
Abstract
Background
Tumor-associated macrophages (TAMs) are the predominant immune cells in the tumor microenvironment and portend poor prognosis. However, the molecular mechanisms underlying the tumor promotion of TAMs have not been fully elucidated.
Methods
Coculture of gastric cancer cells with U937 cells was performed to investigate the impact of TAMs on cancer cell behavior. MicroRNA (miRNA) microarray and bioinformatics were applied to identify the involved miRNAs and the functional target genes. The regulation of the miRNA on its target gene was studied using anti-miRNA and miRNA mimic.
Results
Coculture with CD204+ M2-like TAMs increased proliferation, migration, and epithelial-mesenchymal transition of gastric cancer cells. MiR-210 was the most upregulated miRNA in cancer cells identified by miRNA microarray after coculture. In gastric cancer tissues, miR-210 expression was positively correlated with CD204+ M2-like TAM infiltration. Inactivation of miR-210 by antimir attenuated CD204+ M2-like TAMs-induced cancer cell migration. Using pharmacological inhibitors and neutralizing antibodies, CD204+ M2-like TAMs-secreted TNFα was found to upregulate miR-210 through NF-κB/HIF-1α signaling. Bioinformatics analysis showed netrin-4 (NTN4) as a potential target of miR-210 to suppress gastric cancer cell migration. We also found an inverse expression between miR-210 and NTN4 in cancer cells after coculture or in tumor xenografts. Anti-miR-210 increased NTN4 expression, while miR-210 mimics downregulated NTN4 in cancer cells. Reporter luciferase assays showed that MiR-210 mimics suppressed NTN4 3’ untranslated region-driven luciferase activity in cancer cells, but this effect was blocked after mutating miR-210 binding site.
Conclusions
CD204+ M2-like TAMs can utilize the TNF-α/NF-κB/HIF-1α/miR-210/NTN4 pathway to facilitate gastric cancer progression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic inflammatory insults are currently believed to be a precursor of malignancy development and correlates with cancer progression [1]. Gastric cancer, one of the most common cancers worldwide, arises in close association with chronic inflammation [2]. Atrophic gastritis, persistent chronic inflammation of the gastric mucosa, reportedly has a 5.7-fold increased risk for developing gastric cancer [3, 4]. Tumor-associated macrophages (TAMs) are a major component of the inflammatory infiltrate in the tumor microenvironment (TME), which has been considered a key player in connecting inflammation and cancer [5]. TAM infiltration has been widely accepted as a negative prognostic parameter for most solid tumors [6].
TAMs can be classified into M1 and M2 phenotypes. M1 TAMs, characterized by the production of IL-12, IL-23, and inducible nitric oxide synthase (iNOS), can be activated by IFN-γ, lipopolysaccharides (LPS), and TNF-α to exert cytotoxic effects against cancer [7]. On the other hand, M2 TAMs, arising from exposure to T helper type 2 (Th2) cytokines, exhibit protumor properties and express high levels of IL-10 and scavenger receptors [7]. Clinicopathological studies have shown that the infiltration of M2 TAM in gastric cancer was associated with more malignant phenotypes, such as higher microvascular density, increased depth of invasion, more lymph node involvement, and advanced tumor stages [8, 9]. A high M2 TAM content in solid tumors rendered the prognosis dismal [10]. However, despite increasing evidence for a protumor role of TAMs, the way in which these cells contribute to cancer aggressiveness still needs full elucidation.
MicroRNAs (miRNAs) are a class of short and noncoding endogenous RNA molecules that are able to hinder gene expression at both the transcription and translation levels by binding to the 3’ untranslated region (3’ UTR) of the target genes [11]. Since miRNAs regulate up to 30% of all human genes, a single miRNA may govern expression of multiple target genes or pathways involving in complex signaling networks [12]. Therefore, dysregulation of miRNAs may contribute to the pathogenesis of many human disorders including cancer. Recent studies have highlighted the important role of miRNAs in cancer that nearly 50% of known miRNA genes are located in cancer-associated genomic regions and frequently deleted or amplified in tumorigenesis [13]. Aberrant expression of miRNAs in human malignancies has also been documented, [14, 15] suggesting their potential as diagnostic and prognostic markers and therapeutic targets. Recently, inflammation has been implicated in the regulation of miRNAs. For instance, STAT3, activated by IL-6, has been demonstrated to activate miR-21 and miR-181b-1 to target PTEN and CYLD, respectively, consequently resulting in cell transformation [16]. The tumor suppressor miR-7 was down-regulated in gastritis and gastric tumors in response to activated macrophages-derived small molecules [17]. Accordingly, miRNAs may act as mediators of inflammation-driven carcinogenesis. In this study, the miRNA microarray data showed that TAMs induced miR-210 expression in gastric cancer cells; therefore, we were interested in studying the role of miR-210 in the interaction between TAMs and gastric cancer cells.
Results
CD204+ M2-like TAMs enhance the malignant behavior of gastric cancer cells and confer poor patient survival
High M2 TAM infiltration frequently occurs in gastric cancer tissues and is strongly associated with poor patient survival [18, 19]. IHC staining for the specific M2 marker CD204 in 142 gastric cancer specimens was performed, and the staining was scored as grade 0 (no cells with positive staining), grade 1 (less than 25% cells with positive staining), grade 2 (25–50% cells with positive staining), and grade 3 (more than 50% cells with positive staining). Kaplan-Meier survival analysis showed that patients with high CD204 expression (CD204 staining positivity > 50%) had worse overall survival than those with low CD204 expression (Fig. 1A). In order to study the interaction between inflammatory cells and cancer cells, the in vitro macrophages-cancer cells coculture system was used to mimic the inflammatory microenvironment. U937 cells are able to differentiate into M2 TAMs in response to tumor-derived factors [20]. Coculture with AGS cells for 72 h induced the polarization of U937 cells into M2-like TAMs characterized by increased expression of the macrophage marker CD68 and the M2 marker CD204 (Fig. 1B), which in turn increased the growth (Fig. 1C) and the migration ability of AGS cells (Fig. 1D and E). A more mesenchymal-like morphology (Fig. 1F), down-regulation of the epithelial marker Occludin (Fig. 1G), and increased expression of the mesenchymal markers α-SMA and Vimentin (Fig. 1G) were also seen in cancer cells.

