
Abstract
Radiolabelled receptor-binding peptides targeting receptors (over)expressed on tumour cells are widely under investigation for tumour diagnosis and therapy. The concept of using radiolabelled receptor-binding peptides to target receptor-expressing tissues in vivo has stimulated a large body of research in nuclear medicine. The 111In-labelled somatostatin analogue octreotide (OctreoScan™) is the most successful radiopeptide for tumour imaging, and was the first to be approved for diagnostic use. Based on the success of these studies, other receptor-targeting peptides such as cholecystokinin/gastrin analogues, glucagon-like peptide-1, bombesin (BN), chemokine receptor CXCR4 targeting peptides, and RGD peptides are currently under development or undergoing clinical trials. In this review, we discuss some of these peptides and their analogues, with regard to their potential for radionuclide imaging of tumours.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Radiolabelled receptor-binding peptides have emerged as an important class of radiopharmaceuticals for tumour diagnosis and therapy. The concept of using radiolabelled receptor-binding peptides to target receptor-expressing tissues in vivo has stimulated a large body of research in nuclear medicine. Small peptides for receptor imaging and targeted radiotherapy have advantages over proteins, antibodies and antibody fragments. Peptides are small molecules and show rapid diffusion in target tissue. Due to their low molecular weight they clear rapidly from the blood and non-target tissues, resulting in high tumour-to-background ratios. In addition, peptides generally are non-immunogenic. Peptides have been labelled for use with SPECT and PET. Commonly used gamma emitters used for peptide labelling are 111In and 99mTc. For PET imaging, peptides can be radiolabelled with positron emitting radionuclides such as 68Ga, 18F and 64Cu. Radiolabelled receptor-binding peptides can be used to noninvasively visualize receptor-expressing tissues, a technique referred to as peptide-receptor radionuclide imaging. To date, the 111In-labelled somatostatin analogue octreotide (OctreoScan™) is the most successful radiopeptide for tumour imaging, and was the first to be approved for diagnostic use. It is considered the diagnostic gold standard for imaging several types of neuroendocrine tumours. Other receptor-targeting peptides such as cholecystokinin (CCK)/gastrin analogues, glucagon-like peptide-1 (GLP-1), BN, chemokine receptor CXCR4 targeting peptides, and RGD peptides are currently under development or undergoing clinical trials. Here, we discuss some of the most widely studied peptides, other than peptides targeting somatostatin receptors (SSTR) and their potential for radionuclide imaging of tumours.
CCK peptides
CCK is a peptide hormone, originally discovered in the gastrointestinal tract by Ivy and Oldberg [1]. In 1975, CCK was described as a gastrin-like immunoreactive peptide and as one of the most widespread neuropeptides in the central nervous system [2]. CCK exerts various physiological actions in the gastrointestinal tract and in the central nervous system. CCK was initially characterized as a 33-amino acid sequence, but the peptide was shown to be present in a variety of biologically active molecular forms such as CCK39, CCK33, CCK8 and CCK4, all derived from a 115-amino acid precursor molecule. The most abundant peptide in the brain is CCK8 (Asp-Tyr-Met-Gly-Trp-Met-Asp-Phe-NH2) [3, 4].
Receptors for CCK have been pharmacologically classified based on their affinity for the endogenous peptide CCK and gastrin. These two ligands share the same amidated C-terminal pentapeptide sequence but differ in sulphation of the tyrosine residue at position 6 (gastrin) or 7 (CCK) [5, 6]. Three types of CCK receptors have been identified. The CCK1 (formerly known as CCK-A) receptor was first characterized in pancreatic acinar cells [7], and is mainly located in the periphery. The CCK2 (formerly known as CCK-B) receptor was discovered in the brain [8] and is expressed in the brain and in the stomach, pancreas and gallbladder. In the gastrointestinal tract, activation of this receptor by gastrin stimulates gastric acid secretion [4]. The third type of CCK receptor is the CCK2i4sv receptor, a splice variant of the CCK2 receptor, first isolated and characterized by Hellmich et al. [9]. This receptor is generated by intron 4 retention during RNA processing, resulting in a 69-amino acid insert in the third intracellular loop domain of the receptor [9]. The CCK2i4sv receptor was discovered in human colorectal cancer cells and stimulates cell growth through a gastrin-independent mechanism.
The CCK1 and CCK2 receptor have been shown to differ by their affinity for gastrin binding, their differential distribution and their molecular structure. The CCK1 receptor binds sulphated CCK with a 500- to 1,000-fold higher affinity than nonsulphated CCK. The CCK2 receptor binds gastrin and CCK with almost the same affinity and does not discriminate between the sulphated and nonsulphated CCK analogues [3]. The CCK2 receptor is therefore also referred to as the gastrin receptor.
Tumour expression of CCK receptors
CCK1 and CCK2/gastrin receptors have been identified in several normal tissues and in various tumours. Reubi and Waser identified an unexpectedly high incidence (>90%) of CCK2 receptors in medullary thyroid carcinomas (MTC), whereas differentiated thyroid cancers do not express CCK2 receptors [10]. MTCs comprise 3–12% of all thyroid cancers. In addition, CCK2 receptors are frequently found in astrocytomas (65%) and stromal ovarian cancers (100%). CCK1 receptors are expressed rarely in human tumours [11]. The splice variant of the CCK2 receptor, the CCK2i4sv receptor, is expressed in human colorectal cancers and pancreatic cancers, but not in normal colorectal mucosa [9, 12]. However, expression levels of the CCK2i4sv receptor may be too low to allow efficient targeting with radiolabelled peptides, as a recent study showed rarely any expression of CCK2i4sv mRNA in pancreatic, gastric and colorectal carcinomas [13].
Several research groups have aimed to develop suitable radioligands for targeting the CCK2 receptor in vivo. A variety of radiolabelled CCK/gastrin-related peptides have been synthesized and characterized. All peptides have in common the C-terminal CCK receptor-binding tetrapeptide sequence Trp-Met-Asp-Phe-NH2 or derivatives thereof. The presence of an intact C-terminal sequence has been shown to be crucial for receptor binding, although the methionine may be replaced by leucine or norleucine without affecting receptor binding affinity [5, 14].
Preclinical studies with radiolabelled CCK2R-binding ligands
In the late 1990s, Behr et al. showed promising results with 131I-radioiodinated human gastrin-I in diagnostic and therapeutic applications [14]. This synthetic heptadecapeptide has a low nanomolar affinity for CCK2 receptors, whereas its affinity to CCK1 receptors is at least four orders of magnitude lower. Behr et al. investigated a series of 18 radioiodinated gastrin and CCK derivatives for targeting CCK receptors in vivo [5]. They found that sulphated CCK analogues and some nonsulphated gastrin analogues displayed the highest affinities (IC50 values in the nanomolar range), whereas desulphation or the complete removal of the N-terminally located tyrosine of the peptide led to a loss of affinity.
Reubi et al. developed a series of nonsulphated CCK8 analogues which were N-terminally conjugated with diethylenetriaminepentaacetic acid (DTPA) or 1,4,7,10-tetraazacyclododecane-1,4,7,10 tetraacetic acid (DOTA) to allow radiometal binding [15]. They found a high specificity towards CCK2 receptors, which was determined by the presence of nonsulphated tyrosine. Analogues in which methionine in position 3 and 6 was replaced by norleucine—to prevent oxidation causing a loss of affinity—had similar binding properties (IC50 1.5 nM) to those of native CCK8 (IC50 2.3 nM) and showed increased plasma stability. These analogues showed promising results in non-tumour-bearing rats: rapid clearance by renal excretion, increased plasma stability and low uptake and retention in the main peripheral soft tissues. Based on this study, Reubi et al. concluded that nonsulphated CCK analogues are highly promising for CCK2 receptor scintigraphy.
Subsequently, de Jong et al. investigated the potential of 111In-DOTA-CCK8[Nle3,6] for peptide receptor radionuclide therapy (PRRT) [16]. Internalization, biodistribution and tumour targeting were studied and a receptor-specific and time- and temperature-dependent internalization of 111In-DOTA-CCK8 in AR42J cells (rat pancreatic tumour) was demonstrated. Evaluation in a syngeneic rat tumour model showed good targeting of CA20948 tumours (rat pancreatic tumour). Uptake of 111In-DOTA-CCK8[Nle3,6] in CCK2 receptor-expressing tissues was specific and uptake in receptor-negative organs was low.
We studied two 99mTc-labelled CCK8 analogues for scintigraphic imaging of CCK receptors, nonsulphated CCK8 (nsCCK8) and sulphated CCK8 (sCCK8) [17]. We demonstrated that uptake of the sulphated analogue, 99mTc-tricine/HYNIC-sCCK8, in both CCK1 and CCK2 receptor-expressing tumours in mice was approximately 15-fold higher than that of the nonsulphated analogue. More recently, we showed that 111In-labelled DOTA-sCCK8 and DOTA-minigastrin (MG0) also have affinity for the splice variant of the CCK2 receptor, the CCK2i4sv receptor [18]. Tumour uptake of 111In-labelled DOTA-sCCK8 in CCK2i4sv receptor-positive tumours was similar to the uptake in CCK2 receptor-expressing tumours.
In 2004, Aloj et al. reported results obtained with 111In-DTPA-Glu-Gly-CCK8 using DTPA as a chelator [19, 20]. They used the same chelator Béhé et al. used in their earlier studies with minigastrin [21]. A glycine residue was introduced as a spacer between the chelator and the peptide. The peptide conjugate showed good affinity for the CCK2 receptor. In line with the results of Béhé et al., chelation of 111In was found to be more stable than with conventional DTPA. Tumour uptake of 111In-DTPA-Glu-G-CCK8 in A431-CCK2R xenografts was 4%ID/g at 30 min after injection [19].
Three 99mTc-labelled minigastrin analogues were investigated by Nock et al. for targeting the CCK2/gastrin receptor [22]. They derivatized minigastrin with an open-chain tetraamine either directly (Demogastrin 1) or via different spacers (Demogastrin 2 and 3), to achieve stable labelling with 99mTc. After injection in mice, tumour-to-nontarget ratios were favourable for 99mTc-Demogastrin 2 (with a glycine between the chelator and the peptide). The high kidney uptake could be reduced by coinjection of poly-Glu-containing peptides. Similar observations were reported by Béhé et al. after intraperitoneal injection of poly-Glu peptides in rats [23].
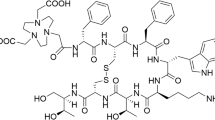
Mather et al. aimed to identify a radioligand that combined the relatively high tumour uptake of peptides belonging to the gastrin family with the low renal uptake seen with CCK derivatives [24]. They prepared a library of DOTA and DTPA peptide conjugates based on the C-terminal structure of minigastrin. Removal of the pentaglutamate sequence present in minigastrin resulted in a strong reduction in kidney uptake from 60%ID/g to 3%ID/g, but also reduced tumour uptake by a factor of 3. Replacement of the pentaglutamate sequence in minigastrin with a hexahistidine tag resulted in a similar reduction in kidney and tumour uptake. They found that a dihistidine analogue (DOTA-His-His-Glu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2) showed the best results in terms of tumour-to-kidney ratio, with a K i value of 3.9 nM. Radiolabelling of these analogues, however, needed heating to 100°C, resulting in a high level of oxidation of the methionine residues. The most effective, clinically acceptable antioxidant to overcome this problem was monothioglycerol. Substitution of the methionine residue with nonoxidizable norleucine lowered both in vitro receptor affinity and in vivo tumour uptake. These findings are in contrast with those of Reubi et al. in their study on CCK8 analogues [15] and with our findings [25]. In the search for stabilized sCCK8 analogues, we synthesized peptides in which the methionine residues were replaced by either norleucine or homopropargylglycine to prevent oxidation. Furthermore, the sulphated tyrosine was replaced by a stable synthetic isostere, phenylalanine sulphonate. In vitro studies showed that the peptides were resistant to oxidation, whereas the affinity was retained in the low nanomolar range. Biodistribution studies in AR42J tumour-bearing mice showed a tumour uptake of 111In-DOTA-sCCK8[Phe2(p-CH2SO3H),Nle3,6] comparable to that of 111In-DOTA-sCCK8. Imaging was performed with 111In-DOTA-sCCK8[Phe2(p-CH2SO3H),Nle3,6] and with the lead compound 111In-DOTA-sCCK8 (Fig. 1).

SPECT/CT images of mice with subcutaneous A431-CCK2R tumours 1 h after injection of 111In-labelled (a) DOTA-sCCK8, (b) DOTA-sCCK8 [Phe2(p-CH2SO3H),Nle3,6] and DOTA-MG0 as a reference. Radiotracer uptake is clearly visible in the A431-CCK2R tumours (arrows), whereas no uptake is observed in the mock-transfected A431 tumours (arrowheads)
Good et al. reported a decreased circulatory half-life and lower tumour uptake of 111In-DOTA-MG11 (D-Glu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2) in mice in comparison with 111In-DTPA-MG0 (D-Glu-(Glu)5-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2) [26]. Although tumour-to-kidney ratios were higher for 111In-DOTA-MG11, the absolute tumour uptake in AR42J tumours was lower. Béhé et al. showed that the high renal uptake of 111In-DTPA-MG0 in mice could be significantly reduced by coinjection of polyglutamic acids, whereas tumour uptake was not impaired [23].
Linear gastrin analogues exist under various folded conformations in solution. As cyclization was shown to improve the in vivo characteristics of other peptides such as RGD analogues [27, 28], von Guggenberg et al. designed and evaluated two cyclic minigastrin analogues (cyclo-MG), based on MG11 [29]. They synthesized two MG analogues containing unnatural amino acids in the peptide chain and a cyclic constraint introduced through an internal amide bond. The peptides were N-terminally conjugated with HYNIC to allow 99mTc labelling. In vitro studies showed that receptor binding was impaired by cyclization, possibly related to the reduced flexibility of the peptide backbone. However, internalization of both cyclic MG analogues in AR42J cells was similar to that of linear 99mTc-HYNIC-MG11. Tumour uptake of >3%ID/g at 1 h after injection was observed in nude mice with subutaneous AR42J tumours for both analogues, whereas the linear MG1 (99mTc-HYNIC-γ-D-Glu-Ala-Tyr-D-Lys-Trp-Met-Asp-Phe-NH2) analogue showed a very low tumour uptake of <0.3%ID/g. More recently, these cyclized peptides were conjugated with DOTA, to allow radiolabelling with 68Ga and 111In [30]. