
Abstract
NY-ESO-1 and LAGE-1 are cancer testis antigens with an ideal profile for tumor immunotherapy, combining up-regulation in many cancer types with highly restricted expression in normal tissues and sharing a common HLA-A*0201 epitope, 157–165. Here, we present data to describe the specificity and anti-tumor activity of a bifunctional ImmTAC, comprising a soluble, high-affinity T-cell receptor (TCR) specific for NY-ESO-1157–165 fused to an anti-CD3 scFv. This reagent, ImmTAC-NYE, is shown to kill HLA-A2, antigen-positive tumor cell lines, and freshly isolated HLA-A2- and LAGE-1-positive NSCLC cells. Employing time-domain optical imaging, we demonstrate in vivo targeting of fluorescently labelled high-affinity NYESO-specific TCRs to HLA-A2-, NY-ESO-1157–165-positive tumors in xenografted mice. In vivo ImmTAC-NYE efficacy was tested in a tumor model in which human lymphocytes were stably co-engrafted into NSG mice harboring tumor xenografts; efficacy was observed in both tumor prevention and established tumor models using a GFP fluorescence readout. Quantitative RT-PCR was used to analyze the expression of both NY-ESO-1 and LAGE-1 antigens in 15 normal tissues, 5 cancer cell lines, 10 NSCLC, and 10 ovarian cancer samples. Overall, LAGE-1 RNA was expressed at a greater frequency and at higher levels than NY-ESO-1 in the tumor samples. These data support the clinical utility of ImmTAC-NYE as an immunotherapeutic agent for a variety of cancers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Understanding of anti-tumor immunity has increased significantly over the past decade, including fundamental insights into the role of innate and adaptive immune responses in targeting and eliminating tumors [1, 2]. In both animal studies and, more recently, clinical studies, CD8+ cytotoxic T cells (CTLs) have been shown to be major effector cells involved in the eradication of tumor cells [3–5]. A requisite step for the targeting of tumors by CTL is the binding of T-cell receptors (TCRs) to peptides derived from the proteome of tumors and presented on the cell surface in the context of MHC Class I molecules (pMHCs) [6]. A major limitation for the effective targeting of established tumors by T cells results from central tolerance, whereby T cells with strong affinity for self-antigens are deleted in the thymus [7]. In addition, the natural selection process inherent in the growth and establishment of tumors in immune-competent hosts leads to the selection of tumors with decreased antigen and MHC expression and alterations in antigen processing and presentation, reducing the surface density of pMHC complexes [8]. Finally, peripheral tolerance mediated through host and tumor-driven immunosuppressive mechanisms contributes to the ultimate inability to mount an effective T-cell immune response to target tumors [9–11].
One approach to overcome the lack of potent anti-tumor T-cell immunity is the ex vivo genetic modification of T cells to target tumors through the use of affinity-enhanced receptors generated from either T cell or antibody-derived receptors; recent clinical data have provided evidence that such approaches hold significant clinical promise [12, 13]. A complementary approach that does not require ex vivo manipulation of T cells involves the use of fusion proteins that combine tumor recognition and T cell engaging domains to redirect T cells to target tumors [14]. Among this class of reagents are several monoclonal antibody-anti CD3 scFv fusion reagents, some of which have undergone clinical evaluation with strong evidence of efficacy [14–17]. The use of antibody-based T-cell targeting agents is restricted to targeting of antigens expressed on the cell surface. To overcome this limitation, we have recently developed a class of bi-specific fusion protein reagents termed ImmTACs (Immune-mobilizing monoclonal TCRs against Cancer), which target proteosomally processed epitopes derived from intracellular, surface bound, and secreted antigens [18]. ImmTACs are comprised of soluble, high-affinity (pico-Molar) monoclonal TCRs (mTCRs) specific for antigen-derived pMHC complexes, combined with a humanized anti-CD3 scFv domain, which activates a potent anti-tumor T-cell response. The first clinical candidate ImmTAC product, IMCgp100, specific for gp100280–288 is currently undergoing Phase I clinical testing in melanoma patients (trial number NCT01211262).
NY-ESO-1 (CT6, also known as LAGE-2) and LAGE-1 are among the prototypic cancer testis antigens [19, 20]. Although little is known about their functions, normal tissue expression of both of these antigens is principally limited to immune privileged sites [20–22]. Like other CT antigens, both NY-ESO-1 and LAGE-1 are expressed during development as well as by a wide range of tumors including myeloma [23, 24] and a variety of solid tumors such as ovarian [25], non-small-cell lung cancer (NSCLC) [26, 27], and melanoma [28, 29]. NY-ESO-1 has been targeted extensively in clinical trials using a variety of approaches including vaccines and gene-modified T cells [13, 30]. Multiple studies have demonstrated that NY-ESO-1 is antigenic in human patients [21, 31], although vaccination approaches alone rarely lead to a clinically significant response [32, 33].
We have previously described both a high-affinity mTCR and ImmTAC reagent (ImmTAC-NYE) specific for the immunogenic HLA-A2 presented peptide NY-ESO-1157–165 (SLLMWITQC) [18, 34]. Notably, since the same epitope is processed from the LAGE-1 antigen [20, 35], ImmTAC-NYE can be used to redirect T cells against HLA-A*0201-positive tumors that express either NY-ESO-1 or LAGE-1 antigens. In this report, we demonstrate that ImmTAC-NYE redirects potent antigen-restricted T-cell activity against NY-ESO-1- and/or LAGE-1-positive tumors, including established cell lines shown previously to present low levels of cell surface epitope [34] and primary lung cancer cells. We show in vivo targeting of high-affinity NYE TCR to tumors presenting the NY-ESO epitope via time-domain optical imaging of fluorescently labelled mTCR. Importantly, we demonstrate ImmTAC-NYE-mediated inhibition of tumor growth in vivo, in a model co-xenografted with both human peripheral blood lymphocytes and cancer cells. The results provide rationale to support the further preclinical and clinical development of ImmTAC-NYE as a bi-specific immunotherapeutic agent to treat HLA-A*0201-positive and NY-ESO-1- and/or LAGE-1-expressing solid cancers.
Materials and methods
Human tissues, cells, and cell lines
Primary tumor tissue from lung and ovarian cancer patients was collected at the University of Pennsylvania under an IRB approved protocol. Pathologic review of biopsies confirmed diagnosis in each case. Tumor biopsies were processed as described [36] to obtain single-cell suspensions which were viably frozen.
Cell lines J82 (urothelial), SK-Mel-37, Mel526, Mel624, A375 (melanoma), IM9 (EBV B), and U266 (multiple myeloma) were obtained from American Type Culture Collection or as described previously [34] and cultured in R-10 medium. Cell lines were acquired between 2004 and 2010; HLA genotype and/or phenotype and expression of various cancer-associated antigens were confirmed by DNA sequencing, RT-PCR, and antibody staining, and lines displayed phenotypic characteristics of the tumor of origin. HEP2, HA2, and CM12 (normal human cells grown in culture) were purchased from ScienCell and NHEM10 cells from PromoCell.
Construction of J82-NY-ESO157–165 and SK-Mel-37-NY-ESO157–165 minigene-transfected cell lines has been described previously (as SLLMWITQC-minigene transfectants) [34].
J82-NY-ESO157–165 cells stably expressing enhanced green fluorescent protein (EGFP) (Clontech Laboratories, Inc., Mountain View, CA) were engineered using the pCGFP retroviral vector [37]. Production of infectious retroviral vector particles in Phoenix A cells and infection of cells were carried out as described [38]. This procedure was repeated twice, producing cells expressing high levels of GFP as determined by fluorescence microscopy (Leica Microsystems, GmbH, Wetzlar, Germany). The 5 % brightest cell population was isolated by a modified FACSAria II flow cytometer (BD BioSciences).
Peripheral blood lymphocytes (PBLs) were obtained from human healthy volunteers following Ficoll-Hypaque density gradient separation (Lymphoprep, Nycomed Pharma AS, Oslo, Norway).
Magnetic bead immunodepletion (Dynal) removed CD19+, CD4+, and CD14+ cells to obtain CD8+ ve T-cell populations.
Quantitative RT-PCR on normal and neoplastic human cells
RNA was isolated from viably frozen specimens or cell lines growing in logarithmic phase using RNAqueous-4 PCR kits (Ambion corp, AM1914). cDNA was synthesized using iScript cDNA synthesis kits (Biorad Corp, 170-8891). qRT-PCR analysis was performed using standard Taqman—MGB technology and amplification conditions using an ABI 7500 FAST instrument (ABI-Life technologies), and the following ABI inventoried primer-probe sets: NY-ESO-1: HS00265824_m1; LAGE-1: HS00535628_m1; Gus-B: HS99999908_m1; β-Actin: HS99999903_m1. Amplifications were performed in triplicate; individual Ct values were determined (minimum of 2/3 replicates with % CV < 15 %) and average values reported. RQ (relative quantification) values for NY-ESO-1 and LAGE-1 transcripts were determined according to the formula RQ = 2−ΔΔCt, with ΔΔCt = ΔCtsample − ΔCtreference, with ΔCtsample = Ctsample − Ctsample normalizer and ΔCtreference = Ctreference − Ctreference normalizer [39]. For all analyses, the melanoma cell line A375 (positive for NY-ESO-1 and LAGE-1) served as a reference; either Gus-B or β-Actin (for a subset of analyses) was used as the normalizer “housekeeping” gene.
Engineering high-affinity TCRs and bi-specific ImmTACs
TCR isolation and engineering to produce ImmTAC reagents has been described previously [18, 40].
Cytotoxicity and cytokine release assays
Cytotoxicity (LDH release) and IFNγ ELISpot assays were performed as previously described [18]. Multiple cytokine release (TNFα, IFNγ, and IL-2) was measured by Meso Scale Discovery (MSD) immunoassay following overnight incubation of CD8+ effector T cells and targets at a 1:1 ratio in the presence of 1 nM ImmTAC-NYE, in accordance with manufacturer’s instructions.
IncuCyte and real-time quantification of cell killing
Target tumor cells were incubated with effector CD8+ T cells (5:1 E:T) in the presence or absence of ImmTAC-NYE. Images were taken every 10 min and the number of apoptotic cells per mm2 was quantified using the CellPlayer 96-well Kinetic Caspase 3/7 reagent and the IncuCyte-FLR-Platform (Essen Biosciences). The reagent is cleaved by activated Caspase 3/7 upon target cell apoptosis resulting in the release of the dye and green fluorescent staining of nuclear DNA. The cocktail of apoptotic drugs used as a positive control contains 10 μM stausporine, 2 μg/ml anisomycin, and 10 μM etoposide.
TCR fluorescence labelling
TCR-NYE-wt, TCR-NYE-(29 nM), and TCR-NYE-(0.048 nM) were labelled with Alexa® Fluor 680 carboxylic acid, succinimidyl ester as previously described [41]. Labelling was restricted to 2 fluors per molecule. Protein concentration and degree of labelling were determined by spectrophotometric analysis using a NanoDrop spectrophotometer (Thermo Scientific, Rockford, USA).
Animal studies
Animal experiments were approved by The Norwegian Animal Research Authority and performed in accordance with the European Convention for the Protection of Vertebrates Used for Scientific Purposes. NOD-scid and NOD-scid IL2rγnull (NSG) mice (6–8 weeks old) were originally a gift of Prof. L. Shultz, Jackson Laboratories, Bar Harbour, USA, and were bred at the Gades Institute, University of Bergen. 5 × 106 tumor cells were injected subcutaneously (s.c.) in a solution of PBS/Matrigel (1:1) bilaterally in flanks of mice (n = 2 per mouse). Tumor volumes were measured by digital calliper measurements using the formula: Volume = π (length × width × height)/6. Blood samples (100 μL) were acquired by submandibular bleeding.
TCR imaging studies
When J82-NY-ESO157–165 tumors reached an average volume of 100–150 mm3, background fluorescence images of the mice (n = 4 per group) were acquired prior to intravenous (i.v.) injection with Alexa680 labelled TCRs (0.6 mg/kg) at 0 h and the mice imaged for TCR fluorescence at 4, 8, 24, 48, and 96 h.
Tumor protection study in mice harboring human PBL xenografts
NSG mice were inoculated i.v. with 20 × 106 human PBLs and randomized into study groups (equally distributed per percentage hu-CD45+CD3+). When mice demonstrated at least 10 % hu-CD3+ cells by flow cytometry (approx. week 3), SK-Mel-37-NY-ESO157–165 cells [34] were injected s.c. and mice treated i.v. with either ImmTAC-NYE (0.04 mg/kg, Q.D.) or PBS (equivalent volume) on days 1–5.
ImmTAC efficacy studies in mice harboring cancer and human PBL xenografts
When J82-NY-ESO GFP157–165 tumors reached 50 mm3, 20 × 106 human PBL per mouse were injected i.v. After mice demonstrated engraftment of human lymphocytes (at least 10 % hu-CD3+ cells) and tumor volumes reached 100–150 mm3, they were imaged for GFP fluorescence and randomized into study groups. Mice were then treated with ImmTAC-NYE, control ImmTAC-GAG (0.04 mg/kg, Q.D., i.v.), or PBS (equivalent volume, i.v.) for days 1–5 and tumors monitored weekly by time-domain optical imaging. The study was terminated 25 days following the first ImmTAC treatment.
Optical imaging
Time-domain optical imaging was performed with Optix® MX2 (ART Inc., Saint-Laurent, QC, Canada) as described previously [42]. Fluorescence images were acquired and analyzed with Optix® Optiview™ (version 2.00; ART Inc.) and Optix® Optiview™ (version 2.02.00, ART Inc.).
Flow cytometric analysis of blood
Peripheral blood cells were analyzed by FACSCanto II (BD) using mouse anti-huCD3 (PE conjugate; clone SK7) and hu CD45 (FITC conjugate; clone 2D1) monoclonal antibodies (BD Immunocytometry). Human T-cell engraftment in peripheral blood was defined as the percentage of huCD45+CD3+ mononuclear cells (MNCs) in murine peripheral blood.
Results
Expression of NY-ESO-1 in normal and in neoplastic cells and tissues
We evaluated the expression of NY-ESO-1 and LAGE-1 in a panel of normal tissues and established cell lines, as well as a panel of primary tumor material from lung and ovarian cancer patients using quantitative real-time PCR. With the exception of low level LAGE expression in one normal tissue sample (SMC3), we did not detect transcripts for either NY-ESO-1 or LAGE-1 in 15 HLA-A2+ normal tissue cells, including melanocytes, hepatocytes, astrocytes, endothelial cells (aortic and dermal microvascular), fibroblasts (dermal and pulmonary), and epithelial cells (renal and ciliary) (Supplementary Table 1). Both NY-ESO-1 and LAGE-1 were broadly expressed at various levels and across a large number of tumor cell lines, including melanoma, myeloma, mesothelioma, lung, ovarian, and prostate cancer; among the tumor cell lines tested, only the colon cancer cell line Colo205 and the colorectal carcinoma HCT116 were negative for both antigens (Table 1 and Supplementary Table 2).
We further evaluated the expression of NY-ESO-1 and LAGE-1 in primary tumor specimens from ten NSCLC and ten ovarian cancer patients (Table 1). In NSCLC specimens, NY-ESO-1 and LAGE-1 transcripts were detected in 3 of 10 samples. In the ovarian cancer specimens, NY-ESO-1 was detected in 6 of 10 and LAGE-1 in 8 of 10 ovarian cancer specimens. NY-ESO-1 and LAGE-1 expression was not always concordant. Notably, as assessed by average Ct values, in some cases (NSCLC-#81, ovarian #23, #96), LAGE-1 mRNA was expressed at significantly higher levels than NY-ESO-1. Overall, 30 % of the NSCLC samples and 80 % of the ovarian specimens were positive for one or both antigens.
Short-term redirected T-cell killing of NY-ESO-1 tumor-derived cell lines by ImmTAC-NYE
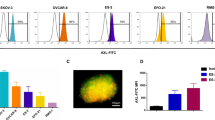
ImmTAC-NYE has a binding affinity in the low pico-Molar range (approximately 50 pM) with a binding half-life of several hours; it has previously been shown to target two NY-ESO-1-/LAGE-1-positive tumor lines and activate T cells resulting in redirected killing [18]. The affinity-enhanced TCR is completely specific for the NY-ESO157–165 epitope presented by HLA-A*0201 demonstrating that specificity has not been lost during engineering (Supplementary Table 3). The ability of ImmTAC-NYE to redirect killing of additional HLA-A2*0201+, NY-ESO-1/LAGE-1+ tumor cell lines over 24 h was assessed by measuring LDH release from the target cells. Figure 1a shows a dose titration of ImmTAC-NYE in the presence of target tumor cells and CD8+ effector T cells. With the exception of the SK-Mel-37 cells, dose-dependent lysis of all the target cells was detected. No lysis was detected in the absence of ImmTAC-NYE. Figure 1b shows that ImmTAC-NYE induces the secretion of multiple cytokines (TNFα, IFNγ, and IL-2) from T cells in response to the IM9 cell line (NY-ESO-1/LAGE-1 high).

Activation and killing by ImmTAC-NYE redirected T cells against NY-ESO-1/LAGE-1 tumor-derived cell lines and freshly isolated tumor cells. a Redirected lysis of HLA-A2+, NY-ESO-1/LAGE+ tumor cell lines (as indicated in the figure key) by ImmTAC-NYE with purified CD8+ effector T cells. Controls: effectors and targets in the absence of ImmTAC-NYE, with the open symbol shapes matching the target cells indicated in the figure key. SK-Mel-37 cells present an average of 25 epitopes per cell, Mel624 cells an average of 45 and U266 cells an average of 5–10. b ImmTAC-NYE induces multiple cytokine release (TNFα, IFNγ, and IL-2) from CD8+ T cells in response to NY-ESO-positive IM9 cells; control cells are Mel526 (NY-ESO low). c IFNγ release from PBMCs in response to ImmTAC-NYE and freshly isolated LAGE-1+ lung cancer sample NSCLC#29 measured by ELISpot; Colo205 cells (antigen negative, HLA-A2+) are the specificity control. Open symbols represent targets and effectors without ImmTAC-NYE, with the symbol shapes matching the target cells indicated in the figure key. d ImmTAC-NYE does not lyse HLA-A2+, NY-ESO-1-/LAGE-1-negative primary human cells derived from normal tissues and expanded in culture. Purified CD8+ T cells were incubated with hepatocytes (HEP2), astrocytes (HA2), melanocytes (NHEM10), and cardiac myocytes (CM12) in the presence of 0.1, 1, and 10 nM ImmTAC-NYE. Antigen-positive U266 cells were included as a positive control. In a and d, lysis was determined by LDH release from target cells after 24 h, with an E:T of 10:1; error bars depict mean values ± SEM
ImmTAC-NYE activates T cells in the presence of freshly isolated tumor cells
To confirm that ImmTAC-NYE recognizes freshly isolated tumor cells in addition to tumor cell lines, we measured T-cell activation in response to lung cancer sample NSCLC#29, which expresses high levels of LAGE-1 (Table 1). Figure 1c shows that IFNγ is released from CD8 T cells in the presence of NSCLC#29 and ImmTAC-NYE, with a similar potency to redirected killing of tumor cell lines. Insufficient numbers of tumor cells were isolated to carry out a killing assay.
ImmTAC-NYE is specific for its target peptide
The specificity of ImmTAC-NYE was investigated using HLA-A2*0201+, NY-ESO-1-/LAGE-1-negative primary human cells derived from normal tissues. The LDH release assay data shown in Fig. 1d demonstrate the lack of lysis of hepatocytes, cardiac myocytes, and melanocytes by concentrations of ImmTAC-NYE up to 1 nM; at 10 nM, a low level of lysis is observed. We have observed a slight loss of epitope specificity at high concentrations (10 nM and above) with other HLA-A2-specific ImmTACs, which is HLA-A2-dependent ([18] and unpublished data).