CD204+ M2-like TAMs increase cancer cell migration and miR-210 expression. (A) Representative images show CD204+ cells in human gastric tumor specimens (left). Staining positivity was graded as 0 (no cells with positive staining), 1 (less than 25% cells with positive staining), 2 (25–50% cells with positive staining), and 3 (more than 50% cells with positive staining). Kaplan-Meier analysis was performed to determine the association between CD204 expression and the overall survival in 142 gastric cancer patients. (B) U937 cells were monocultured or cocultured with AGS cells for 72 h. Cell lysates of U937 cells were subjected to Western blot analysis of CD68 and CD204. (C) Seventy-two hours after coculture with U937 cells, AGS cell growth was measured using the cell counters. ∗ P < 0.05 versus monoculture control, Student’s t-test. (D) Seventy-two hours after coculture with U937 cells, the migration of AGS cells was analyzed by the Transwell migration assay (left). The bar graph indicates the numbers of migrating cells (right). ∗ P < 0.05 versus monoculture control, Student’s t-test. (E) After 72 h of coculture with U937 cells, AGS cells were collected and grown in 2-well culture-insert to generate wounds. The wound area was measured from 0 to 16 h. The bar graph indicates the percentages of wound areas (right). ∗ P < 0.05 versus monoculture control, Student’s t-test. (F) Morphology of AGS cells after 72 h of U937 coculture was observed with an optical microscope. (G) Expression of EMT markers in cancer cells cocultured with or without U937 cells for 72 h was measured by Western blotting
MiR-210 contributes to CD204+ M2-like TAMs-induced gastric cancer cell migration
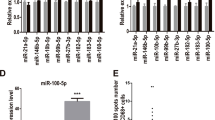
To identify the factors involved in CD204+ M2-like TAMs-induced cancer cell migration, microarray-based miRNA profiling analysis in AGS cells after coculture was conducted. Among the top differentially expressed miRNAs, miR-210 was the most upregulated one with 12.8-fold difference compared to the control group (Fig. 2A). We further validated the microarray data by performing quantitative real-time PCR in human gastric cancer cell lines and tumor tissues. Expression of miR-210 in cancer cell lines after coculture was higher than those in monocultures (Fig. 2B). Similar results could also be seen in AGS cells after coculture with cytokine-induced M2-like macrophages from U937 cells, monocytic THP-1 cells, or monocyte-derived macrophages (Supplementary Figure S1). Elevated expression of miR-210 was more frequently detected in gastric cancer tissues than in the adjacent normal gastric tissues (Fig. 2C). Furthermore, the correlation between miR-210 expression and CD204+ M2-like TAM density was verified in 142 human gastric tumors. MiR-210 expression and CD204 expression were detected by ISH and IHC staining, respectively. The result show that miR210-expressing tumor cells were surrounded by CD204+ M2-like TAMs in close juxtaposition (Fig. 2D). After the staining was scored into 4 grades, the scatter plot of scoring revealed a positive correlation between miR-210 and CD204 expression (P = 0.039; Fig. 2E), suggesting that miR-210 expression in gastric cancer cells could be regulated by CD204+ M2-like TAM TAMs. Because miR-210 expression has been associated with cell migration and metastasis in several types of cancers, [21,22,23,24,25,26] we sought to ascertain whether miR-210 mediates CD204+ M2-like TAMs-induced cancer cell migration. After coculture, the migration ability of AGS cells was increased, and inactivation of miR-210 by the miR-210-specific hairpin inhibitor effectively counteracted the promotion (Fig. 2F), demonstrating that CD204+ M2-like TAMs promote cancer cell migration by upregulating miR-210.

CD204+ M2-like TAMs-induced cancer cell migration is mediated through miR-210. (A) MiRNA expression microarray analysis shows the top 20 downregulated and upregulated miRNAs in AGS cells cocultured with U937 cells in comparison with AGS monocultures. (B) Expression of miR-210 in gastric cancer cells cocultured with or without U937 cells was determined by qPCR. The bar graph depicts the relative miR-210 expression. (C) Expression of miR-210 in gastric tumor tissues and their matched adjacent noncancerous tissues were determined by qPCR. The bar graph depicts the relative miR-210 expression. (D) Representative dual IHC-ISH staining for CD204 (brown) and miR-210 (indigo) in human gastric tumor tissues. Magnification: 100X (left). The image (right) is high-magnification (400X) of the areas outlined by black squares. Red arrows indicate M2 TAMs. Yellow arrows indicate cancer cells expressing miR-210. (E) Representative images show miR-210-positive cells in human gastric tumor specimens (left). Staining positivity was graded as 0 (no cells with positive staining), 1 (less than 25% cells with positive staining), 2 (25–50% cells with positive staining), and 3 (more than 50% cells with positive staining). The correlation between CD204 and miR-210 expression was evaluated using Kendall’s tau correlation (Kendall’s tau-c = 0.151, P = 0.039; right). (F) AGS cells were treated with or without miR-210-specific hairpin inhibitor for 24 h prior to coculture with U937 cells for 72 h. The migration of AGS cells was determined by the Transwell migration assay (left). The bar graph indicates the numbers of migrating cells (right). NS, not significant; ∗ P < 0.05; ∗∗ P < 0.01; ∗∗∗ P < 0.001, significant differences between groups, one-way ANOVA. (G) Expression of EMT markers in AGS cells that were treated with or without miR-210-specific hairpin inhibitor for 24 h prior to coculture with U937 cells for 72 h was measured by Western blotting
CD204+ M2-like TAMs induce miR-210 expression via the TNF-α/NF-κB/HIF-1α pathway
MiR-210 has been demonstrated to be the most consistently and robustly induced miRNA under hypoxia in different types of tumor cells and normal cells [27]. The transcription factor HIF-1 is a master regulator in controlling the adaptive response to hypoxia in cells. Besides hypoxia, HIF-1 protein is also upregulated by the inflammatory mediator NF-κB to promote tumor growth and metastasis, suggesting that HIF-1 links inflammation to oncogenesis [28]. We therefore sought to determine whether the upregulation of miR-210 induced by CD204+ M2-like TAMs is mediated by NF-κB and HIF-1α in cancer cells. Co-culture with U937 cell indeed increased HIF-1α expression (Fig. 3A) and enhanced nuclear translocation of NF-κB (Fig. 3B) in AGS, AZ521, and MKN45 cells. Inhibition of HIF-1α using CAY10585 reduced miR-210 expression (Fig. 3C). Treatment with BAY 11-7082 to suppress NF-κB activity downregulated not only miR-210 (Fig. 3D) but also HIF-1α (Fig. 3E), suggesting NF-κB is an upstream regulator of HIF-1α. Furthermore, neutralizing TNF-α with adalimumab could reduce coculture-induced nuclear translocation of NF-κB (Fig. 3F), suggesting the involvement of TNF-α in the interaction between gastric cancer cells and CD204+ M2-like TAMs. We also identified the cell source of TNF-α in the coculture, and found that after 72 h of coculture, expression of TNF-α mRNA was not changed in AGS cells compared with AGS monoculture. However, expression of TNF-α mRNA was greatly upregulated in U937 cells after coculture with AGS cells for 72 h compared with U937 monoculture (Fig. 3G), suggesting that CD204+ M2-like TAMs were the major source of TNF-α release. In addition, we added exogenous TNF-α to AGS cells and found that p65 could be indeed activated (Fig. 3H). Taken together, these results demonstrate that the TNFα/NF-κB/HIF-1α pathway may contribute to CD204+ M2-like TAMs-dependent induction of miR-210 expression in gastric cancer cells.

