
Abstract
Objective
Naturally occurring regulatory T (TR) cells suppress autoreactive T cells whereas adaptive TR cells, induced in the periphery, play an important role in chronic viral diseases and cancer. Several studies indicate that cyclooxygenase (COX) inhibitors prevent cancer development of colon adenomas and delay disease progression in patients with colorectal cancer (CRC). We have shown that adaptive TR cells express COX-2 and produce PGE2 that suppress effector T cells in a manner that is reversed by COX-inhibitors.
Methods and results
Here we demonstrate that CRC patients have elevated levels of PGE2 in peripheral blood, and CRC tissue samples and draining lymph nodes display increased numbers of FOXP3+ TR cells. Depletion of TR cells from PBMC enhanced anti-tumor T-cell responses to peptides from carcinoembryonic antigen. Furthermore, the COX inhibitor indomethacin and the PKA type I antagonist Rp-8-Br-cAMPS significantly improved the anti-tumor immune activity.
Conclusion
We suggest that adaptive TR cells contribute to an immunosuppressive microenvironment in CRC and inhibit effector T cells by a COX-2–PGE2-dependent mechanism and thereby facilitate tumor growth. Therapeutic strategies targeting TR cells and the PGE2–cAMP pathway may be interesting to pursue to enhance anti-tumor immune activity in CRC patients.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) ranks second in incidence and mortality among malignant disease in both men and women in developed countries, although the recent advances in surgical technique and use of neo-adjuvant chemotherapy have improved clinical outcomes [4, 23, 46]. Most cases of CRC are sporadic, and both genetic background and environmental factors are important. Increasing evidence suggests that chronic inflammation plays a role in the pathogenesis of cancer, including CRC [1, 10]. Epidemiologic studies indicate that regular use of aspirin or nonsteroidal anti-inflammatory drugs (NSAIDs) can reduce the incidence of CRC in humans by 40–50% [17, 20, 39]. The chemopreventive role of NSAIDs and aspirin is thought to be exerted through the inhibition of cyclooxygenases (COXs), specifically COX-2, and prostaglandin (PG) production [7]. Exogenous PGE2 treatment protects small intestinal adenomas from NSAID-induced regression in APCmin mice, a murine model for familial adenomatous polyposis (FAP), directly demonstrating that the anti-tumor effect of NSAIDs is related to reduction of PGE2 levels [18]. However, use of COX-2 inhibitors to prevent CRC in humans has been connected with an increased frequency of adverse cardiovascular events [3, 34].
COX exists in two isoforms, named COX-1 and COX-2. COX-1 is constitutively expressed in a broad range of cells and tissues, and its enzymatic activity is important for renal function, gastric mucosal integrity and platelet aggregation. In contrast, COX-2 expression is normally absent in most cells and tissues, but is induced in response to proinflammatory cytokines [13]. The COX-2 derived PGE2 plays a predominant role in inflammatory diseases such as arthritis. PGE2 exerts its cellular effects by binding to its cognate receptors (EP1–EP4) that belong to the family of seven transmembrane G protein coupled receptors.
Several reports have demonstrated that PGE2 promotes tumor growth through stimulation of EP receptor signaling with subsequent enhancement of cellular proliferation, promotion of angiogenesis, inhibition of apoptosis, stimulation of tumor invasion, and suppression of immune responses [44]. Possible mechanisms involved are activation of peroxisome proliferator-activated receptors δ (PPARδ), epidermal growth factor receptor (EGFR), Ras-MAPK pathway, nuclear factor kappa B (NFκB), and Wnt signaling [44]. The central role of PGE2 in tumorigenesis has been further confirmed through homozygous deletion of its receptors. Mice with deletions in EP1 and EP4 receptors, but not in EP3, were partially resistant to colon carcinogen mediated induction of abnormal crypt foci [29, 45]. Furthermore, EP2 disruption in APCΔ716 KO mice leads to decreased size and number of polyps [40]. Both PGE2 production and level of COX-2 expression is increased in CRC compared with normal colonic mucosa [14, 21]. A recent study has shown association between high levels of a urinary PGE2 metabolite and CRC risk [5].
Carcinoembryonic antigen (CEA) is a 180 kD glycoprotein that is highly expressed in colon epithelial cells during embryonic development. The level of expression is low in adult tissues, but is overexpressed in almost all CRC. Several reports have shown that CEA is immunogenic, making it an attractive target for immunotherapeutic purposes and cancer vaccine development [19]. CEA-epitopes that induce anti-tumor immune responses in both CD4+ and CD8+ T cells have been identified [6, 19].
The role of CD4+CD25+ regulatory T (TR) cells in suppressing anti-tumor immune activity is subject to considerable interest [30, 36]. TR cells express the forkhead transcription factor FOXP3 and maintain immunological tolerance by suppression of autoreactive T cells [12, 15, 35]. Naturally occurring TR cells develop in the thymus, however CD4+CD25- T cells in the periphery convert to CD4+CD25+FOXP3+ adaptive TR cells and acquire regulatory function under certain circumstances [28, 43]. Accumulating evidence indicates that adaptive TR cells are enriched in tumor tissue, draining lymph nodes, malignant effusions and peripheral blood from patients with various cancers, suggesting that the tumor microenvironment may foster immune tolerance by attracting and/or inducing dominant inhibitory cells [50].
We have shown that continuous antigenic stimulation of human peripheral CD4+CD25- T cells gives rise to CD4+CD25+FOXP3+ adaptive TR cells which suppress autologous CD4+CD25- responder T cells [28]. Furthermore, we have reported a novel mechanism of immune suppression whereby adaptive TR cells express COX-2 and suppress effector T cells in a PGE2-dependent fashion that can be reversed with COX-2 inhibitors or PGE2-receptor antagonists [28]. In addition, we and others have implicated PGE2 in induction of FOXP3 in CD4+CD25- T cells that contributes to modulation of T cell function [2, 28]. In a murine lung cancer model, inhibition of COX-2 enhanced anti-tumor immune responses [37, 38]. In line with these observations, we hypothesized that chronic immune stimulation in a cancer microenvironment induces TR cells that inhibit anti-tumor immune responses in a COX-2–PGE2–cAMP-dependent manner. In this study we demonstrate that CRC patients have high levels of PGE2 in plasma, and that TR cells suppress anti-CEA immune responses in a manner that can be reversed by COX-inhibitor or PKA-antagonist to the same extent as depletion of TR cells.
Materials and methods
Patients and clinical study control
The study protocol was approved by the Regional Committee for Medical Research Ethics, Norway. Patients with CRC at Ullevaal University Hospital were enrolled in the study after written informed consent. Only patients with first presentation of an adenocarcinoma were included (TNM stage I–IV; Dukes’ stage A–D). Patients (n = 29; 15 men and 14 women; mean age 68 years; range 41–88 years) and healthy blood donors (n = 8) were screened for PGE2 levels in the plasma. A subgroup of 12 patients (seven men and five women; mean age 74 years; range 49–88 years) was selected for analysis of anti-CEA immune responses. Patient data for the subgroup are presented in Table 1. CEA level was routinely determined in serum preoperatively in all patients, except two. Blood samples from healthy blood donors at Blood Bank, Ullevaal University Hospital, Norway were used as controls.
Isolation of cells
30–50 ml of peripheral blood was drawn from patients or controls in EDTA tubes. Peripheral blood mononuclear cells (PBMC) were isolated by Isopaque-Ficoll (Lymphoprep, Nycomed Pharma AS, Oslo) gradient centrifugation. CD4+CD25+ T cells were isolated using a CD4+CD25+ regulatory T cell isolation kit according to the manufacturer’s manual (Miltenyi Biotec, Auburn, CA). The cells were routinely analyzed by flow cytometry and the purity of CD4+CD25+ and CD4+CD25- T cell populations were consistently >98%. In anti-tumor immune assays, CD25+ cells were depleted directly from PBMC after incubation with CD25 microbeads (Miltenyi Biotec, Auburn). Cells were cultured in RPMI 1640 (Gibco, Paisley, UK) supplemented with 10% heat-treated FCS, 100 U/ml penicillin/streptomycin, 1 mM sodium pyruvate and 1:100 non-essential amino acids (further described as complete medium).
Peptides
We selected six sequences of the CEA protein for peptide synthesis, based on previously published data [19]. The peptides were synthesized using an in-house multipep automated peptide synthesizer (INTAVIS Bioanalytical Instruments AG) following standard Fmoc-chemistry protocol with following sequences: CEA 1–6D [YLSGADLNL] [48], CEA 61–69 [HLFGYSWYK] [24], CEA 268–278 [QYSWFVNGTF], CEA 318–327 [TYACFVSNL] [31], CEA 570–579 [SYLSGANLNL] [19], and CEA 571–579 [YLSGANLNL] [42]. Scrambled control peptides were generated the same way with the sequences: [SNYLLALGN], [SGYLLALDN], [LGSYHFWKY], [NYSLSLALGN], [YWVGFQFSNT], and [NVLFSTCYA]. The peptides were subjected to high performance capillary electrophoresis (HPCE), isolated with >90% purity and quality assessed by mass spectrometry. They were dissolved in DMSO at a concentration of 5 mg/ml and further diluted in complete medium and used at a final concentration of 5 μg/ml.