Direct visualization of redirected tumor cell killing mediated by ImmTAC-NYE
Killing of tumor cell lines over longer time periods was investigated using IncuCyte-FLR technology, enabling visualization of caspase 3/7-dependent apoptosis by microscopy at 37 °C in real time. Target tumor cells were incubated with effector CD8+ T cells in the presence or absence of ImmTAC-NYE and images taken at intervals of 10 min. The killing time course for SK-Mel-37 cells (HLA-A2*0201+, LAGE-1+), a cell line relatively resistant to lysis after 24 h (see Fig. 1a) is shown in Fig. 2a) in the presence of 0.1 nM and 1 nM ImmTAC-NYE. In agreement with the LDH assay (Fig. 1a), no significant killing was observed after 24 h. However, many apoptotic cells were present at 72 h, indicating that for some target cells (e.g., those that are resistant to lysis or with very low epitope levels), ImmTAC-NYE-induced killing requires in excess of 24 h. Figure 2b shows the killing time courses for Mel624 cells, again indicating that cell death occurs mainly after 24 h; the images taken at 48 h (Fig. 2h–i) confirm that close to 100 % of the cells have been killed at this time point. Figure 2c shows killing time courses for normal kidney epithelial cells (REN2) incubated with 0.1 and 1 nM ImmTAC-NYE over 72 h, confirming that normal cells are not killed in response to the ImmTAC even after 3 days. This is the first reported use of IncuCyte-FLR technology to measure CTL activity.

Target cell apoptosis induced by ImmTAC-NYE redirected T cells measured in real time. a SK-Mel-37, b Mel624 melanoma cells, or c REN2 normal renal epithelial cells were incubated with CD8+ cells in the presence or absence of ImmTAC-NYE and images taken at 10-min intervals. In c REN2, cells were also treated with a cocktail of drugs to induce apoptosis (apoptosis cocktail) to control for the presence of live cells. Representative end point images are shown for SK-Mel-37 (d–f, 72 h) and Mel624 (g, h, i 48 h) targets at the indicated concentrations of ImmTAC-NYE. White flashes marked with red-crosses represent cells undergoing apoptosis and are recorded as objects. Control: effectors and targets in the absence of ImmTAC-NYE
In vivo imaging of tumors by soluble NY-ESO-specific TCRs
In vivo tumor targeting of NY-ESO-1 antigen was visualized using Alexa-fluor 680 labelled un-fused TCRs (rather than ImmTAC fusion proteins) in a xenograft model of J82-NY-ESO157–165, a human bladder cell line engineered to express the NY-ESO157–165 epitope [34]. Three TCRs were employed, all binding specifically to this epitope but differing in affinity: a) the wild-type TCR-NYE-wt, b) medium-affinity TCR-NYE-(29 nM), and c) high-affinity TCR-NYE-(0.048 nM); the affinities of the engineered TCRs are indicated in brackets, that is, 29 nM and 48 pM. Time-domain optical imaging of engrafted tumors was performed at intervals following administration of Alexa 680 labelled TCRs. The resulting images (Fig. 3a) were gated for fluorescence lifetimes of the TCR-Alexa 680 conjugates facilitating the distinction of specific mTCR-targeted fluorescence from background fluorescence. Mice inoculated with TCR-NYE-wt exhibited only transient tumoral fluorescence at the 4- and 8-h imaging time points before a return to background fluorescence levels at 48 h (Fig. 3b, c). Imaging of TCR-NYE-(29 nM)-treated mice demonstrated a similar pattern, although fluorescence intensities were higher (24 and 48 h) than those observed for the TCR-NYE-wt-treated mice. Only with mice injected with Alexa 680 labelled high-affinity TCR-NYE-(0.048 nM) were higher than background fluorescence levels observed throughout the time course; significantly, higher fluorescence intensities were noted in these mice at 24 and 48 h (p < 0.05), and even at 96 h, intensities were 2.5 times higher than background.

TCR-Alexa 680 targeting studies. a Mice xenografted with J82-NY-ESO157–165 cells were injected with Alexa 680 labelled TCR and analyzed at intervals using time-domain optical imaging. Images were then gated for the fluorescence lifetime of TCR-Alexa 680 and TCR-Alexa 680 photons quantified. b Representative gated fluorescence images of mice injected with TCR-NYE-wt, TCR-NYE-(29 nM), and TCR-NYE-(0.048 nM) over the time course. Fluorescence scale is the same for all mice. c Histogram of gated fluorescence normalized on 4-h results. Error bars present SEM. *p < 0.05
ImmTAC-NYE therapy prevents the growth of NY-ESO epitope+ tumors in a xenografted mouse model
The evaluation of immuno-therapeutic agents in immunodeficient preclinical models typically requires subcutaneous (s.c.) co-injection of cancer and T cells or repeated injections [s.c. or intravenous (i.v.)] of high numbers of T cells in mice with pre-established tumors. However, these assays do not accurately reflect the potential of endogenous T cells to target cancer cells; for example, imaging studies over 4 days from the time of injection of T cells into tumor-bearing mice reported that less than 1 % of i.v. injected T cells home to tumors [42]. Recent studies have demonstrated the NSG mouse model to be sufficiently immunodeficient to permit the xenografting of functional immune systems [43]. Thus, we investigated whether this host would permit co-engraftment of both cancer cell lines and hu-PBLs, in order to develop a model in which the efficacy of ImmTAC-NYE can be evaluated in vivo. NSG mice were inoculated with hu-PBLs, and flow cytometry analyses showed the presence of robust populations of hu-CD45+CD3+ cells in xenografted recipients from week 3, with all mice exhibiting a hu-CD45+CD3+ cell population of at least 10 % of total peripheral blood lymphocytes. SK-Mel-37-NY-ESO157–165 cells were injected subcutaneously and treatment with either ImmTAC-NYE or PBS was initiated on the same day (Fig. 4a, b); a total of five doses of ImmTAC-NYE were administered.

Tumor protection study in mice harboring human PBL xenografts. a, b Human PBL-engrafted mice were injected subcutaneously with SK-Mel-37-NY-ESO157–165 cells and treated intravenously with ImmTAC-NYE (n = 6) or PBS (n = 6) days 1–5. c Tumor volumes plotted against time. Error bars present SEM. ***p < 0.001
While the tumors in PBS-treated mice underwent progressive growth with average volumes reaching 368 mm3 by day 50, palpable tumors were only just evident in the ImmTAC-NYE-treated mice at this time point (Fig. 4c; p < 0.001). Furthermore, once these tumors had been established, they developed with demonstrably slower growth kinetics only reaching average tumor volumes of 141 mm3 3 months after treatment (data not shown).