CD204+ M2-like TAMs upregulate miR-210 in cancer cells by activating NF-κB and HIF-1α via TNF-α. (A) Cancer cells were cocultured with U937 cells for 72 h. The cancer cell lysates were subjected to Western blotting to detect HIF-1α expression. (B) Seventy-two hours after coculture with U937 cells, cancer cells were lysed and fractionated. Cytoplasmic (Cyto) and nuclear (Nu) extracts were subjected to Western blot analysis for NF-κB p65 detection. Lamin A/C and β-actin were used as a nuclear marker/control and a cytoplasmic marker/control, respectively. (C) After U937 coculture, cancer cells were treated with CAY10585 (10 µM) to inhibit HIF-1α. Expression of miR-210 in cancer cells was assessed by qPCR. The bar graphs depict the relative expression of miR-210. ∗ P < 0.05; ∗∗ P < 0.01 versus control, Student’s t-test. (D-E) After U937 coculture, cancer cells were treated with the NF-κB inhibitor BAY 11-7082 (5 µM). Expression of miR-210 in cancer cells was assessed by qPCR, and HIF-1α protein expression in cancer cells was measured by Western blot analysis. The bar graphs depict the relative expression of miR-210. ∗ P < 0.05; ∗∗ P < 0.01 versus control, Student’s t-test. (F) AGS cells were treated with conditioned medium (CM) from AGS/U937 coculture in the presence or absence of the TNF-α neutralizing antibody adalimumab (10 µg/mL) for 3 h. Nuclear (Nu) extracts of AGS cells were subjected to Western blot analysis for NF-κB p65 detection. The nuclear marker Lamin A/C was used as an internal control. (G) AGS cells and U937 cells were monocultured or cocultured for 72 h. Expression of TNF-α mRNA in AGS cells and U937 cells after monoculture or coculture was measured by qPCR. The bar graphs depict the relative expression of TNF-α mRNA. ∗∗∗ P < 0.001 versus monoculture control, Student’s t-test. (H) AGS cells were treated with TNF-α recombinant protein (10 ng/mL) for 3 h, and expression of phosphorylated p65 and total p65 protein was detected by Western blotting
MiR-210 expression is inversely correlated with NTN4 expression
To identify the gene targets of miR-210, microarray analysis was carried out to search for the downregulated genes in AGS cells after coculture compared with that in monoculture. Among the genes with altered expression, a set of 105 genes (Table S3) were predicted to be putative targets of miR-210 by using several well-known miRNA target gene prediction programs, including TargetScan, MicroCosm, miRanda, PicTar, mirBD and microRNA.org (Fig. 4A). In order to investigate the modulatory effect of miR-210 on M2 TAMs-mediated cancer progression, enrichment analysis using the MetaCore™ platform was performed to identify the biological functions and pathways related to the 105 putative target genes of miR-210. The data show that the 105 potential target genes had a strong correlation with cell migration, and of these genes, the laminin-related secreted protein NTN4 is one of the most downregulated migration-associated genes (Fig. 4B). We then determined the function of NTN4 in gastric cancer cells. Both mRNA and protein levels of NTN4 were much lower in cancer cells after coculture than in cancer cell monocultures (Fig. 4C and D). the 72-hour time course experiment showed that there existed a negative correlation between miR-210 and NTN4 expression during coculture (Fig. 4E). The inverse correlation could be also observed in vivo. After the cell mixture of AGS cells and U937 cells was inoculated into mice to form tumors, stronger miR-210 expression and weaker NTN4 expression were found in the mice with bigger tumors and poor prognosis (#2) than in the mice with small tumors (#1) (Fig. 4F). When we used another gastric cancer cell line SCM-1 in animal experiments, similar results could also be observed (Supplementary Figure S2). We further verified function of NTN4 in gastric cancer cells and found that treatment with human recombinant NTN4 inhibited coculture-induced migration of AGS cells (Fig. 4G). Gene expression of the mesenchymal markers α-SMA, Vimentin, Twist, and Slug could be decreased by NTN4 (Fig. 4H). Gelsolin (GSN), an actin binding protein, functions as a switch to control epithelial-mesenchymal transition (EMT) in cancer. Previous studies have shown that decreased GSN expression enhanced mesenchymal marker expression to promote cell migration, invasion, and metastasis in several types of cancers such as breast cancer, glioblastoma, and gastric cancer [29,30,31]. We also found that, after NTN4 addition, GSN expression was increased in AGS cells (Fig. 4I), suggesting the contribution of GSN to the effect of NTN4 on cell migration. Collectively, these results indicate that NTN4 is a negative regulator of cell migration and EMT.

NTN4 expression is repressed by CD204+ M2-like TAMs and has a negative correlation with miR-210. (A) Schema of the candidate genes identified by two independent approaches. The left circle represents a bioinformatics prediction of putative target genes of miR-210. The right circle indicates the genes downregulated in AGS-U937 coculture, which were identified by mRNA microarray analysis. Through the intersection of these two gene sets, 105 genes were obtained. (B) Metacore analysis of putative target genes. (C) After monoculture and U937 coculture, mRNA expression of NTN4 in cancer cells was determined by qPCR. The bar graph depicts the relative NTN4 mRNA expression. (D) After monoculture and U937 coculture, protein expression of NTN4 in cancer cells was analyzed by Western blotting. (E) AGS cells were cocultured with U937 cells for the indicated time points. NTN4 mRNA and miR-210 levels were measured by qPCR, and their expression relative to the time point of 0 h was represented using a line graph. (F) Expression of miR-210 and NTN4 protein was detected by ISH and IHC, respectively, in AGS xenografts in mice with ascites (#2) or without ascites (#1). (G) AGS cells were cocultured with U937 cells. After exposure to human recombinant NTN4 (100 ng/mL), the migration of AGS cells was analyzed with the Transwell migration assay (left). The bar graph indicates the numbers of migrating cells (right). ∗∗ P < 0.01 versus control, Student’s t-test. (H) After treatment with NTN4 (100 ng/mL), mRNA expression of the mesenchymal markers including α-SMA, Vimentin, Twist, and Slug in AGS cells was determined by qPCR. The bar graph depicts the relative mRNA expression. ∗ P < 0.05; ∗∗∗ P < 0.001 versus control, Student’s t-test. (I) After treatment with NTN4 (100 ng/mL), GSN expression in AGS cells was measured by Western blotting. (J) Expression of NTN4 mRNA in human gastric tumor specimens and the matched adjacent noncancerous tissues was assessed by qPCR. (K) Kaplan-Meier analysis was conducted to examine the association between NTN4 protein expression and the overall survival in 142 gastric cancer patients. (L) The Kaplan-Meier method was performed to evaluate the association of combined expression of miR-210 and NTN4 with the overall survival in 142 gastric cancer patients
The combination of low NTN4 expression and high miR-210 expression portends poor patient survival
To determine the significance of miR-210 and NTN4 in gastric cancer, we measured their expression in tumor tissues, and carried out Kaplan–Meier to estimate overall survival. NTN4 mRNA expression was lower in gastric tumor tissues than in noncancerous parts (Fig. 4J). After IHC staining for NTN4, we further divided patients into the NTN4high group (staining positivity > 50%) and the NTN4low group (staining positivity ≤ 50%). NTN4 expression was marginally associated with patient outcomes (Fig. 4K). However, patients with simultaneously high miR-210 expression and low NTN4 expression had significantly worst overall survival among all groups (Fig. 4L). Taken together, NTN4 might be a potential prognostic factor for gastric cancer.
MiR-210 suppresses NTN4 expression by directly binding to the 3’-UTR of NTN4 transcript
To verify whether NTN4 expression regulated by miR-210, we manipulated the expression of miR-210 in cancer cells by transient transfection with the miR-210 hairpin inhibitors or artificial miR-210 mimics. Suppression of miR-210 expression by the miR-210 hairpin inhibitors (Fig. 5A) increased both mRNA (Fig. 5B) and protein expression (Fig. 5C) of NTN4 in AGS, AZ521, and MKN45 cells. In contrast, transfection with synthetic miR-210 mimics to enforce expression of miR-210 (Fig. 5D) diminished NTN4 mRNA expression in AGS, AZ521 and MKN45 cells (Fig. 5E). Furthermore, to examine whether miR-210 represses NTN4 expression by directly targeting NTN4 mRNA transcripts, we used the MicroCosm prediction program to search for potential miR-210 binding sites on NTN4 mRNA. We found that the 3’-UTR of NTN4 mRNA houses a sequence that matches the 7-mer seed sequence of miR-210 (Fig. 5F). To test the prediction, the NTN4 3’UTR was cloned into luciferase reporter vectors. After transfection of reporter into cancer cells, we treated the cells with miR-210 mimics or non-targeting control miRNAs. The luciferase activity was reduced by approximately 40% in AGS cells and 50% in AZ521 cells in comparison with control, whereas the introduction of point mutations into the miR-210 seed sequence of the NTN4 3′UTR could prevent this effect (Fig. 5G). Taken together, these results clearly demonstrated NTN4 as a direct target of miR-210.

NTN4 is a direct target of miR-210. (A) Cancer cells were transiently transfected with miR-210 hairpin inhibitors or scrambled control for 48 hours. Expression of miR-210 was measured by qPCR. The bar graphs depict the relative expression of miR-210. ∗ P < 0.05; ∗∗ P < 0.01 versus scrambled controls, Student’s t-test. (B-C) Expression of NTN4 mRNA and protein in cancer cells transfected with miR-210 hairpin inhibitors or scrambled control was measured by qPCR and Western blotting, respectively. The bar graphs depict the relative expression of NTN4 mRNA. ∗ P < 0.05; ∗∗ P < 0.01 versus scrambled controls, Student’s t-test. (D) Cancer cells were transiently transfected with miR-210 mimics or scrambled control miRNAs for 48 hours. Expression of miR-210 was measured by qPCR. The bar graphs depict the relative expression of miR-210. ∗ P < 0.05; ∗∗P < 0.01; ∗∗∗ P < 0.001 versus scrambled controls, Student’s t-test. (E) Expression of NTN4 mRNA in cancer cells transfected with miR-210 mimics or scrambled control miRNAs was determined by qPCR. The bar graphs depict the relative expression of NTN4 mRNA. ∗ P < 0.05; ∗∗ P < 0.01 versus scrambled controls, Student’s t-test. (F) Construction of the wild-type and the mutant NTN4 3’-UTR reporters. (G) Luciferase activity in cancer cells cotransfected with miR-210 mimics and luciferase reporters harboring wild-type (wt) or mutant NTN4 3’-UTR (mut) were analyzed with the luciferase reporter assay. The bar graph depicts the relative luciferase activity. ∗ P < 0.05; ∗∗ P < 0.01 vs. scrambled controls, Student’s t-test
Discussion
Highly plastic macrophages originating from circulating monocytes are capable of differentiating into TAMs in the TME to facilitate cancer progression and metastasis; [32] which has been well documented but remains a poorly understood phenomenon at the molecular level. Because both TAMs and miRNAs are highly involved in cancer-related inflammation, the correlation between TAMs and miRNA regulation in cancer cells was investigated in this study. We found that miR-210 in cancer cells was profoundly upregulated in the presence of CD204+ M2-like TAMs. Increased miR-210 expression promoted migration ability of cancer cells by inhibiting NTN4 expression. We further identified that the induction of miR-210 expression by CD204 + M2-like TAMs was mediated through the NF-κB/HIF-1α pathway.
In response to hypoxic stress, miR-210 can modulate many cellular adaptations such as cell proliferation, DNA repair, metabolism, cell migration, and angiogenesis [33]. Given that hypoxia commonly occurs in solid tumors, hypoxia-inducible miR-210 has also been implicated in tumor development and progression. It has been observed that miR-210 is highly expressed in several types of cancers, such as breast cancer, lung cancer, oral cancer, pancreatic cancer, and melanoma [34,35,36,37,38]. Increased expression of miR-210 predicts unfavorable survival in cancer patients [39]. High miR-210 expression levels are positively correlated with lymph node metastasis in pancreatic ductal adenocarcinoma patients [40]. Here, we also found that miR-210 was frequently upregulated in gastric tumors, which was associated with poor patient outcomes. In addition, CD204+ M2-like TAMs promoted tumor progression by regulating miR-210. Taken together, miR-210 upregulation in cancers is a common event and can be as an effective biomarker and prognostic tool in gastric cancer.
Recent research reveals that miRNAs not only exist inside cells but also detect in extracellular space. Intriguingly, miRNAs in plasma or serum remain stable under severe conditions, such as high temperatures, extreme pH values, repeated freeze-thaw cycles, and long-term storage [41,42,43]. The high stability of the circulating miRNAs may result from their smaller size and the protection provided by exosomes, RNA-binding proteins, or lipoprotein complexes [44,45,46]. Because exosomes deliver messages from donor cells to nearby or distant cells, the existence of miRNAs in exosomes suggests a role for circulating miRNAs in cell-to-cell communication. Previously, only direct contact or secretion of cytokine/chemokine is considered to mediate the communication between macrophages and tumor cells; however, increasing evidence has shown that macrophages are capable of secreting exosomes containing both genetic and protein materials to regulate tumor progression [47, 48].
NTN4 belongs to the Netrin family and acts as a regulator of neurite outgrowth and branching and cell migration [49]. Recently, increasing evidence has suggested that NTN4 plays a suppressor role in cancer. In breast cancer, only about 40% of tumors expressed detectable amounts of NTN4 at the protein level, and the expression of NTN4 was associated with better overall survival and disease-free survival [50]. Overexpression of NTN-4 impaired tumor growth and angiogenesis in a mouse xenograft model of colon cancer. Similarly, we also found lower NTN4 mRNA expression in gastric tumor tissues compared with corresponding noncancerous tissues, and gastric cancer patients with high NTN4 expression had favorable survival, suggesting that NTN4 exerts a protective function against cancer progression.
In conclusion, we have clearly demonstrated that CD204+ M2-like TAMs are able to suppress NTN4 expression by upregulating miR-210 in gastric cancer cells, thereby leading to increased cell migration. CD204 + M2-like TAMs induce miR-210 expression mainly through TNF-α that activates the NF-κB/HIF-1α pathway. This study provides new insight into the molecular function of miR-210 in the communication between gastric tumor cells and M2-like TAMs, which represents a viable therapeutic target.
Materials and methods
Cell culture
The human gastric cancer cell lines, AGS and NCI-N87 were purchased from Food Industry Research and Development Institute (Hsinchu, Taiwan). The human gastric cancer cell lines, HR and MKN45, and the duodenal cancer cell line AZ521 were provided by Prof. Chia-Jui Yen (Department of Medical Oncology, National Cheng Kung University Hospital, Tainan, Taiwan). The human leukemic monocyte lymphoma cell line U937 was a kind gift from Prof. Ming-Derg Lai (Department of Biochemistry and Molecular Biology, College of Medicine, National Cheng Kung University, Tainan, Taiwan). All the cell lines were maintained in RPMI-1640 medium (HyClone) supplemented with 10% fetal bovine serum (HyClone), 15 mM HEPES (Biological Industries), 2 mM L-glutamine (Caisson), and 1x antibiotic-antimycotic solution (1,000 units/L penicillin, 2.5 µg/L amphotericin B, and 1000 µg/L streptomycin) (Caisson) and were cultured at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air.
In vitro coculture assay
Transwell inserts with 0.4 μm pore size (BD Bioscience) were used for coculture assays. AGS, NCI-N87, AZ521, HR, or MKN45 cells (1 × 105/well) were seeded in the upper chamber inserts to attach overnight respectively. The next day, 1 × 105 U937 cells were placed in the lower chamber. The coculture was incubated for 72 h.
Total protein extraction
Cells were collected and lysed in RIPA lysis buffer (Millipore) supplemented with protease inhibitor cocktail (Roche). The samples were incubated for 30 min at 4 °C with constant agitation followed by centrifugation at 14,000 rpm for 10 min at 4 °C. The supernatants were transferred to new 1.5 mL tubes and stored at -80 °C. Protein concentration was determined by the BCA protein assay (Pierce).
Isolation of nuclear and cytoplasmic proteins
Cells were harvested and washed once with PBS, followed by resuspension in low-salt permeabilization buffer (10 mM HEPES, pH 7.4, 10 mM KCl, 50 µg/mL digitonin) containing protease inhibitors for 30 min at 4 °C. After centrifugation at 3,000 g for 5 min at 4 °C, the supernatants were collected and saved as cytosolic extracts. The pallets were washed with low-salt permeabilization buffer and then extracted with RIPA lysis buffer, followed by centrifugation at 14,000 rpm for 10 min. The supernatants were saved as nuclear protein. Protein concentration was measured by the BCA protein assay (Pierce).
Western blot analysis
Mixtures containing 30 µg total protein and 6× loading dye were heated at 95 °C for 5 min before being loaded onto SDS-polyacrylamide gels to separate proteins. The proteins were transferred to PVDF membrane (Millipore). The membrane was blocked with 5% milk diluted in 0.05% TBST for 1 h at room temperature followed by incubation with primary antibodies diluted in 0.05% TBST buffer overnight at 4 °C. After washing three times for 10 min each in 0.05% TBST, the membrane was incubated with HRP-conjugated secondary antibodies diluted in 0.05% TBST buffer for 1 h at room temperature. The blots were developed using Immobilon Western Chemiluminescent HRP Substrate (Millipore) according to the manufacturer’s instruction and were captured by the Biospectrum Imaging System (UVP Ltd.). All antibodies used in this study were listed in Table S1.
RNA extraction
Total RNA of cell lines and gastric cancer specimens was extracted using TRIzol reagent (Invitrogen) and incubated at room temperature for 10 min. After addition of 200 µL chloroform, the samples were mixed gently by hand and incubated at room temperature for 5 min, followed by centrifugation at 12,000 g at 4 °C for 15 min. The aqueous phase supernatant was carefully transferred to a new tube and mixed gently with 700 µL isopropyl alcohol. After incubation on ice for 2 h, the samples were centrifuged at 12,000 g at 4 °C for 10 min. The supernatant was discarded, and the RNA pellet was washed with 75% ethanol and was air-dried at room temperature for few minutes. The dry pellet was resuspended in nuclease-free water and the RNA concentration was determined by the ratio of spectrophotometric absorbance of the sample at 260 nm to that of 280 nm.
Reverse transcription
One µg total RNA was reverse transcribed for each cDNA. Firstly, each RNA sample was mixed with 1 µL oligo-dT primer (0.5 µg/µL; Promega) and heated at 95 °C for 5 min to disrupt the secondary structure of mRNA. The samples were immediately cooled on ice and mixed with 1 µL dNTP mix (10 mM; Promega), 0.5 µL recombinant RNasin ribonuclease inhibitor (40 units/µL; Promega), 4 µL M-MLV 5× reaction buffer (Promega), 1 µL M-MLV reverse transcriptase (200 units/µL; Promega) and nuclease-free water to bring the final volume to 20 µL. The reaction mixtures were incubated in a thermocycler (Bio-Rad) at 42 °C for 60 min and 70 °C for 15 min and were held at 4 °C. The product was stored at -20 °C for further use.
Quantitative real time polymerase chain reaction (qPCR) for mRNAs
One µL cDNA template was added to 9 µL mixed reagent containing 1 µL forward primers (10 µM), 1 µL reverse primers (10 µM), 5 µL GoTaq qPCR master mix (2×; Promega), and 2 µL nuclease-free water in a 96-well plate (Applied Biosystems). The plate was sealed and centrifuged at low speed for 1 min to collect all reaction components at the bottom. The reaction was performed using the StepOne Real-Time PCR System (Applied Biosystems), and the protocol was set as follows: initial denaturation at 95 °C for 5 min, 40 cycles of denaturation at 95 °C for 10 s, annealing at 60 °C for 10 s, and extension at 72 °C for 10 s. During the last cycle, the length of the extension step was increased to 7 min, and finally the temperature was held at 4 °C.
qPCR assay for mature miRNAs
Each reverse transcription reaction for miRNA detection was carried out using 30 ng of total RNA extracted from cancer cell lines or human tumor specimens. The miRNA qPCR was performed using the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems), TaqMan Universal PCR Master Mix (Applied Biosystems), and TaqMan MicroRNA Assay primers for human miR-210 (Applied Biosystems), according the manufacturer’s instructions. Amplification and detection of the qPRC products were performed with the StepOne Real-Time PCR System (Applied Biosystems). The levels of miRNAs were normalized to U44 controls.
In situ hybridization (ISH)
ISH for miR-210 in deparaffinized human tumor tissues was performed according to a previously published protocol [51]. Tissue sections were fixed in 4% paraformaldehyde for 20 minutes at room temperature and treated with proteinase K (Clontech) for 15 minutes at 37°C. The sections were then incubated with pre-hybridization solution (Biochain) for 4 hours at 60°C followed by hybridization with the probe cocktail containing 10 nM digoxigenin-conjugated probe (Exiqon) in hybridization solution (Biochain) at 60°C overnight. The sequences of the probes containing the dispersed locked nucleic acid modified bases with digoxigenin conjugated to the 5’ end were: miR-210 5’-TCAGCCGCTGTCACACGCACAG-3’. After hybridization, the sections were incubated with blocking solution (Biochain) for 1 h at room temperature and then AP-conjugated antidigoxigenin antibodies (Roche) for 1 h at room temperature. After washing with alkaline phosphatase buffer (Biochain), the probe/target complexes were visualized by chromogen nitroblue tetrazolium and bromochloroindolyl phosphate (Biochain).
Immunohistochemistry (IHC)
A total of 142 gastric tumor tissue specimens were collected from patients who underwent gastrectomy in National Cheng Kung University Hospital. The study protocol was reviewed and approved by the Institutional Review Board of National Cheng Kung University Hospital. The slides of paraffin-embedded tissue sections were put into xylene for 10 min three times to remove paraffin, followed by 100% ethanol for 5 min twice, 95% ethanol for 5 min twice, and 75% ethanol for 5 min twice for rehydration. After PBS wash, slides were incubated in sodium citrate buffer (10 mM, pH 6.0) and heated by autoclave for 10 min at 121 °C for antigen retrival. The endogenous peroxidase activity was quenched by incubation in methanol containing 3% hydrogen peroxide for 30 min. The sections were incubated with blocking buffer (Thermo) at room temperature for 30 min and then incubated with primary antibodies diluted in antibody diluents (DAKO) overnight at 4 °C. After washing three times with PBS, the slides were incubated with biotinylated secondary antibodies (DAKO) for 30 min at room temperature. The immunoreaction products were visualized using the DAB chromogen system (DAKO), and the slides were counterstained with hematoxylin.
Transient transfection with oligonucleotides
Cells at 70% confluence in 6-well plates were transfected with miRNA mimics or miRNA hairpin inhibitors (100 pmol; Ambio) using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer’s instruction. Cells were harvested 48 h after transfection.
Construction of the 3’-UTR-luciferase plasmids and reporter assay
The netrin-4 (NTN4) 3’UTR possessing the predicted miR-210 target site was amplified from human genomic DNA by PCR using the primers listed in Table S2. The NTN4 3’UTR was then inserted downstream of the luciferase in the pMIR-REPORT luciferase vector which was a kind gift from Dr. Tse-Ming Hong (National Cheng Kung University, Taiwan). Site-directed mutagenesis of the miR-210 target site within the NTN4 3’UTR was carried out using the primers NTN4-UTR-mut-F 5’-TCTGTACCTGTATGTGACCACTATACATAGTTTCTTTTGTAC-3’ and NTN4-UTR-mut-R 5’-AGTGGTGGTCACATACAGGTACAGAAGATGAATAATAATGAAA-3’. For reporter assays, cells were transiently transfected with the reporter plasmids and the artificial mimic of miR-210 (Applied Biosystems) using Lipofectamine 2000 (Invitrogen). To correct for transfection efficiency, the Renilla luciferase encoding plasmids were cotransfected into cells with the reporter plasmids. Forty-eight hours after transfection, luciferase activity was measured by chemoluminescence assay using the Dual-Luciferase Reporter Assay system (Promega).
Migration assay
In vitro cell migration assays were performed using Transwell chambers (24-well, 8-µm pore size; BD Biosciences). The lower chamber was filled with 600 µL of growth medium containing 20 µg/mL fibronectin (Sigma). 1 × 105 cells in 100 µL of serum free medium were seeded in the upper chamber and incubated at 37 °C for 8 h. Cells in the upper side of the membrane were removed gently with a cotton swab and rinsed. Cells that migrated through the membrane and attached to the bottom of the membrane were fixed in methanol for 10 min at room temperature, followed by staining with hematoxylin (Merck).
Wound healing assay
AGS cells were cultured alone or cocultured with U937 cells for 72 h, and were collected for would healing assay. The collected AGS cells were grown in the ibidi Culture-Insert 2 well (Thermo). When both wells were filled with adherent cells, a cell-free wound was generated after removing the insert. Cell migration into the wound area was monitored and photographed at 0 and 16 h using an optical microscope.
Tumor formation in BALB/c nude mice
CAnN.Cg-Foxn1nu/Crl (BALB/c Nude) mice were purchased from the BioLASCO Taiwan (Bltw). The mixtures of AGS cells and U937 cells (1:1; 1 × 106) were subcutaneously injected into the mice (age of 6–8 weeks old) to form tumors. After 4 weeks of tumor growth, tumors from the group of mice with ascites (n = 5 at least) and the group of mice without ascites (n = 5 at least) were collected for further histological examination. All procedures were approved by the Institutional Animal Care and Use Committee of NCKU.
Statistical analysis
Statistical analysis of the data performed using the Student’s t-test and ANOVA. Values expressed as means ± SEM of triplicate determinations. Overall survival curves were plotted using the Kaplan-Meier method and the differences were calculated using log-rank test. P value less than 0.05 was considered statistically significant.
Data and code availability
The authors declare that all data supporting the findings of this study are available within the paper and its supplemental information files.
Abbreviations
- EMT:
-
Epithelial-mesenchymal transition
- LPS:
-
Lipopolysaccharides
- IHC:
-
Immunohistochemistry
- iNOS:
-
inducible nitric oxide synthase
- ISH:
-
In situ hybridization
- miRNA:
-
microRNA
- NTN4:
-
Netrin-4
- qPCR:
-
Quantitative real time polymerase chain reaction
- TAM:
-
Tumor-associated macrophage
- Th2:
-
T helper type 2
- TME:
-
Tumor microenvironment
- 3’ UTR:
-
3’ Untranslated region
References
Greten FR, Grivennikov SI (2019) Inflammation and Cancer: triggers, mechanisms, and consequences. Immunity 51:27–41. https://doi.org/10.1016/j.immuni.2019.06.025
Wessler S, Krisch LM, Elmer DP, Aberger F (2017) From inflammation to gastric cancer - the importance of Hedgehog/GLI signaling in Helicobacter pylori-induced chronic inflammatory and neoplastic Diseases. Cell Commun Signal 15:15. https://doi.org/10.1186/s12964-017-0171-4
Tatsuta M, Iishi H, Nakaizumi A, Okuda S, Taniguchi H, Hiyama T, Tsukuma H, Oshima A (1993) Fundal atrophic gastritis as a risk factor for gastric cancer. Int J Cancer 53:70–74
Ito M, Haruma K, Kamada T et al (2002) Helicobacter pylori eradication therapy improves atrophic gastritis and intestinal metaplasia: a 5-year prospective study of patients with atrophic gastritis. Aliment Pharmacol Ther 16:1449–1456
Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L (2019) Macrophages and metabolism in the Tumor Microenvironment. Cell Metab 30:36–50. https://doi.org/10.1016/j.cmet.2019.06.001
Komohara Y, Fujiwara Y, Ohnishi K, Takeya M (2016) Tumor-associated macrophages: potential therapeutic targets for anti-cancer therapy. Adv Drug Deliv Rev 99:180–185. https://doi.org/10.1016/j.addr.2015.11.009
Caux C, Ramos RN, Prendergast GC, Bendriss-Vermare N, Menetrier-Caux C (2016) A milestone review on how macrophages affect Tumor Growth. Cancer Res 76:6439–6442. https://doi.org/10.1158/0008-5472.CAN-16-2631
Ohta M, Kitadai Y, Tanaka S, Yoshihara M, Yasui W, Mukaida N, Haruma K, Chayama K (2003) Monocyte chemoattractant protein-1 expression correlates with macrophage infiltration and Tumor vascularity in human gastric carcinomas. Int J Oncol 22:773–778
Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Okumura H, Matsumoto M, Miyazono F, Hokita S, Aikou T (2003) Tumor-associated macrophage (TAM) infiltration in gastric cancer. Anticancer Res 23:4079–4083
Aras S, Zaidi MR (2017) TAMeless traitors: macrophages in cancer progression and Metastasis. Br J Cancer 117:1583–1591. https://doi.org/10.1038/bjc.2017.356
Inui M, Martello G, Piccolo S (2010) MicroRNA control of signal transduction. Nat Rev Mol Cell Biol 11:252–263. https://doi.org/10.1038/nrm2868
Fabbri M, Croce CM, Calin GA (2008) MicroRNAs Cancer Journal 14:1–6. https://doi.org/10.1097/PPO.0b013e318164145e
Calin GA, Sevignani C, Dumitru CD et al (2004) Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA 101:2999–3004. https://doi.org/10.1073/pnas.0307323101
Lu J, Getz G, Miska EA et al (2005) MicroRNA expression profiles classify human cancers. Nature 435:834–838. https://doi.org/10.1038/nature03702
Volinia S, Calin GA, Liu CG et al (2006) A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA 103:2257–2261. https://doi.org/10.1073/pnas.0510565103
Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K (2010) STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell 39:493–506. https://doi.org/10.1016/j.molcel.2010.07.023
Kong D, Piao YS, Yamashita S et al (2012) Inflammation-induced repression of Tumor suppressor miR-7 in gastric Tumor cells. Oncogene 31:3949–3960. https://doi.org/10.1038/onc.2011.558
Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z (2019) Tumor-associated macrophages: an accomplice in solid Tumor progression. J Biomed Sci 26:78. https://doi.org/10.1186/s12929-019-0568-z
Lin CN, Wang CJ, Chao YJ, Lai MD, Shan YS (2015) The significance of the co-existence of osteopontin and tumor-associated macrophages in gastric cancer progression. BMC Cancer 15:128. https://doi.org/10.1186/s12885-015-1114-3
Kuang DM, Wu Y, Chen N, Cheng J, Zhuang SM, Zheng L (2007) Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood 110:587–595. https://doi.org/10.1182/blood-2007-01-068031
Rothe F, Ignatiadis M, Chaboteaux C et al (2011) Global microRNA expression profiling identifies MiR-210 associated with Tumor proliferation, invasion and poor clinical outcome in Breast cancer. PLoS ONE 6:e20980. https://doi.org/10.1371/journal.pone.0020980
Ying Q, Liang L, Guo W et al (2011) Hypoxia-inducible microRNA-210 augments the metastatic potential of Tumor cells by targeting vacuole membrane protein 1 in hepatocellular carcinoma. Hepatology (Baltimore, Md). 54:2064–2075. https://doi.org/10.1002/hep.24614
Redova M, Poprach A, Besse A et al (2013) MiR-210 expression in Tumor tissue and in vitro effects of its silencing in renal cell carcinoma. Tumour Biology: The Journal of the International Society for Oncodevelopmental Biology and Medicine 34:481–491. https://doi.org/10.1007/s13277-012-0573-2
Liu Y, Han Y, Zhang H, Nie L, Jiang Z, Fa P, Gui Y, Cai Z (2012) Synthetic miRNA-mowers targeting mir-183-96-182 cluster or miR-210 inhibit growth and migration and induce apoptosis in Bladder cancer cells. PLoS ONE 7:e52280. https://doi.org/10.1371/journal.pone.0052280
Liu T, Zhao L, Chen W, Li Z, Hou H, Ding L, Li X (2014) Inactivation of Von Hippel-Lindau increases Ovarian cancer cell aggressiveness through the HIF1alpha/miR-210/VMP1 signaling pathway. Int J Mol Med 33:1236–1242. https://doi.org/10.3892/ijmm.2014.1661
Qu A, Du L, Yang Y et al (2014) Hypoxia-inducible MiR-210 is an Independent prognostic factor and contributes to Metastasis in Colorectal cancer. PLoS ONE 9:e90952. https://doi.org/10.1371/journal.pone.0090952
Huang X, Ding L, Bennewith KL, Tong RT, Welford SM, Ang KK, Story M, Le QT, Giaccia AJ (2009) Hypoxia-inducible mir-210 regulates normoxic gene expression involved in Tumor initiation. Mol Cell 35:856–867. https://doi.org/10.1016/j.molcel.2009.09.006
Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L (2003) IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 17:2115–2117. https://doi.org/10.1096/fj.03-0329fje
Tanaka H, Shirkoohi R, Nakagawa K et al (2006) siRNA gelsolin knockdown induces epithelial-mesenchymal transition with a cadherin switch in human mammary epithelial cells. Int J Cancer 118:1680–1691. https://doi.org/10.1002/ijc.21559
Zhang J, Furuta T, Sabit H et al (2020) Gelsolin inhibits malignant phenotype of glioblastoma and is regulated by mir-654-5p and miR-450b-5p. Cancer Sci 111:2413–2422. https://doi.org/10.1111/cas.14429
Wang HC, Chen CW, Yang CL, Tsai IM, Hou YC, Chen CJ, Shan YS (2017) Tumor-Associated macrophages promote epigenetic silencing of Gelsolin through DNA methyltransferase 1 in gastric Cancer cells. Cancer Immunol Res 5:885–897. https://doi.org/10.1158/2326-6066.CIR-16-0295
Solinas G, Germano G, Mantovani A, Allavena P (2009) Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol 86:1065–1073. https://doi.org/10.1189/jlb.0609385
Huang X, Le QT, Giaccia AJ (2010) MiR-210–micromanager of the hypoxia pathway. Trends Mol Med 16:230–237. https://doi.org/10.1016/j.molmed.2010.03.004
Radojicic J, Zaravinos A, Vrekoussis T, Kafousi M, Spandidos DA, Stathopoulos EN (2011) MicroRNA expression analysis in triple-negative (ER, PR and Her2/neu) breast cancer. Cell cycle (Georgetown, Tex.). 10: 507 – 17
Puissegur MP, Mazure NM, Bertero T et al (2011) miR-210 is overexpressed in late stages of Lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ 18:465–478. https://doi.org/10.1038/cdd.2010.119
Scapoli L, Palmieri A, Lo Muzio L, Pezzetti F, Rubini C, Girardi A, Farinella F, Mazzotta M, Carinci F (2010) MicroRNA expression profiling of oral carcinoma identifies new markers of Tumor progression. Int J ImmunoPathol Pharmacol 23:1229–1234
Greither T, Grochola LF, Udelnow A, Lautenschlager C, Wurl P, Taubert H (2010) Elevated expression of microRNAs 155, 203, 210 and 222 in pancreatic tumors is associated with poorer survival. Int J Cancer 126:73–80. https://doi.org/10.1002/ijc.24687
Satzger I, Mattern A, Kuettler U, Weinspach D, Voelker B, Kapp A, Gutzmer R (2010) MicroRNA-15b represents an Independent prognostic parameter and is correlated with Tumor cell proliferation and apoptosis in malignant Melanoma. Int J Cancer 126:2553–2562. https://doi.org/10.1002/ijc.24960
Wang J, Zhao J, Shi M, Ding Y, Sun H, Yuan F, Zou Z (2014) Elevated expression of miR-210 predicts poor survival of cancer patients: a systematic review and meta-analysis. PLoS ONE 9:e89223. https://doi.org/10.1371/journal.pone.0089223
Wang J, Raimondo M, Guha S et al (2014) Circulating microRNAs in Pancreatic Juice as candidate biomarkers of Pancreatic Cancer. J Cancer 5:696–705. https://doi.org/10.7150/jca.10094
Mitchell PS, Parkin RK, Kroh EM et al (2008) Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA 105:10513–10518. https://doi.org/10.1073/pnas.0804549105
Brase JC, Wuttig D, Kuner R, Sultmann H (2010) Serum microRNAs as non-invasive biomarkers for cancer. Mol Cancer 9:306. https://doi.org/10.1186/1476-4598-9-306
Chen X, Ba Y, Ma L et al (2008) Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other Diseases. Cell Res 18:997–1006. https://doi.org/10.1038/cr.2008.282
Arroyo JD, Chevillet JR, Kroh EM et al (2011) Argonaute2 complexes carry a population of circulating microRNAs Independent of vesicles in human plasma. Proc Natl Acad Sci USA 108:5003–5008. https://doi.org/10.1073/pnas.1019055108
Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9:654–659. https://doi.org/10.1038/ncb1596
Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT (2011) MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol 13:423–433. https://doi.org/10.1038/ncb2210
Yang M, Chen J, Su F, Yu B, Su F, Lin L, Liu Y, Huang JD, Song E (2011) Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into Breast cancer cells. Mol Cancer 10:117. https://doi.org/10.1186/1476-4598-10-117
Menck K, Klemm F, Gross JC, Pukrop T, Wenzel D, Binder C (2013) Induction and transport of wnt 5a during macrophage-induced malignant invasion is mediated by two types of extracellular vesicles. Oncotarget 4:2057–2066
Koch M, Murrell JR, Hunter DD, Olson PF, Jin W, Keene DR, Brunken WJ, Burgeson RE (2000) A novel member of the netrin family, beta-netrin, shares homology with the beta chain of laminin: identification, expression, and functional characterization. The Journal of cell biology. 151: 221 – 34
Esseghir S, Kennedy A, Seedhar P, Nerurkar A, Poulsom R, Reis-Filho JS, Isacke CM (2007) Identification of NTN4, TRA1, and STC2 as prognostic markers in Breast cancer in a screen for signal sequence encoding proteins. Clin cancer Research: Official J Am Association Cancer Res 13:3164–3173. https://doi.org/10.1158/1078-0432.ccr-07-0224
Obernosterer G, Martinez J, Alenius M (2007) Locked nucleic acid-based in situ detection of microRNAs in mouse tissue sections. Nat Protoc 2:1508–1514. https://doi.org/10.1038/nprot.2007.153
Acknowledgements
We thank the staff of Clinical Medicine Research Center, National Cheng Kung University Hospital, Tainan, Taiwan for the assistance in providing careful review and insightful comments regarding study design, analysis, manuscript wiring, submission and revision. We are also grateful for the support from the Human Biobank, Clinical Medicine Research Center, National Cheng Kung University Hospital. This work was supported by Kaohsiung Veterans General Hospital Yong-Kang Branch (grant number VHYK102-03, VHYK103-04, and VHYK104-01) and MOST (grant number 99-2314-B-006-020-MY2). This study was approved by Institute of Review Board of National Cheng Kung University Hospital with IRB number: A-ER-102-071. The informed consent was waived. All the animal experiments were carried out in compliance with national legislation and was approved by the Institutional Animal Care and Use Committee of NCKU and with related codes of practice.
Author information
Authors and Affiliations
Contributions
CWC, HCW, and YSS conceived and designed the study; HCW, IMT, YLC, and CJC performed the experiments; CWC, HCW, ISC, YLC, YCH, and CJC analyzed the data and prepared the figures; CWC, HCW, IMT, and YSS wrote and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, CW., Wang, HC., Tsai, IM. et al. CD204-positive M2-like tumor-associated macrophages increase migration of gastric cancer cells by upregulating miR-210 to reduce NTN4 expression. Cancer Immunol Immunother 73, 1 (2024). https://doi.org/10.1007/s00262-023-03601-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00262-023-03601-5