen Z, Egen JG, Zhang Z, Yu X (2020) Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell 181(2):442-459.e29. https://doi.org/10.1016/j.cell.2020.03.048
Min B (2017) Heterogeneity and stability in Foxp3+ regulatory T Cells. J Interferon Cytokine Res 37(9):386–397. https://doi.org/10.1089/jir.2017.0027
Zhang R, Miao J, Zhu P (2021) Regulatory T cell heterogeneity and therapy in autoimmune diseases. Autoimmun Rev 20(5):102715. https://doi.org/10.1016/j.autrev.2020.102715
Wing JB, Tanaka A, Sakaguchi S (2019) Human FOXP3+ regulatory T cell heterogeneity and function in autoimmunity and cancer. Immunity 50(2):302–316. https://doi.org/10.1016/j.immuni.2019.01.020
Shi A-P, Tang X-Y, Xiong Y-L, Zheng K-F, Liu Y-J, Shi X-G, Lv Y, Jiang T, Ma N, Zhao J-B (2022) Immune Checkpoint LAG3 and Its Ligand FGL1 in Cancer. Front Immunol. https://doi.org/10.3389/fimmu.2021.785091
Deng J, Zhao S, Zhang X, Jia K, Wang H, Zhou C, He Y (2019) OX40 (CD134) and OX40 ligand, important immune checkpoints in cancer. Onco Targets Ther 12:7347–7353. https://doi.org/10.2147/ott.S214211
Schaefer W, Regula JT, Bähner M, Schanzer J, Croasdale R, Dürr H, Gassner C, Georges G, Kettenberger H, Imhof-Jung S, Schwaiger M, Stubenrauch KG, Sustmann C, Thomas M, Scheuer W, Klein C (2011) Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci 108(27):11187–11192. https://doi.org/10.1073/pnas.1019002108
Li Y, Hickson JA, Ambrosi DJ, Haasch DL, Foster-Duke KD, Eaton LJ, DiGiammarino EL, Panchal SC, Jiang F, Mudd SR, Zhang C, Akella SS, Gao W, Ralston SL, Naumovski L, Gu J, Morgan-Lappe SE (2018) ABT-165, a dual variable domain immunoglobulin (DVD-Ig) targeting DLL4 and VEGF, demonstrates superior efficacy and favorable safety profiles in preclinical models. Mol Cancer Ther 17(5):1039–1050. https://doi.org/10.1158/1535-7163.Mct-17-0800
Schanzer J, Jekle A, Nezu J, Lochner A, Croasdale R, Dioszegi M, Zhang J, Hoffmann E, Dormeyer W, Stracke J, Schäfer W, Ji C, Heilek G, Cammack N, Brandt M, Umana P, Brinkmann U (2011) Development of tetravalent, bispecific CCR5 antibodies with antiviral activity against CCR5 Monoclonal antibody-resistant HIV-1 strains. Antimicrob Agents Chemother 55(5):2369–2378. https://doi.org/10.1128/aac.00215-10
Ribas A, Hanson DC, Noe DA, Millham R, Guyot DJ, Bernstein SH, Canniff PC, Sharma A, Gomez-Navarro J (2007) Tremelimumab (CP-675,206), a cytotoxic T lymphocyte-associated antigen 4 blocking monoclonal antibody in clinical development for patients with cancer. Oncologist 12(7):873–883. https://doi.org/10.1634/theoncologist.12-7-873
Stewart R, Hammond SA, Oberst M, Wilkinson RW (2014) The role of Fc gamma receptors in the activity of immunomodulatory antibodies for cancer. J ImmunoTher Cancer 2(1):29. https://doi.org/10.1186/s40425-014-0029-x
Cohen Saban N, Yalin A, Landsberger T, Salomon R, Alva A, Feferman T, Amit I, Dahan R (2023) Fc glycoengineering of a PD-L1 antibody harnesses Fcγ receptors for increased antitumor efficacy. Sci Immunol. https://doi.org/10.1126/sciimmunol.add8005
Gan X, Shan Q, Li H, Janssens R, Shen Y, He Y, Chen F, van Haperen R, Drabek D, Li J, Zhang Y, Zhao J, Qin B, Jheng MJ, Chen V, Wang J, Rong Y, Grosveld F (2022) An anti-CTLA-4 heavy chain-only antibody with enhanced T(reg) depletion shows excellent preclinical efficacy and safety profile. Proc Natl Acad Sci U S A. 119(32):e2200879119. https://doi.org/10.1073/pnas.2200879119