Flow cytometric analysis of lymphocytes
Cells were fixed in 4% paraformaldehyde (PFA) and permeabilized in FACS permeabilizing solution (BD BioSciences, San Jose, CA) prior to staining with CD4 APC, CD8 APC, CD3 PerCP, CD25 PE, anti-human IFN-γ PE, anti-human TNF-α FITC, (BD BioSciences Pharmingen, Cayman Chemicals). The CD25+ T cells were tested for FOXP3 expression by staining with APC anti-human FOXP3 staining kit, according to manufacturer’s instructions (clone PCH-101, eBioscience). The cells were washed once in PBS containing 1% BSA before acquiring data on a flow cytometer (FACSCalibur, BD Biosciences, San Jose, CA) and analyzed using FlowJo software (Tree Star, San Carlos, CA).
Peptide stimulation and cytokine assays
The PBMC, PBMC depleted of CD25+ cells, or PBMC co-cultured with autologous CD25+ T cells were stimulated with 5 μg/ml staphylococcal enterotoxin B (SEB) (Sigma-Aldrich), CEA-peptides (5 μg/ml), or complete medium for 18–20 h. Brefeldin A (Sigma-Aldrich) was added to a final concentration of 5 μM for the last 6 h of incubation. When used, COX inhibitor indomethacin (25 μM) (Sigma-Aldrich) or PKA type I antagonist Rp-8-Br-cAMPS (1 mM) (Lauras AS, Oslo, Norway) were added 90 min prior to activation with SEB or CEA-peptides. In all co-culture experiments, CD25+ T cells were prestained with 2 μM CFSE in order to separate TR cell population from responding effector T cells. The cells were fixed, permeabilized, stained for CD3, CD4, IFN-γ and TNF-α and analyzed by flow cytometry as described above. The SEB was used as a positive control for activation in the assay. The cytokine levels in un-stimulated cells were subtracted from the levels of activated cells to look at the induction in cytokine secretion.
Prostagandin E2 measurement by ELISA
Peripheral blood from patients and controls was centrifuged and plasma isolated and stored at −80°C, thawed, and subsequently analyzed for PGE2 concentration according to the manufacturer’s instructions (R&D, London, UK).
Preparation of tissue specimens and multicolor immunostaining
Large intestine resection specimens were fixed in 10% buffered formalin and processed by standard methods for pathologic evaluation. Relevant specimens were obtained from the tumor and unaffected mucosa (n = 11) as well as from regional draining lymph nodes (n = 12). Formalin-fixed and paraffin-embedded tissue was cut at 4 μm, deparaffinized, rehydrated and then subjected to antigen retrieval by cooking for 20 min in 0.01 M citrate buffer (pH 6.0) in a water bath at 98°C. Two-color immunofluorescence staining was performed as described elsewhere [22]. To determine the density and phenotype of FOXP3+ T cells a rat mAb specific for FOXP3 (clone PCH101, IgG2a, 1/20; eBiosciences) was combined with mouse mAbs to either CD25 (clone 4C9, IgG2b, 1/100; Novocastra, UK) or COX-2 (#160112, IgG1, 1 μg/ml; Cayman Chemicals, Ann Arbor, MI). Primary antibody combinations were incubated overnight at +4°C followed by incubation with a mixture of Cy3-labeled donkey anti-rat IgG (2.5 μg/ml; Jackson ImmunoResearch Laboratories, West Grove, PA) and biotinylated subclass-specific goat anti-mouse IgG2b (10 μg/ml) or biotinylated goat anti-mouse IgG1 (2.5 μg/ml) (both from Jackson ImmunoResearch Laboratories) for 3 h, and finally Cy2-streptavidin (1 μg/ml; Amersham, UK) for 1 h. In addition, a rabbit antiserum to CD3 (1/50; DAKO, Denmark) was incubated overnight either with anti-FOXP3 (clone PCH101) or anti-COX2 (# 160112) followed by Alexa 488-labeled goat anti-rabbit IgG (2 μg/ml; Molecular Probes Invitrogen) combined with either Cy3-labeled donkey anti-rat IgG or Cy3-labeled goat anti-mouse IgG1 (Jackson ImmunoResearch Laboratories) for 3 h. In all staining experiments, negative controls were obtained both by omission of primary mAbs and by parallel incubation with irrelevant isotype- and concentration-matched primary mAbs. The specimens were examined at ×40 magnification by confocal microscopy (Leica, TCS SP, Heidelberg, Germany) and images were recorded with the confocal acquisition program (Leica TCS-NT). The images were further processed using Adobe Photoshop and Adobe Illustrator.
