
Abstract
Background
Radiotherapy (RT) synergizes with immune checkpoint blockade (ICB). CD1c(BDCA-1)+/CD141(BDCA-3)+ myeloid dendritic cells (myDC) in the tumor microenvironment are indispensable at initiating effector T-cell responses and response to ICB.
Methods
In this phase II clinical trial, anti-PD-1 ICB pretreated oligometastatic patients (tumor agnostic) underwent a leukapheresis followed by isolation of CD1c(BDCA-1)+/CD141(BDCA-3)+ myDC. Following hypofractionated stereotactic body RT (3 × 8 Gy), patients were randomized (3:1). Respectively, in arm A (immediate treatment), intratumoral (IT) ipilimumab (10 mg) and avelumab (40 mg) combined with intravenous (IV) pembrolizumab (200 mg) were administered followed by IT injection of myDC; subsequently, IV pembrolizumab and IT ipilimumab/avelumab were continued (q3W). In arm B (contemporary control arm), patients received IV pembrolizumab, with possibility to cross-over at progression. Primary endpoint was 1-year progression-free survival rate (PFS). Secondary endpoints were safety, feasibility, objective response rate, PFS, and overall survival (OS).
Results
Thirteen patients (10 in arm A, eight non-small cell lung cancer, and five melanoma) were enrolled. Two patients crossed over. One-year PFS rate was 10% in arm A and 0% in arm B. Two patients in arm A obtained a partial response, and one patient obtained a stable disease as best response. In arm B, one patient obtained a SD. Median PFS and OS were 21.8 weeks (arm A) versus 24.9 (arm B), and 62.7 versus 57.9 weeks, respectively. An iatrogenic pneumothorax was the only grade 3 treatment-related adverse event.
Conclusion
SBRT and pembrolizumab with or without IT avelumab/ipilimumab and IT myDC in oligometastatic patients are safe and feasible with a clinically meaningful tumor response rate. However, the study failed to reach its primary endpoint.
Trial registration number: Clinicaltrials.gov: NCT04571632 (09 AUG 2020). EUDRACT: 2019-003668-32. Date of registration: 17 DEC 2019, amendment 1: 6 MAR 2021, amendment 2: 4 FEB 2022.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Immune checkpoint blockade (ICB) has significantly improved survival in melanoma and non-small-cell lung cancer (NSCLC); yet, response rates remain low [1, 2]. Managing oligoprogression, i.e., the emergence of a few lesions post-initial systemic therapy success, is challenging [3].
The cancer-immunity cycle underscores dendritic cells' (DC) pivotal role in priming CD4+ and CD8+ T-cells through presentation of tumor-associated antigens (TAAs) in tumor-draining lymph nodes (TDLN) [4]. In the tumor microenvironment (TME), DC plays a crucial role, divided into plasmacytoid DC (pDC) and conventional (cDC) or myeloid DC (myDC), further categorized as CD141 (BDCA-3)+ or cDC1 and CD1c (BDCA-1)+ or cDC2 [5]. While cDC1 primarily drives CD8+ cytotoxic T-lymphocytes (CTL) responses and cDC2 activates CD4+ T-lymphocytes within the TME, inadequate DC recruitment under tumor growth conditions hampers anti-tumor immunity [4, 6]. Excluding cDC from the TME diminishes ICB efficacy, resulting in defective CTL activation and facilitating metastases evasion [7]. Furthermore, an immunosuppressive phenotype marked by PD-L1 expression (referred to as cDC3, LAMP3 + DCs, or mregDCs) has been identified [5]. Limited (pre-)clinical trials have explored the isolation and intratumoral/intranodal administration of myDC and myDC-derived cellular vaccines [8].
Stereotactic body radiation therapy (SBRT) has emerged as a treatment for oligoprogression, particularly in NSCLC patients [9]. Preclinical studies indicate that SBRT induces a decrease in CD8+ T-cells and cDC1 and an increase in cDC2 and CD4+/Treg T-cell ratio supporting intratumoral immune cell repertoire boosting, with little understanding of optimal dose and fractionation [10]. Combining low-dose-fractionated radiotherapy (3 × 8 Gy) with anti-CTLA-4 antibodies has shown promise in inducing abscopal effects through cDC1 activation in preclinical models. Despite SBRT's local immunosuppressive effect, it can trigger a systemic anti-tumor immune response, enhancing both priming and effector stages [11]. SBRT induces immunogenic cell death (ICD), releasing TAAs that activate DC, especially the cDC1 subset, which migrate to TDLN to prime CD8+ T-cells [12]. SBRT-induced cell death activates toll-like receptor 4 and type 1 interferon signaling and boosts antigen processing and presentation by increasing major histocompatibility complex class I levels, suggesting the potential of combining DC treatment with SBRT as a strategy to enhance anti-tumor immune response [13, 14].
This study combines the cDC presence in the TME with the immunogenic effect of SBRT through ICD induction. We investigated the efficacy and toxicity of SBRT (3 × 8 Gy) combined with an intratumoral (IT) injection of CD1c(BDCA-1)+/CD141(BDCA-3)+ myDC, anti-PD-L1, and anti-CTLA-4. Previously, we reported the combination of myDC and IT ICB to be safe [15]. We aimed to assess whether this combination enhances responses in advanced solid tumor patients with oligoprogression on anti-PD-1 therapy compared to SBRT alone.
Methods
Patients
Patients with unresectable advanced solid tumors, presenting with oligoprogressive disease (defined as ≤ 7 sites) on anti-PD1 therapy (pembrolizumab or nivolumab) alone or in combination (anti-CTLA-4 ICB or chemotherapy), were eligible. Initially limited to NSCLC, the trial expanded to all solid tumors in January 2022 due to slow recruitment. At least one lesion (> 1 cm) needed to be injectable clinically or imaging-guided and be eligible for SBRT. Other criteria included age ≥ 18, ECOGps < 2, normal hematological/liver/renal function, and negative HIV/syphilis/hepatitisserology. Exclusions comprised untreated central nervous system metastases, autoimmune disease, immunosuppressive treatment, grade 3–4 immune-related adverse events (AEs), malignancy in the past 5 years, and contraindication to stop anticoagulation.
Study design and treatment
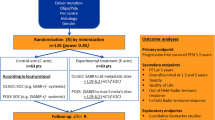
This phase II randomized trial investigated the combination of SBRT and pembrolizumab with or without intratumoral avelumab/ipilimumab plus autologous CD1c (BDCA-1)+/CD141 (BDCA-3)+ myDCs (Supplementary Fig. S1). After screening, patients underwent leukapheresis. Following SBRT to progressive lesions (3 × 8 Gy), patients were randomized (3:1) to an immediate treatment arm (A) or a contemporary control arm (B). In arm A, 10 mg of ipilimumab (Yervoy®) and 40 mg of avelumab (Bavencio®) were injected in the chosen lesion along with intravenous (IV) administration of 200 mg pembrolizumab (KeytrudaTM). In the following day, autologous myDCs (suspension volume based on lesion size) were injected in the same lesion. Patients continued IV pembrolizumab along with IT ipilimumab and avelumab Q3W. In arm B, patients received IV pembrolizumab 200 mg Q3W, with possibility to start IT injections upon progressive disease (PD). Approval was obtained of Universitair Ziekenhuis Brussel ethics committee (2019-440) and the Belgian Federal Agency for Medicines and Health Products (EudraCT2019-003668-32).
Leukapheresis, isolation, and cryopreservation of myeloid dendritic cells
Leukapheresis and myDC isolation were previously described in J. Tijtgat et al., 2024. The cryopreservation procedure is described in supplementary methods.
Assessment of response and toxicity
Tumor responses were evaluated using whole-body 18-fluorodeoxyglucose positron emission tomography-computed tomography (18FFDG-PET/CT) every 12 weeks, with objective response rates (ORR) defined by Response Evaluation Criteria in Solid Tumors for Immunotherapy (iRECISTv1.1). Clinical examinations and blood analyses were conducted before each treatment cycle, and AEs were documented using CTCAE (v5.0) by type, frequency, and severity.
Determination of lymphocyte subsets and HMGB1 in peripheral blood
Peripheral blood was collected from all patients at baseline (= during leukapheresis), at D-5 (start of SBRT), D1 (start of IT and IV treatment) and every Q3W thereafter. The determination of lymphocyte subsets and HMGB1 is described in the supplementary methods.
Tumor biopsies and immunohistochemistry
When safe and feasible, core-needle tumor biopsies were collected on D1 and every Q3W. Immunohistochemistry (IHC) stainings for CD3 (clone 2GV6) and CD8 (clone SP57) were routinely performed while SOX10 (clone SP267), PD-L1 (clone 22C3, SP263), and, in case of lung tissue, CK 7 (clone SP52), were stained occasionally (all clones Roche®).
Statistical analysis
The study's primary endpoint is 1-year progression-free survival (1YPFS) rate following randomization. Based on a historical 1YPFS rate of 45% (as reported by Collen et al., 2014) in a cohort of 26 oligometastatic (≤ 5) NSCLC patients treated with SBRT after induction chemotherapy (n = 17) or as primary treatment (n = 9), the desired 1YPFS rate for arm A was set at 65% [16]. Using a Fleming one-stage design (null hypothesis p0 45%, alternative hypothesis p1 65%, α = 0.1 and β = 0.2), the required sample size of arm A was 27 patients and nine in arm B. Safety, feasibility, ORR by iRECIST, response of (non-)injected tumor sites, duration of response, PFS, and overall survival (OS) were secondary endpoints.
Descriptive statistics were used for baseline characteristics, treatment administration, efficacy endpoints, and AE. Demographics and study drug administration were tabulated, including cycle count, dose intensity, modifications, and reason for deviations. 1YPFS rate and OS were determined using Kaplan–Meier survival estimates. Simple linear regression analysis was applied to study correlations. Data were analyzed using GraphPad Prism v10.
Database was locked on November 24, 2023
Results
Patient characteristics
Thirteen patients (nine male and four female) were recruited (CONSORT diagram in Fig. 1). The trial was terminated prematurely because of slow recruitment, partly related to the COVID-19 pandemic. Additionally, patient inclusion was hindered by advanced disease stages, rapid disease progression, and prior radiation therapy to target lesions.

CONSORT diagram: 13 patients were recruited between September 2020 and June 2023
Ten patients were randomized to arm A and three to arm B. The baseline characteristics per treatment arm are in Table 1. Individual patient tumor types and stages are reported in Supplementary Table S1.
Treatment disposition
Patients received a median of 4 IT (arm A) and 7 IV administrations. One patient in arm A discontinued IT injections, while two patients could not receive any due to clinical deterioration or lesion shrinkage post-radiation. Additionally, two patients crossed over to arm A at progression (median of 3.5 IT and 15 IV administrations).
Isolation and characterization of myDC
All patients were able to undergo leukapheresis, and no unexpected AEs were observed. Isolation of CD1c (BDCA-1)+ and CD141 (BDCA-3)+ myDC was successful in all but one patient due to a technical issue. Median numbers and phenotypes of myDC (CD1c/CD141) are summarized in Supplementary Table S1.
Safety and adverse events (AEs)
Most common AEs were fatigue (38%), nausea (38%), and pneumonitis (30%). Three NSCLC patients developed grade 3 pneumonitis, requiring therapy discontinuation. Other discontinuation reasons included cerebrovascular ischemia (n = 1) and hypophysitis (n = 1). IT injections were generally well-tolerated. Grades 2 and 3 AEs are detailed in Table 2 (full list in Supplementary Table S2).
Efficacy/clinical outcome
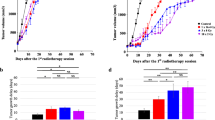
All patients (n = 13) were evaluable for tumor response. In this mixed population of melanoma (n = 4) and NSCLC (n = 8) patients, we observed two partial responses (PR) (patient 2A NSCLC and 14A melanoma, ongoing for 15.6 and 6 months, respectively), yielding an ORR of 20% in arm A. No objective responses were observed in arm B. Stable disease (SD) was objectified in two patients of arm A (patient 5A NSCLC and 10A melanoma) and one patient in arm B (patient 9B melanoma) (Fig. 2a). The 1YPFS rate was 10% in arm A and 0% in arm B. Median PFS and OS were 21.8 and 62.7 weeks in arm A, respectively, and 24.9 and 57.9 weeks in arm B (Fig. 2b, c). Two of three patients in arm B progressed and crossed over to IT injections, with a median PFS of 11.14 weeks thereafter. A case illustration is shown in Fig. 3.

Swimmer plot (a) of randomized patients, Kaplan–Meier curve of progression-free survival (PFS) (b), and overall survival (OS) (c). Abbreviations: stable disease (SD), partial response (PR), progressive disease (PD), progressive disease after cross-over (PD2)

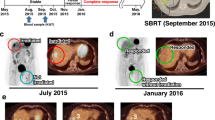
Case illustration of patient 2 (arm A), who had a PR at week 25, but encountered IT treatment interruptions (at week 33) due to pain and fibrosis. Hospitalization for fever, elevated inflammatory markers, and widespread lung activity on PET-CT was managed by high-dose corticosteroids. A relapse of the latter followed by a COVID-19 infection led to further treatment interruption with disease progression by week 70. Upper row images: CT, second row: PET/CT maximum intensity projections (MIP) of disease locations, third row (single image): fused PET/CT image illustrating diffuse PET activity in both lungs, suggestive of (ir-)pneumonitis. SD = stable disease, PR = partial response, PD = progressive disease, PEMBRO = pembrolizumab, AVE = avelumab, IPI = ipilimumab
Effect of treatment on blood immune cell composition and immunohistochemistry
At every IT injection, five lymphocyte subsets of PBMCs were analyzed. Two patients (1A, 6A) exhibited PD shortly after treatment initiation (Fig. 2), halting further longitudinal sample collection. Baseline naive T-cell levels (defined as CD45RA+CD45RO−CD27+CCR7+CD62L+) were low, likely due to prior treatments. No significant changes were observed in naive, effector (CD45RA+CD45RO−CD27−CCR7−CD62L−), effector memory (CD45RA−CD45RO+CD27+CCR7+CD62L+), and central memory T-cells (CD45RA−CD45RO+CD27+CCR7+CD62L+) between arm A and B due to high interpatient variability. Additionally, no significant changes were observed when comparing T-cell subsets at baseline (D-5) and post-treatment (D1 and Q3W thereafter) (data not shown). A variable increase in Tregs was observed directly post-SBRT (D1) as compared to pre-SBRT (D-5), with a subsequent normalization of Treg levels (Supplementary Fig. S2).
Supplementary figures 3a and 3b provide an overview of immune checkpoint expression on CD8+ and CD4+ T-cells, respectively.
We hypothesized that adding SBRT induces ICD, enhancing anti-tumoral immune responses. Plasma HMGB1 levels, an ICD marker, remained stable in eight patients but significantly increased post-SBRT in four patients (5A, 10A (both SD), 11A, and 13A (both PD)) (Supplementary Fig. S4). No correlation was found between irradiated volumes and HMGB1 increase (Supplementary Fig. S5). Surprisingly, Patient 1A showed decreased HMGB1 levels post-SBRT despite initial high levels. Some patients showed increased PD-L1 and CD8+ T-cell levels in tumor biopsies (Supplementary Fig. S6).
Discussion
This randomized phase II clinical trial investigated the combination of SBRT, IV and IT ICB, and IT administration of myDC in anti-PD-1 pretreated patients. Despite demonstrating safety and feasibility, the observed anti-tumor activity in the experimental arm did not meet expectations, leading to recruitment termination. However, clinically meaningful activity was noted in a minority of patients. A challenge was identifying eligible candidates, with some excluded due to early clinical deterioration, and others having undergone prior radiation therapy, particularly in the NSCLC population, making the intended radiation dose unsafe and unfeasible.
While leukapheresis was successful in all but one patient, IT administration feasibility was suboptimal. Limited efficacy was seen in three patients in arm A, with a shorter PFS compared to the historical cohort [16]. High pneumonitis incidence, by combining ICB and radiotherapy, particularly in NSCLC patients, was noted, hindering tumor biopsy collection [17].
Combining ICB and SBRT aims to reduce immunosuppression and induce ICD in the TME, enhancing the anti-tumor immune response. However, prospective trials have not consistently shown significant efficacy improvements [18]. Implementing stricter selection criteria, like excluding patients with prior radiation at disease progression sites and ensuring optimal performance scores before treatment, may enhance response rates.
Determining the best timing and dosage of SBRT alongside ICB remains elusive, as shown in the Belgian CHEERS trial, where SBRT (3 × 8 Gy) addition did not improve PFS or OS [19]. Optimal timing of ICB with radiation varies based on the immunotherapy mechanism, while conflicting data on ideal radiation dosage for synergy with ICB underscores the need for preclinical exploration [14, 20]. Moreover, despite reported instances of an abscopal effect, its consistency and interaction with ICB merit further investigation, as our study did not observe such outcomes.
Conclusion
Combining SBRT, ICB, and IT myDC proved safe and feasible in select patients, with promising activity. However, the primary 1YPFS endpoint was not met, leading to early study termination. Further investigation into SBRT timing and dosage, along with patient selection, is crucial to enhance the therapeutic effectiveness of this approach.
Abbreviations
- 1YPFS:
-
1-year progression-free survival rate
- AEs:
-
Adverse events
- CD:
-
Cluster of differentiation
- cDC:
-
Conventional or circulating dendritic cell(s)
- CTL:
-
Cytotoxic T-lymphocytes
- DC:
-
Dendritic cell(s)
- ECOGps:
-
Eastern cooperative oncology group performance status
- ICB:
-
Immune checkpoint blockade
- iRECIST:
-
Immunotherapy response evaluation criteria in solid tumors
- IT:
-
Intratumoral
- IV:
-
Intravenous
- myDC:
-
Myeloid dendritic cell(s)
- NSCLC:
-
Non-small-cell lung carcinoma
- ORR:
-
Objective response rate
- OS:
-
Overall survival
- PBMC:
-
Peripheral blood mononuclear cells
- PD:
-
Progressive disease
- pDC:
-
Plasmacytoid dendritic cell(s)
- PFS:
-
Progression-free survival
- RT:
-
Radiotherapy
- SBRT:
-
Stereotactic body radiation therapy
- SD:
-
Stable disease
- TAAs:
-
Tumor-associated antigens
- TDLN:
-
Tumor-draining lymph nodes
- TME:
-
Tumor microenvironment
References
Larkin J et al (2015) Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 373(1):23–34
Gandhi L et al (2018) Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med 378(22):2078–2092
Guckenberger M et al (2020) Characterisation and classification of oligometastatic disease: a European Society for Radiotherapy and Oncology and European Organisation for Research and Treatment of Cancer consensus recommendation. Lancet Oncol 21(1):e18–e28
Broz ML et al (2014) Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26(6):938
Pittet MJ et al (2023) Dendritic cells as shepherds of T cell immunity in cancer. Immunity 56(10):2218–2230
Gardner A, de Mingo Pulido A, Ruffell B (2020) Dendritic cells and their role in immunotherapy. Front Immunol 11:924
Salmon H et al (2016) Expansion and activation of CD103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity 44(4):924–938
Schwarze JK et al (2023) Current “state of the art” on dendritic cell-based cancer vaccines in melanoma. Curr Opin Oncol 35(2):87–93
Kagawa Y et al (2020) Efficacy of local therapy for oligoprogressive disease after programmed cell death 1 blockade in advanced non-small cell lung cancer. Cancer Sci 111(12):4442–4452
Reijmen E et al (2021) Fractionated radiation severely reduces the number of CD8+ T cells and mature antigen presenting cells within lung tumors. Int J Radiat Oncol Biol Phys 111(1):272–283
Dewan MZ et al (2009) Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res 15(17):5379–5388
Di Blasio S et al (2016) Human CD1c(+) DCs are critical cellular mediators of immune responses induced by immunogenic cell death. Oncoimmunology 5(8):e1192739
Janopaul-Naylor JR, et al (2021) The Abscopal effect: a review of pre-clinical and clinical advances. Int J Mol Sci 22(20)
Dagoglu N et al (2019) Abscopal effect of radiotherapy in the immunotherapy era: systematic review of reported cases. Cureus 11(2):e4103
Schwarze JK, et al (2020) Intratumoral combinatorial administration of CD1c (BDCA-1)(+) myeloid dendritic cells plus ipilimumab and avelumab in combination with intravenous low-dose nivolumab in patients with advanced solid tumors: a phase IB clinical trial. Vaccines (Basel) 8(4)
Collen C et al (2014) Phase II study of stereotactic body radiotherapy to primary tumor and metastatic locations in oligometastatic nonsmall-cell lung cancer patients. Ann Oncol 25(10):1954–1959
Tyczynski JE et al (2021) Incidence and risk factors of pneumonitis in patients with non-small cell lung cancer: an observational analysis of real-world data. Oncol Ther 9(2):471–488
Turchan WT, Chmura SJ (2021) The role of immunotherapy in combination with oligometastasis-directed therapy: a narrative review. Ann Palliat Med 10(5):6028–6044
Spaas M, et al (2023) Checkpoint inhibitors in combination with stereotactic body radiotherapy in patients with advanced solid tumors: the CHEERS Phase 2 randomized clinical trial. JAMA Oncol
Young KH et al (2016) Optimizing timing of immunotherapy improves control of tumors by hypofractionated radiation therapy. PLoS ONE 11(6):e0157164
Acknowledgements
Research Foundation Flanders (FWO).
Author information
Authors and Affiliations
Contributions
Manon Vounckx (80%) and Jens Tijtgat wrote the main manuscript text. Ines Dufait and Latoya Stevens wrote the translational part of the main manuscript. Manon Vounckx and Jens Tijtgat prepared the three main figures. Manon Vounckx, Latoya Stevens, and Inès Dufait prepared all supplementary figures and tables. All authors reviewed the manuscript. Manon Vounckx wrote the revision to shorten the article to a brief report. Manon Vounckx and Inès Dufait wrote the revisions of the brief report. Since the work was mostly provided by Manon Vounckx, the two last authors decided to provide her full single first authorship.
Corresponding author
Ethics declarations
Competing interests
Conflict of interest: Manon Vounckx: Research funding from Kom op tegen Kanker Jens Tijtgat: Since January 2024 Disease Area Specialist (Medical Lead) Thoracic Oncology at Bristol Myers Squibb Iris Dirven: Research funding: Kom op tegen Kanker; Travel, Accomodations, Expenses: AstraZeneca, Pierre Fabre Julia Katarina Schwarze: JKS reports non-financial support from MSD and Amgen; personal fees from Novartis.Lore Decoster: payments to institution for advisory boards, consultancy, lectures, travel from Astra Zeneca, Roche, MSD, BMS, Servier, Lilly Bart Neyns: Personal financial compensation for public speaking, consultancy, and participation in advisory board meetings from Roche, Bristol Myers Squibb, Merck Sharp & Dohme, Novartis. My institution (UZ Brussel) received research funding related to research projects conducted by my team from Pfizer, Novartis, Roche, and Merck-Serono
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vounckx, M., Tijtgat, J., Stevens, L. et al. A randomized phase II clinical trial of stereotactic body radiation therapy (SBRT) and systemic pembrolizumab with or without intratumoral avelumab/ipilimumab plus CD1c (BDCA-1)+/CD141 (BDCA-3)+ myeloid dendritic cells in solid tumors. Cancer Immunol Immunother 73, 167 (2024). https://doi.org/10.1007/s00262-024-03751-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00262-024-03751-0