Korman AJ, Garrett-Thomson SC, Lonberg N (2022) The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat Rev Drug Discov 21(7):509–528. https://doi.org/10.1038/s41573-021-00345-8
Pol J, Kroemer G (2018) Anti-CTLA-4 immunotherapy: uncoupling toxicity and efficacy. Cell Res 28(5):501–502. https://doi.org/10.1038/s41422-018-0031-9
Liu Y, Zheng P (2018) How does an anti-CTLA-4 antibody promote cancer immunity? Trends Immunol 39(12):953–956. https://doi.org/10.1016/j.it.2018.10.009
Waight JD, Chand D, Dietrich S, Gombos R, Horn T, Gonzalez AM, Manrique M, Swiech L, Morin B, Brittsan C, Tanne A, Akpeng B, Croker BA, Buell JS, Stein R, Savitsky DA, Wilson NS (2018) Selective FcgammaR co-engagement on APCs modulates the activity of therapeutic antibodies targeting T cell antigens. Cancer Cell 33(6):1033–1047. https://doi.org/10.1016/j.ccell.2018.05.005
Acknowledgements
We gratefully acknowledge the support provided by colleagues at Mabwell (Shanghai) Bioscience Co., Ltd.
Funding
This work was supported by Mabwell (Shanghai) Bioscience Co., Ltd.
Author information
Authors and Affiliations
Contributions
Xun Gui and Cuicui Guo played pivotal roles in initiating the study and overseeing the research activities. Cuicui Guo and Xiaodong Dai were responsible for designing the experiments and analyzing the data collected throughout the study. Xiaodong Dai and Xiumei Xiong carried out the majority of the experimental work and contributing significantly to the empirical findings of the research. Yulei Du was instrumental in conducting the bioinformatic analysis and providing critical insights into the data interpretation. The manuscript was primarily written by Cuicui Guo and Xiaodong Dai, with both also responsible for the creation of the figures. Xun Gui and Cuicui Guo provided essential direction for the project and were involved in the manuscript's revision process to ensure accuracy and clarity.
Corresponding author
Ethics declarations
Conflict of interest
Cuicui Guo, Xiaodong Dai, Xiumei Xiong, Yulei Du, and Xun Gui are employees of Mabwell (Shanghai) Bioscience Co., Ltd. Cuicui Guo and Xun Gui may hold shares in Mabwell (Shanghai) Bioscience Co., Ltd, suggesting a financial interest in the outcomes of the research. No competing interests were declared by the other authors, indicating that beyond their employment, they do not have any additional financial stakes that could be perceived as influencing the research outcomes.
Ethical approval
All mouse experiments were performed according to the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC) of Yicon (Beijing) BioMedical Technology Inc. Experiments involving cynomolgus monkeys were conducted at Suzhou Guochen Biotechnology Co., Ltd., adhering to ethical guidelines as dictated by the relevant committees.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, C., Dai, X., Du, Y. et al. Preclinical development of a novel CCR8/CTLA-4 bispecific antibody for cancer treatment by disrupting CTLA-4 signaling on CD8 T cells and specifically depleting tumor-resident Tregs. Cancer Immunol Immunother 73, 210 (2024). https://doi.org/10.1007/s00262-024-03794-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00262-024-03794-3