Statistical analysis
The data were analyzed using SPSS for Windows. The plasma levels of PGE2 in patients and controls were compared using Mann–Whitney U test. Anti-CEA responses in patients and controls were compared by Wilcoxon signed ranks test. Differences with two-sided P < 0.05 were considered significant.
Results
CRC patients have elevated levels of PGE2 in plasma
To assess the level of PGE2 in peripheral blood, we collected plasma from CRC patients prior to surgery (n = 29) and from healthy blood donors (n = 8). CRC patients had elevated levels of PGE2 in peripheral blood compared to normal blood donors (Fig. 1). However, the levels of PGE2 did not directly correlate with the size of the tumor or disease stage (Table 1). The level of CEA was also independent of tumor size or PGE2 in the plasma.

Increased levels of PGE2 in patients with CRC. Levels of PGE2 were measured in plasma from patients with CRC prior to surgery (n = 29) and healthy blood donors (n = 8). Freshly isolated plasma was frozen at −80°C before PGE2 was analyzed by ELISA. Box plots with median; 25–75% (box) and 2.5–97.5% (bar) percentiles are shown (P < 0.01, Mann–Whitney U test, two-tailed)
FOXP3+ TR cells accumulate in CRCs and regional lymph nodes
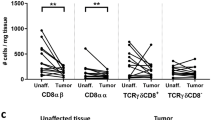
We next assessed tumor samples in a subgroup of patients (n = 12) described in Table 1. One patient in this subgroup presented with disseminated carcinomatosis upon laparotomy and was not subject to surgical resection. Numerous FOXP3+ cells were observed both in the center and in the invasive margin of all CRCs examined (n = 11). The FOXP3 expression was restricted to CD3+ T cells and FOXP3+ cells constituted up to 30% of all CD3+ T cells (Fig. 2a). In contrast, FOXP3+CD3+ T cells only occurred in very low numbers in the unaffected healthy mucosa (Fig. 2d). FOXP3+ T cells invariably coexpressed CD25, both within and outside the tumor (Fig 2b, e). However, some reports have shown that FOXP3 can also be expressed in activated T cells, so the presentation of both regulatory T cells and activated T cells can not be ruled out [33]. In the regional draining lymph nodes, both those with and without metastases, high numbers of FOXP3+CD25+ T cells were observed (Fig. 3a, b). Almost none of the FOXP3+ T cells coexpressed COX-2 at the tumor site (Fig. 2c), but a fraction of FOXP3+ cells in the lymph nodes expressed COX-2 (Fig. 3c). Consistent with previous reports, the tumor cells strongly expressed COX-2 whereas the normal colonic epithelial cells were virtually unreactive to COX-2 antibodies (Fig. 2c, f).

Regulatory T cells accumulate within CRCs. Dual immunofluorescence staining for FOXP3 (a, b, d, e; red) and CD3 (a, d; green) or CD25 (b, e; green); or COX−2 (c, f; red) and CD3 (c, f; green) in representative fields from formalin-fixed and paraffin-embedded tumor tissue (a–c) and unaffected colonic mucosa (d–f). Note numerous CD3+CD25+FOXP3+ cells within the tumor (a, b). Scale bars: 50 μm (a, b, d, e) and 100 μm (c, f)

Regulatory T cells in draining lymph nodes express COX-2. Paired immunofluorescence staining for FOXP3 (a, b; red) and CD3 (a; green) or CD25 (b; green); or COX-2 (c; red) and CD3 (c; green) in representative fields from formalin-fixed and paraffin-embedded tissue from a representative draining lymph node. Scale bars: 50 μm
TR cells inhibit anti-tumor immune responses in patients with CRC
To assess the anti-tumor immune activity in CRC patients, we examined the effect of a selected series of CEA-peptides (Fig. 4a, bottom) on TNF-α and IFN-γ expression in freshly isolated PBMC in the presence or absence of CD25+ T cells. Depletion of CD25+ T cells dramatically augmented both CEA-induced TNF-α and IFN-γ expression in T cells (Fig. 4a, b patients; one representative of n = 3 patients shown). In control experiments with PBMC from healthy blood donors, depletion of CD25+ T cells did not demonstrate any increase in TNF-α or IFN-γ expression (Fig. 4a, b controls). Stimulation with scrambled control peptides did not show any increase in immune responses upon depletion of CD25+ T cells (data not shown). These data suggest that TR cells are antigen-specific leading to suppression of anti-CEA immune responses in effector T cells from patients with CRC. The PBMC depleted of CD25+ cells did not contain any FOXP3+ cells as these were only present in the isolated CD25+ T cells (Fig. 4c).

Regulatory T cells suppress anti-CEA immune responses in patients with colon cancer. Different CEA-peptides were synthesized as indicated in the figure. PBMC or PBMC depleted of CD25+ cells from colon cancer patients or healthy blood donors were stimulated with the peptides for 18 h. Immune responses were measured as production of TNF-α (a) and IFN-γ (b) by CD3+ T cells. The purity of PBMC depleted of CD25+ cells was analyzed by FACS staining for CD25 and FOXP3 (c, gated on CD4+ cells). Note: FOXP3+ cells were only present in CD25+ cells. Data are from one representative of n = 3 patients examined
TR cells inhibit anti-CEA responses in CRC patients in a COX-2–PGE2-dependent manner
From the pool of CEA-peptides shown in Fig. 4, we selected one CEA-peptide (CEA 61–69) which appeared to elicit responses in most patients examined, and investigated whether the inhibitory effects of TR cells on anti-tumor immune activity were mediated by the COX-2–PGE2–cAMP pathway. Freshly-isolated PBMCs from CRC patients (n = 12; Table 1) were stimulated with CEA 61–69, before and after depletion of TR cells. Addition of the COX-inhibitor indomethacin or the PKA type I antagonist Rp-8-Br-cAMP to PBMC prior to activation elicited anti-CEA responses in both the CD4+ and CD8+ T cell populations, which were similar in magnitude as were those induced by depletion of TR cells (Fig. 5). These responses were not seen in healthy blood donors (data not shown). However, in several experiments there was a greater induction of T cell responses by COX-inhibitor than by depletion of TR cells, suggesting that PGE2 from other immune cells may also be involved in the immune suppression. Furthermore, when PBMC depleted of CD25+ T cells were co-cultured with autologous, purified CD25+ T cells in a 3:1 ratio and polyclonally activated with SEB, the CD25+ T cells suppressed both TNF-α and IFN-γ production in effector T cells, as previously reported by our group and others ([28] and data not shown). Moreover, when availability of material allowed, we also co-incubated PBMC depleted of CD25+ cells with autologous TR cells and observed that this also suppressed anti-CEA peptide responses in CRC patients to low levels (Fig. 5; n = 7).

Inhibition of COX significantly increases CEA-mediated immune responses in patients with colon cancer. The PBMCs from patients with CRC were stimulated for 18 h with CEA-antigen (CEA 61–69; HLFGYSWYK) presence and absence of CD25+ T cells. COX-inhibitor (indomethacin) and PKA antagonist (Rp-8-Br cAMPS) were added 90 min prior to activatian with CEA-peptide. Immune responses were measured as production of TNF-α (left) and IFN-γ (right) by CD4+ (a) and CD8+ (b) T cells. Mean ± SEM are shown (n = 12). Co-culture of PBMC depleted of CD25+ cells with autologous TR cells at a 3:1 ratio also suppressed anti-CEA immune responses (# n = 7). The cytokine levels of unstimulated cells were subtracted from the CEA peptide-activated cells. The cytokine levels in unstimulated cells were for TNF-α in CD4+ T cells 1, 16 ± 0, 47; TNF-α in CD8+ T cells 0, 76 ± 0, 31; IFN-γ in CD4+ T cells 1, 44 ± 0,50; and IFN-γ in CD8+ T cells 1, 02 ± 0, 30 (mean ± SEM). Note: PBMCs incubated with indomethacin or Rp-8-Br cAMPS increased immune responses to same extent as depletion of CD25+ T cells. *P < 0.005, **P < 0.01, ***P < 0.05; (Wilcoxon signed ranks test, two-tailed)
Discussion
Tumor cells are immunogenic and tumor-infiltrating lymphocytes (TILs) in melanoma, CRC, and ovarian cancer inhibit tumor growth and are associated with improved prognosis [9, 32, 49]. Interestingly, the type, density, and location of immune cells in CRC may have a higher predictive power than the prognosis estimated by the UICC-TNM classification [16]. However, tumor-specific TR cells at the tumor site play a significant role in the suppression of anti-tumor immune activity which may adversely affect the prognosis [16]. Several studies have shown that depletion of TR cells promote anti-tumor immune responses and tumor rejection [8, 41]. Several human cancers, including gastrointestinal, lung, and ovarian tumors, are also associated with a higher proportion of TR cells in the peripheral blood [8, 11, 36, 47]. In ovarian cancer the presence of TR cells in TILs has been shown to be an independent risk factor associated with poor prognosis [11]. Numerous tumor-derived soluble factors such as vascular endothelial growth factor, IL-10, TGF-β and PGE2 may also act in concert to establish an immunosuppressive microenvironment which promotes T cell tolerance and tumor immune evasion [25]. Consequently, modulation of the regulatory function of TR cells may be an important avenue to improve anti-tumor immune activity.
Continuous antigen stimulation of naïve human peripheral CD4+ T cells leads to induction of CD4+CD25+FOXP3+ TR cells that express COX-2 and suppress effector T cells by a PGE2–cAMP dependent mechanism [28]. Here we show that patients with CRC have high plasma levels of PGE2, that TR cells from these patients inhibit anti-CEA immune responses and that this immunosuppression can be reversed ex vivo in an equally effective manner by treatment with the COX-inhibitor indomethacin or the PKA type I antagonist Rp-8-Br-cAMPS as by depletion of TR cells. However, in some patients COX-inhibitor improved the anti-CEA immune responses more than depletion of TR cells. Thus, the role of PGE2 from other immune cells cannot be ruled out. In the present study we have assessed T cell immune responses in PBMC cultures, and PGE2 from activated monocytes may have contributed to the additional immunosuppression unmasked by COX-inhibitor.
Several studies have demonstrated a significant infiltration of FOXP3+ TR cells into CRCs [26, 27]. The presence of TR cells in the tumor microenvironment and in draining lymph nodes with metastasis diminishes anti-tumor immune responses; however, previous studies have not elucidated the mechanism of suppression. In our study, the majority of TR cells at the tumor site were COX-2 negative or their presence were overshadowed by the strong staining in the CRC cells. However, COX-2+ TR cells were present in draining lymph nodes with and without metastasis, suggesting that COX-2+ TR cells contribute to the immune suppression by secretion of PGE2 in the lymph nodes where tumor antigens are presented by APC. The high expression of COX-2 in tumor epithelial cells is probably the main source of the elevated PGE2 level in plasma observed in these patients, which may drive both tumorigenesis, induction of TR cells, and local immune suppression. However, as indomethacin reverses the TR cells-mediated suppression of anti-tumor immune responses in PBMC from CRC subjects ex vivo where plasma PGE2 is washed out, the production of PGE2 in COX-2+ TR cells appears to be sufficient to inhibit immune responses. This argues that TR cells mediated PGE2 production in situ in draining lymph nodes is sufficient to inhibit the immune responsiveness to metastatic cells from the primary tumor. COX-inhibitors prevent development of CRC by several mechanisms as PGE2 affects cell proliferation, migration, apoptosis and angiogenesis. Improved anti-tumor immune function by inhibition of TR cells may be another mechanism that contributes to the beneficial effects of COX-inhbitors.
In conclusion, the present report supports the notion that TR cells suppress anti-tumor immune responses in a COX-2–PGE2–cAMP-dependent manner and that this mechanism may represent a target for future cancer immunotherapy.
Abbreviations
- CEA:
-
Carcinoembryonic antigen
- COX-2:
-
Cyclooxygenase type 2
- CRC:
-
Colorectal cancer
- PGE2 :
-
Prostagandin E2
- TR cells:
-
Regulatory T cells
References
Balkwill F, Charles KA, Mantovani A (2005) Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 7:211–217
Baratelli F, Lin Y, Zhu L, Yang SC, Heuze-Vourc’h N, Zeng G, Reckamp K, Dohadwala M, Sharma S, Dubinett SM (2005) Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J Immunol 175:1483–1490
Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, Tang J, Rosenstein RB, Wittes J, Corle D, Hess TM, Woloj GM, Boisserie F, Anderson WF, Viner JL, Bagheri D, Burn J, Chung DC, Dewar T, Foley TR, Hoffman N, Macrae F, Pruitt RE, Saltzman JR, Salzberg B, Sylwestrowicz T, Gordon GB, Hawk ET (2006) Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med 355:873–884
Boyle P, Ferlay J (2005) Cancer incidence and mortality in Europe, 2004. Ann Oncol 16:481–488
Cai Q, Gao YT, Chow WH, Shu XO, Yang G, Ji BT, Wen W, Rothman N, Li HL, Morrow JD, Zheng W (2006) Prospective study of urinary prostaglandin E2 metabolite and colorectal cancer risk. J Clin Oncol 24:5010–5016
Campi G, Crosti M, Consogno G, Facchinetti V, Conti-Fine BM, Longhi R, Casorati G, Dellabona P, Protti MP (2003) CD4(+) T cells from healthy subjects and colon cancer patients recognize a carcinoembryonic antigen-specific immunodominant epitope. Cancer Res 63:8481–8486
Cha YI, Dubois RN (2007) NSAIDs and Cancer Prevention: Targets Downstream of COX-2. Annu Rev Med 58:239–252
Clarke SL, Betts GJ, Plant A, Wright KL, El-Shanawany TM, Harrop R, Torkington J, Rees BI, Williams GT, Gallimore AM, Godkin AJ (2006) CD4CD25FOXP3 regulatory T cells suppress anti-tumor immune responses in patients with colorectal cancer. PLoS ONE 1:e129
Clemente CG, Mihm MC Jr, Bufalino R, Zurrida S, Collini P, Cascinelli N (1996) Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 77:1303–1310
Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420:860–867
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W (2004) Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10:942–949
Dieckmann D, Plottner H, Berchtold S, Berger T, Schuler G (2001) Ex vivo isolation and characterization of CD4(+)CD25(+) T cells with regulatory properties from human blood. J Exp Med 193:1303–1310
Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, Lipsky PE (1998) Cyclooxygenase in biology and disease. FASEB J 12:1063–1073
Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, Dubois RN (1994) Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 107:1183–1188
Fontenot JD, Gavin MA, Rudensky AY (2003) Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 4:330–336
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoue F, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Pages F (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313:1960–1964
Gupta RA, Dubois RN (2001) Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer 1:11–21
Hansen-Petrik MB, McEntee MF, Jull B, Shi H, Zemel MB, Whelan J (2002) Prostaglandin E(2) protects intestinal tumors from nonsteroidal anti-inflammatory drug-induced regression in Apc(Min/+) mice. Cancer Res 62:403–408
Horig H, Medina FA, Conkright WA, Kaufman HL (2000) Strategies for cancer therapy using carcinoembryonic antigen vaccines. Expert Rev Mol Med 2000:1–24
Jacobs EJ, Thun MJ, Bain EB, Rodriguez C, Henley SJ, Calle EE (2007) A large cohort study of long-term daily use of adult-strength aspirin and cancer incidence. J Natl Cancer Inst 99:608–615
Jaffe BM, Parker CW, Philpott GW (1971) Immunochemical measurement of prostaglandin or prostaglandin-like activity from normal and neoplastic cultured tissue. Surg Forum 22:90–92
Jahnsen FL, Brandtzaeg P, Halstensen TS (1994) Monoclonal antibody EG2 does not provide reliable immunohistochemical discrimination between resting and activated eosinophils. J Immunol Methods 175:23–36
Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ (2006) Cancer statistics, 2006. CA Cancer J Clin 56:106–130
Kawashima I, Tsai V, Southwood S, Takesako K, Sette A, Celis E (1999) Identification of HLA-A3-restricted cytotoxic T lymphocyte epitopes from carcinoembryonic antigen and HER-2/neu by primary in vitro immunization with peptide-pulsed dendritic cells. Cancer Res 59:431–435
Kim R, Emi M, Tanabe K, Arihiro K (2006) Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res 66:5527–5536
Ling KL, Pratap SE, Bates GJ, Singh B, Mortensen NJ, George BD, Warren BF, Piris J, Roncador G, Fox SB, Banham AH, Cerundolo V (2007) Increased frequency of regulatory T cells in peripheral blood and tumour infiltrating lymphocytes in colorectal cancer patients. Cancer Immun 7:7
Loddenkemper C, Schernus M, Noutsias M, Stein H, Thiel E, Nagorsen D (2006) In situ analysis of FOXP3+ regulatory T cells in human colorectal cancer. J Transl Med 4:52
Mahic M, Yaqub S, Johansson CC, Tasken K, Aandahl EM (2006) FOXP3+CD4+CD25+ adaptive regulatory T cells express cyclooxygenase-2 and suppress effector T cells by a prostaglandin E2-dependent mechanism. J Immunol 177:246–254
Mutoh M, Watanabe K, Kitamura T, Shoji Y, Takahashi M, Kawamori T, Tani K, Kobayashi M, Maruyama T, Kobayashi K, Ohuchida S, Sugimoto Y, Narumiya S, Sugimura T, Wakabayashi K (2002) Involvement of prostaglandin E receptor subtype EP(4) in colon carcinogenesis. Cancer Res 62:28–32
Nomura T, Sakaguchi S (2005) Naturally arising CD25+CD4+ regulatory T cells in tumor immunity. Curr Top Microbiol Immunol 293:287–302
Nukaya I, Yasumoto M, Iwasaki T, Ideno M, Sette A, Celis E, Takesako K, Kato I (1999) Identification of HLA-A24 epitope peptides of carcinoembryonic antigen which induce tumor-reactive cytotoxic T lymphocyte. Int J Cancer 80:92–97
Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D, Meatchi T, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Galon J (2005) Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med 353:2654–2666
Pillai V, Ortega SB, Wang CK, Karandikar NJ (2007) Transient regulatory T-cells: a state attained by all activated human T-cells. Clin Immunol 123:18–29
Psaty BM, Potter JD (2006) Risks and benefits of celecoxib to prevent recurrent adenomas. N Engl J Med 355:950–952
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M (1995) Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 155:1151–1164
Sasada T, Kimura M, Yoshida Y, Kanai M, Takabayashi A (2003) CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies: possible involvement of regulatory T cells in disease progression. Cancer 98:1089–1099
Sharma S, Yang SC, Zhu L, Reckamp K, Gardner B, Baratelli F, Huang M, Batra RK, Dubinett SM (2005) Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res 65:5211–5220
Sharma S, Zhu L, Yang SC, Zhang L, Lin J, Hillinger S, Gardner B, Reckamp K, Strieter RM, Huang M, Batra RK, Dubinett SM (2005) Cyclooxygenase 2 inhibition promotes IFN-gamma-dependent enhancement of antitumor responses. J Immunol 175:813–819
Sheng H, Shao J, Kirkland SC, Isakson P, Coffey RJ, Morrow J, Beauchamp RD, Dubois RN (1997) Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J Clin Invest 99:2254–2259
Sonoshita M, Takaku K, Sasaki N, Sugimoto Y, Ushikubi F, Narumiya S, Oshima M, Taketo MM (2001) Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(delta 716) knockout mice. Nat Med 7:1048–1051
Tanaka H, Tanaka J, Kjaergaard J, Shu S (2002) Depletion of CD4+ CD25+ regulatory cells augments the generation of specific immune T cells in tumor-draining lymph nodes. J Immunother 25:207–217
Tsang KY, Zaremba S, Nieroda CA, Zhu MZ, Hamilton JM, Schlom J (1995) Generation of human cytotoxic T cells specific for human carcinoembryonic antigen epitopes from patients immunized with recombinant vaccinia-CEA vaccine. J Natl Cancer Inst 87:982–990
Walker MR, Kasprowicz DJ, Gersuk VH, Benard A, Van LM, Buckner JH, Ziegler SF (2003) Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+. J Clin Invest 112:1437–1443
Wang D, Dubois RN (2006) Prostaglandins and cancer. Gut 55:115–122
Watanabe K, Kawamori T, Nakatsugi S, Ohta T, Ohuchida S, Yamamoto H, Maruyama T, Kondo K, Ushikubi F, Narumiya S, Sugimura T, Wakabayashi K (1999) Role of the prostaglandin E receptor subtype EP1 in colon carcinogenesis. Cancer Res 59:5093–5096
Weitz J, Koch M, Debus J, Hohler T, Galle PR, Buchler MW (2005) Colorectal cancer. Lancet 365:153–165
Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B (2003) Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res 9:606–612
Zaremba S, Barzaga E, Zhu M, Soares N, Tsang KY, Schlom J (1997) Identification of an enhancer agonist cytotoxic T lymphocyte peptide from human carcinoembryonic antigen. Cancer Res 57:4570–4577
Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, Rubin SC, Coukos G (2003) Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 348:203–213
Zou W (2005) Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 5:263–274
Acknowledgments
We are grateful for the routine pathology data made available from Department of Pathology, Ullevaal University Hospital and technical assistance with preparation of tissue specimens and multicolor immunostaining by Linda Kristiansen, Vigdis Wendel, and Aaste Aursjo (The Pathology Clinic, Rikshospitalet-Radiumhospitalet Medical Center). This work was supported by grants from the Norwegian Functional Genomics Programme (FUGE), The Research Council of Norway, The Norwegian Cancer Society, Novo Nordic Foundation Committee, and the European Union grant no. 037189. S. Yaqub and M. Mahic are fellows of the Norwegian Cancer Society.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yaqub, S., Henjum, K., Mahic, M. et al. Regulatory T cells in colorectal cancer patients suppress anti-tumor immune activity in a COX-2 dependent manner. Cancer Immunol Immunother 57, 813–821 (2008). https://doi.org/10.1007/s00262-007-0417-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-007-0417-x