ImmTAC-NYE therapy targets established J82-NY-ESO GFP157–165 tumors in mice engrafted with human PBMC
Having demonstrated in vivo targeting of TCR-NYE to J82-NY-ESO157–165 cells and tumor prevention by ImmTAC-NYE in a xenograft model harboring both SK-Mel-37-NY-ESO157–165 cells and hu-CD45+CD3+ T-cell populations, we sought to evaluate anti-tumor efficacy of ImmTAC-NYE using optical imaging in pre-established tumors. J82-NY-ESO GFP157–165 cells were injected s.c. into the flanks of NSG mice, and when the tumors reached approximately 50 mm3, mice were inoculated with hu-PBL (Supplementary Figure 1). When recipient mice demonstrated at least 10 % T-cell chimerism (Supplementary Table 4) and tumor volumes of 100–150 mm3 (PBS, 124.9 ± 23.8 mm3; ImmTAC-GAG, 133.5 ± 25.8 mm3; ImmTAC-NYE 136.6 ± 4.7 mm3; one-way ANOVA p = 0.98), they were treated with ImmTAC-NYE (0.04 mg/kg), control ImmTAC-GAG (specific for an HLA-A2 presented HIV epitope) (0.04 mg/kg) or PBS for 5 days, and tumor viability monitored by fluorescence imaging (Supplementary Figure 2). Treatment of control groups with either ImmTAC-GAG or PBS did not affect the tumor growth of the J82-NY-ESO GFP157–165 cells as demonstrated by GFP fluorescence intensity (Fig. 5a, b). In contrast, mice treated with ImmTAC-NYE exhibited significantly reduced GFP fluorescence on Day 7 when compared with pre-treatment Day 0 fluorescence and ImmTAC-GAG or PBS controls on Day 7 (p < 0.001). By days 14 and 21, the GFP fluorescence intensity values of J82-NY-ESO GFP157–165 tumors in ImmTAC-NYE-treated mice increased from day 7 nadirs but remained significantly lower than either of the control groups (p < 0.01).

ImmTAC efficacy study in mice co-xenografted with human PBL and J82-NY-ESO GFP157–165 cells. a GFP gated fluorescence time course of representative mice xenografted with human PBL and J82-NY-ESO GFP157–165 cells and treated with ImmTAC-GAG (n = 4), ImmTAC-NYE (n = 4), or PBS (n = 4). b Average fold change in GFP-gated fluorescence for all animals over the time course. Asterisks indicate unpaired Student’s t test statistical analysis of the difference in GFP-gated fluorescence of ImmTAC-NYE- and ImmTAC-GAG-treated mice. Error bars present SEM. *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
The current study was performed to investigate the utility of a bi-specific ImmTAC reagent targeting an epitope common to both NY-ESO-1 and LAGE-1 for cancer immunotherapy. The expression patterns of both NY-ESO-1 and LAGE-1 in tumors identify these proteins as suitable targets for the treatment of a wide variety of malignancies. NY-ESO-1 and LAGE-1 are members of the cancer testis class of tumor-associated antigens, which has the “cleanest” expression profile in normal tissue. Since these antigens are intracellular, they are not accessible to conventional therapeutic antibodies and targeting of MHC-presented peptide epitopes provides an opportunity for tumor-directed immunotherapy [35, 44].
RT-PCR studies have detected NY-ESO-1 or LAGE-1 transcripts in the testis, placenta, and ovary; other normal human tissues completely lacked expression, with the exception of one qRT-PCR study which also identified low levels of NY-ESO-1 mRNA in liver and pancreas [20, 22, 45]. In broad agreement with previous data, qRT-PCR analyses performed in the current study did not detect NY-ESO-1 or LAGE-1 transcripts in several normal human tissues including hepatocytes, melanocytes, and a range of fibroblasts and endothelial cells derived from different organs. In contrast, NY-ESO-1 and/or LAGE-1 transcript expression was detected not only in cell lines derived from a variety of tumor types but also in primary tumors of ovarian cancer and NSCLC. In agreement with other studies [25, 35, 46, 47], there was general overlap in NY-ESO-1 and LAGE-1 mRNA expression, although a subset of samples was identified which expressed only one of the antigens (Table 1). Particularly for the ovarian cancer specimens, LAGE-1 expression was detected at higher RNA levels and at a greater frequency than NY-ESO-1, suggesting that LAGE-1 may be a superior target antigen for cancer immunotherapy in at least some tumor types. Andrade et al. [24] made a similar observation in multiple myeloma (49 % LAGE-1 vs. 33 % NY-ESO). A previous study detected presentation of the NY-ESO-1157–165 epitope by the SK-Mel-37 and Mel 624 cell lines [34], which express far higher levels of RNA for LAGE-1 than for NY-ESO-1 (Table 1), confirming that this peptide can be processed efficiently from LAGE-1.
We have engineered three soluble mTCRs specific for the NY-ESO-1157–165 epitope, TCR-NYE-wt (the wild-type affinity TCR), and two affinity matured variants TCR-NYE-(29 nM) and TCR-NYE-(0.048 nM). Since previous studies have shown the importance of mTCR affinity for ImmTAC potency [18], the highest affinity ImmTAC-NYE was evaluated in vitro and in vivo. ImmTAC-NYE was able to activate and redirect normal CD8+ T cells to a range of cell lines derived from melanoma (A375, Mel624, and SK-Mel-37), myeloma (U266), and EBV transformed B cells (IM9), in addition to freshly isolated lung cancer cells. The demonstration that the redirected cell lysis was limited to those cells expressing endogenous NY-ESO-1 and/or LAGE-1 proteins [34] was essential to exclude the possibility of non-specific toxicity against normal tissues in a clinical setting. While significant lysis of the majority of the tumor cell lines occurred during the first 24 h, the IncuCyte real-time imaging system demonstrated that some cell lines required longer periods for caspase 3/7-dependent apoptosis to occur. One possible explanation for the observed variation in cell lysis is the difference in density of target epitopes on the surface of each cell line; previous studies using TCR-NYE-(0.048 nM) reported the presence of 25 and 45 epitopes/cell on the SK-Mel-37 and Mel624 cell lines respectively [34], and U266 cells have 5–10 epitopes per cell (unpublished data). Other factors which may influence the rate of cell killing are firstly susceptibility of individual tumor cell lines to lysis and secondly the presence of cofactors such as CD80 and CD86 on the tumor cells which may augment killing.
An essential element for the efficacy of targeted TCR therapy is the effective delivery of engineered reagents to the tumor site. Such targeting of biologics has previously been demonstrated in vivo employing molecular imaging in both clinical and preclinical settings [48]. In particular, fluorescence reflectance molecular imaging has typically been employed preclinically owing to its high sensitivity [49], low cost, and potential for rapid in vivo screening. A major challenge for this strategy is to achieve sufficient target-to-background ratios to evaluate specific targeting over endogenous background auto-fluorescence and non-specific binding. However, the density of specific MHC/peptide complexes on the cell surface is usually low (between 10 and 150 epitopes per cell) ([34] and unpublished data), thwarting the use of TCRs in conventional fluorescence imaging.
In contrast to conventional imaging, fluorescence lifetime or time-domain imaging can discriminate between fluorophores with different lifetime decay rates [50]. Time-domain optical imaging has been employed with great success in distinguishing fluorescently labelled monoclonal antibodies bound to target antigens from both auto-fluorescence [41] and background fluorescence from unbound antibodies [51]. In the current study, the administration of three near-infrared-labelled TCR-NYEs of varying affinities, combined with the use of time-domain optical imaging techniques, confirmed targeting of the mTCRs to tumors presenting the NY-ESO-1157–165 epitope.
To evaluate in vivo efficacy of ImmTAC-NYE, we utilized NSG mice engrafted with human PBL in both engraftment and established tumor models. We demonstrated that a short course (5-day administration) of ImmTAC-NYE substantially delayed engraftment of SK-Mel-37-NY-ESO157–165 tumor cells, with tumors just detectable in treated mice 50 days post-implantation, while tumors had reached an average volume of 368 mm3 in the control treated mice. In an established tumor model using J82 tumor cells engineered to express NY-ESO157–165 and GFP, we observed that five daily doses of ImmTAC-NYE resulted in a significant reduction in GFP fluorescence 7 days post-initiation of therapy compared to control treated mice, indicating a substantial reduction in tumor cell viability. An issue with this particular cell line in vivo was its inherent slow growth kinetics, which made differentiation of therapeutic effect in this pilot study impossible by calliper measurements. The significant reduction in fluorescence demonstrated with only five doses of ImmTAC-NYE by time-domain optical imaging endorses both application of this model and fluorescence lifetime gating in further efficacy strategies.
In xenografted animal models which have investigated ImmTAC efficacy (this study and [18]), inhibition of tumor growth continues for long periods of time (over 5 weeks) after the final dose of reagent. ImmTAC-activated T cells release soluble factors including interferon-γ, IL-2 and TNFα (Fig. 1b), which can not only attract additional T cells and other effector immune cells to the tumor site but also promote components of the death receptor pathway in the tumor cells [52], which can result in long-term anti-tumor activity. In cancer patients, it is probable that mechanisms such as epitope spreading (activation of endogenous T cells specific for other tumor epitopes) will occur, following cross-presentation of antigens from lysed tumor cells by dendritic cells. Indeed, we have demonstrated cross-presentation induced by ImmTAC redirected tumor cell killing in vitro (manuscript in preparation). These mechanisms have the potential to stimulate a prolonged anti-tumor immune response capable of breaking tumor tolerance. In addition, we have previously demonstrated serial killing of several tumor cells by a single ImmTAC-activated T cell, which may partly account for the high potency of these reagents [18]. It is not known whether the PBMC-engrafted mice have functioning dendritic cells and therefore whether cross-presentation and epitope spreading can occur in this model system.
ImmTACs have been shown to detect low densities of cell surface epitope (as few as 10 per cell), in contrast to naturally occurring tumor-specific T cells which require higher levels of target epitope to become activated [18]. Thus, ImmTACs have the potential to kill tumors which are not recognized by low-avidity T cells in patients, for example responses induced by cancer vaccines, since higher avidity CTL specific for self-peptides have been removed by negative thymic selection or inactivated by peripheral tolerance mechanisms. The ability of ImmTACs to detect a low density of pMHCs and to activate immune cells independently of co-receptors and other regulatory cells could circumvent immune tolerance in the tumor microenvironment and overcome inhibitory mechanisms such as MHC down-regulation or the presence of Tregs [8, 9]. Decreased levels of MHC (relative to normal tissue cells) are frequently observed in tumors, whereas complete loss of both chromosomal loci encoding MHC and associated processing machinery is a relatively rare event (unpublished observations and [53, 54]). Only patients with tumors positive for both NY-ESO-1/LAGE-1 and HLA-A*0201 are eligible for treatment with ImmTAC-NYE; we are currently isolating NY-ESO-specific TCRs with different HLA restrictions, in order to expand the target patient population. To conclude, we have demonstrated specific and potent ImmTAC-NYE redirected killing of NY-ESO-1 and LAGE-1 tumor cells both in vitro and in vivo, supporting the clinical development of this bi-specific reagent.
References
Borghesi L, Milcarek C (2007) Innate versus adaptive immunity: a paradigm past its prime? Cancer Res 67(9):3989–3993
Ikeda H, Chamoto K, Tsuji T, Suzuki Y, Wakita D, Takeshima T, Nishimura T (2004) The critical role of type-1 innate and acquired immunity in tumor immunotherapy. Cancer Sci 95(9):697–703
Celluzzi CM, Mayordomo JI, Storkus WJ, Lotze MT, Falo LD Jr (1996) Peptide-pulsed dendritic cells induce antigen-specific CTL-mediated protective tumor immunity. J Exp Med 183(1):283–287
Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, Morton KE, Laurencot CM, Steinberg SM, White DE, Dudley ME (2011) Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 17(13):4550–4557
Mackensen A, Meidenbauer N, Vogl S, Laumer M, Berger J, Andreesen R (2006) Phase I study of adoptive T-cell therapy using antigen-specific CD8+ T cells for the treatment of patients with metastatic melanoma. J Clin Oncol 24(31):5060–5069
Davis MM, Boniface JJ, Reich Z, Lyons D, Hampl J, Arden B, Chien Y (1998) Ligand recognition by alpha beta T cell receptors. Annu Rev Immunol 16:523–544
van der Merwe PA, Davis SJ (2003) Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol 21:659–684
Dunn GP, Old LJ, Schreiber RD (2004) The three Es of cancer immunoediting. Annu Rev Immunol 22:329–360
Galgani M, Di Giacomo A, Matarese G, La Cava A (2009) The Yin and Yang of CD4(+) regulatory T cells in autoimmunity and cancer. Curr Med Chem 16(35):4626–4631
Gallimore AM, Simon AK (2008) Positive and negative influences of regulatory T cells on tumour immunity. Oncogene 27(45):5886–5893
Zou W (2005) Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 5(4):263–274
Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH (2011) T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 3(95):95ra73
Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, Kammula US, Hughes MS, Restifo NP, Raffeld M, Lee CC, Levy CL, Li YF, El-Gamil M, Schwarz SL, Laurencot C, Rosenberg SA (2011) Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 29(7):917–924
Baeuerle PA, Reinhardt C (2009) Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res 69(12):4941–4944
Dreier T, Lorenczewski G, Brandl C, Hoffmann P, Syring U, Hanakam F, Kufer P, Riethmuller G, Bargou R, Baeuerle PA (2002) Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int J Cancer 100(6):690–697
Nagorsen D, Bargou R, Ruttinger D, Kufer P, Baeuerle PA, Zugmaier G (2009) Immunotherapy of lymphoma and leukemia with T-cell engaging BiTE antibody blinatumomab. Leuk Lymphoma 50(6):886–891
Topp MS, Kufer P, Gokbuget N, Goebeler M, Klinger M, Neumann S, Horst HA, Raff T, Viardot A, Schmid M, Stelljes M, Schaich M, Degenhard E, Kohne-Volland R, Bruggemann M, Ottmann O, Pfeifer H, Burmeister T, Nagorsen D, Schmidt M, Lutterbuese R, Reinhardt C, Baeuerle PA, Kneba M, Einsele H, Riethmuller G, Hoelzer D, Zugmaier G, Bargou RC (2011) Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol 29(18):2493–2498
Liddy N, Bossi G, Adams KJ, Lissina A, Mahon TM, Hassan NJ, Gavarret J, Bianchi FC, Pumphrey NJ, Ladell K, Gostick E, Sewell AK, Lissin N, Harwood NE, Molloy PE, Li Y, Cameron BJ, Sami M, Baston E, Todorov PT, Paston SJ, Dennis RE, Harper J, Dunn SM, Ashfield R, Johnson A, McGrath Y, Plesa G, June CH, Kalos M, Price DA, Vuidepot A, Williams DD, Sutton DH, Jakobsen BK (2012) Monoclonal TCR-directed tumour cell killing. Nat Med 18:980–987
Hofmann O, Caballero OL, Stevenson BJ, Chen YT, Cohen T, Chua R, Maher CA, Panji S, Schaefer U, Kruger A, Lehvaslaiho M, Carninci P, Hayashizaki Y, Jongeneel CV, Simpson AJ, Old LJ, Hide W (2008) Genome-wide analysis of cancer/testis gene expression. Proc Natl Acad Sci USA 105(51):20422–20427
Lethe B, Lucas S, Michaux L, De Smet C, Godelaine D, Serrano A, De Plaen E, Boon T (1998) LAGE-1, a new gene with tumor specificity. Int J Cancer 76(6):903–908
Chen JL, Dunbar PR, Gileadi U, Jager E, Gnjatic S, Nagata Y, Stockert E, Panicali DL, Chen YT, Knuth A, Old LJ, Cerundolo V (2000) Identification of NY-ESO-1 peptide analogues capable of improved stimulation of tumor-reactive CTL. J Immunol 165(2):948–955
Sato S, Noguchi Y, Wada H, Fujita S, Nakamura S, Tanaka R, Nakada T, Hasegawa K, Nakagawa K, Koizumi F, Ono T, Nouso K, Jungbluth A, Chen YT, Old LJ, Shiratori Y, Nakayama E (2005) Quantitative real-time RT-PCR analysis of NY-ESO-1 and LAGE-1a mRNA expression in normal tissues and tumors, and correlation of the protein expression with the mRNA copy number. Int J Oncol 26(1):57–63
van Rhee F, Szmania SM, Zhan F, Gupta SK, Pomtree M, Lin P, Batchu RB, Moreno A, Spagnoli G, Shaughnessy J, Tricot G (2005) NY-ESO-1 is highly expressed in poor-prognosis multiple myeloma and induces spontaneous humoral and cellular immune responses. Blood 105(10):3939–3944
Andrade VC, Vettore AL, Felix RS, Almeida MS, Carvalho F, Oliveira JS, Chauffaille ML, Andriolo A, Caballero OL, Zago MA, Colleoni GW (2008) Prognostic impact of cancer/testis antigen expression in advanced stage multiple myeloma patients. Cancer Immun 8:2
Odunsi K, Jungbluth AA, Stockert E, Qian F, Gnjatic S, Tammela J, Intengan M, Beck A, Keitz B, Santiago D, Williamson B, Scanlan MJ, Ritter G, Chen YT, Driscoll D, Sood A, Lele S, Old LJ (2003) NY-ESO-1 and LAGE-1 cancer-testis antigens are potential targets for immunotherapy in epithelial ovarian cancer. Cancer Res 63(18):6076–6083
Konishi J, Toyooka S, Aoe M, Omura Y, Washio K, Tsukuda K, Shimizu N (2004) The relationship between NY-ESO-1 mRNA expression and clinicopathological features in non-small cell lung cancer. Oncol Rep 11(5):1063–1067
Gure AO, Chua R, Williamson B, Gonen M, Ferrera CA, Gnjatic S, Ritter G, Simpson AJ, Chen YT, Old LJ, Altorki NK (2005) Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small cell lung cancer. Clin Cancer Res 11(22):8055–8062
Barrow C, Browning J, MacGregor D, Davis ID, Sturrock S, Jungbluth AA, Cebon J (2006) Tumor antigen expression in melanoma varies according to antigen and stage. Clin Cancer Res 12(3 Pt 1):764–771
Vaughan HA, Svobodova S, Macgregor D, Sturrock S, Jungbluth AA, Browning J, Davis ID, Parente P, Chen YT, Stockert E, St Clair F, Old LJ, Cebon J (2004) Immunohistochemical and molecular analysis of human melanomas for expression of the human cancer-testis antigens NY-ESO-1 and LAGE-1. Clin Cancer Res 10(24):8396–8404
Nicholaou T, Ebert L, Davis ID, Robson N, Klein O, Maraskovsky E, Chen W, Cebon J (2006) Directions in the immune targeting of cancer: lessons learned from the cancer-testis Ag NY-ESO-1. Immunol Cell Biol 84(3):303–317
Jager E, Jager D, Knuth A (1999) CTL-defined cancer vaccines: perspectives for active immunotherapeutic interventions in minimal residual disease. Cancer Metastasis Rev 18(1):143–150
Bioley G, Dousset C, Yeh A, Dupont B, Bhardwaj N, Mears G, Old LJ, Ayyoub M, Valmori D (2009) Vaccination with recombinant NY-ESO-1 protein elicits immunodominant HLA-DR52b-restricted CD4+ T cell responses with a conserved T cell receptor repertoire. Clin Cancer Res 15(13):4467–4474
Caballero OL, Chen YT (2009) Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci 100(11):2014–2021
Purbhoo MA, Sutton DH, Brewer JE, Mullings RE, Hill ME, Mahon TM, Karbach J, Jager E, Cameron BJ, Lissin N, Vyas P, Chen JL, Cerundolo V, Jakobsen BK (2006) Quantifying and imaging NY-ESO-1/LAGE-1-derived epitopes on tumor cells using high affinity T cell receptors. J Immunol 176(12):7308–7316
de Carvalho F, Vettore AL, Inaoka RJ, Karia B, Andrade VC, Gnjatic S, Jungbluth AA, Colleoni GW (2011) Evaluation of LAGE-1 and NY-ESO-1 expression in multiple myeloma patients to explore possible benefits of their homology for immunotherapy. Cancer Immun 11:1
Chu CS, Woo EY, Toll AJ, Rubin SC, June CH, Carroll RG, Schlienger K (2002) Tumor-associated macrophages as a source of functional dendritic cells in ovarian cancer patients. Clin Immunol 102(3):291–301
Lorens JB, Bennett MK, Pearsall DM, Throndset WR, Rossi AB, Armstrong RJ, Fox BP, Chan EH, Luo Y, Masuda E, Ferrick DA, Anderson DC, Payan DG, Nolan GP (2000) Retroviral delivery of peptide modulators of cellular functions. Mol Ther 1(5 Pt 1):438–447
Swift S, Lorens J, Achacoso P, Nolan GP (2001) Rapid production of retroviruses for efficient gene delivery to mammalian cells using 293T cell-based systems. Curr Protoc Immunol Chapter 10:Unit 10 7C
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29(9):e45
Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, Dunn S, Liddy N, Jacob J, Jakobsen BK, Boulter JM (2005) Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol 23(3):349–354
Hofmann M, McCormack E, Mujic M, Rossberg M, Bernd A, Bereiter-Hahn J, Gjertsen BT, Wiig H, Kippenberger S (2009) Increased plasma colloid osmotic pressure facilitates the uptake of therapeutic macromolecules in a xenograft tumor model. Neoplasia 11(8):812–822
Bobisse S, Rondina M, Merlo A, Tisato V, Mandruzzato S, Amendola M, Naldini L, Willemsen RA, Debets R, Zanovello P, Rosato A (2009) Reprogramming T lymphocytes for melanoma adoptive immunotherapy by T-cell receptor gene transfer with lentiviral vectors. Cancer Res 69(24):9385–9394
Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, Watanabe T, Akashi K, Shultz LD, Harada M (2005) Development of functional human blood and immune systems in NOD/SCID/IL2 receptor gamma chain(null) mice. Blood 106(5):1565–1573
Jungbluth AA, Chen YT, Stockert E, Busam KJ, Kolb D, Iversen K, Coplan K, Williamson B, Altorki N, Old LJ (2001) Immunohistochemical analysis of NY-ESO-1 antigen expression in normal and malignant human tissues. Int J Cancer 92(6):856–860
Chen DY, Vance BA, Thompson LB, Domchek SM, Vonderheide RH (2007) Differential lysis of tumors by polyclonal T cell lines and T cell clones specific for hTERT. Cancer Biol Ther 6(12):1991–1996
Cuffel C, Rivals JP, Zaugg Y, Salvi S, Seelentag W, Speiser DE, Lienard D, Monnier P, Romero P, Bron L, Rimoldi D (2011) Pattern and clinical significance of cancer-testis gene expression in head and neck squamous cell carcinoma. Int J Cancer 128(11):2625–2634
Rimoldi D, Rubio-Godoy V, Dutoit V, Lienard D, Salvi S, Guillaume P, Speiser D, Stockert E, Spagnoli G, Servis C, Cerottini JC, Lejeune F, Romero P, Valmori D (2000) Efficient simultaneous presentation of NY-ESO-1/LAGE-1 primary and nonprimary open reading frame-derived CTL epitopes in melanoma. J Immunol 165(12):7253–7261
Willmann JK, van Bruggen N, Dinkelborg LM, Gambhir SS (2008) Molecular imaging in drug development. Nat Rev Drug Discov 7(7):591–607
Weissleder R, Pittet MJ (2008) Imaging in the era of molecular oncology. Nature 452(7187):580–589
Karlsen TV, McCormack E, Mujic M, Tenstad O, Wiig H (2012) Minimally invasive quantification of lymph flow in mice and rats by imaging depot clearance of near-infrared albumin. Am J Physiol Heart Circ Physiol 302(2):H391–H401
Ardeshirpour Y, Chernomordik V, Zielinski R, Capala J, Griffiths G, Vasalatiy O, Smirnov AV, Knutson JR, Lyakhov I, Achilefu S, Gandjbakhche A, Hassan M (2012) In vivo fluorescence lifetime imaging monitors binding of specific probes to cancer biomarkers. PLoS One 7(2):e31881
De Geer A, Carlson LM, Kogner P, Levitskaya J (2008) Soluble factors released by activated cytotoxic T lymphocytes interfere with death receptor pathways in neuroblastoma. Cancer Immunol Immunother 57(5):731–743
Giacomini P, Giorda E, Fraioli R, Nicotra MR, Vitale N, Setini A, Delfino L, Morabito A, Benevolo M, Venturo I, Mottolese M, Ferrara GB, Natali PG (1999) Low prevalence of selective human leukocyte antigen (HLA)-A and HLA-B epitope losses in early-passage tumor cell lines. Cancer Res 59(11):2657–2667
Marincola FM, Shamamian P, Alexander RB, Gnarra JR, Turetskaya RL, Nedospasov SA, Simonis TB, Taubenberger JK, Yannelli J, Mixon A et al (1994) Loss of HLA haplotype and B locus down-regulation in melanoma cell lines. J Immunol 153(3):1225–1237
Acknowledgments
We thank M. Popa, L. Vikebø, M. Boge, and K. Jacobsen for expert assistance in all preclinical work and M. Enger for flow cytometry assistance. All imaging and flow cytometry was performed at the Molecular Imaging Center (MIC), the Department of Biomedicine, University of Bergen, Norway. This study has been supported by the Norwegian Cancer Society, the Western Health Board of Norway, and the Bergen Research Foundation.
Conflict of interest
We wish to disclose the following conflicts of interest: Namir Hassan, Nikolai Lissin, Malkit Sami, Deborah Baker, Alex Powlesland, Milos Aleksic, Annelise Vuidepot, Rebecca Ashfield, and Bent Jakobsen are all currently employees of Immunocore Ltd; Tereza Osdal is an employee of KinN Therapeutics AS; Emmet McCormack and Bjørn Tore Gjertsen are shareholders in KinN Therapeutics.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
McCormack, E., Adams, K.J., Hassan, N.J. et al. Bi-specific TCR-anti CD3 redirected T-cell targeting of NY-ESO-1- and LAGE-1-positive tumors. Cancer Immunol Immunother 62, 773–785 (2013). https://doi.org/10.1007/s00262-012-1384-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-012-1384-4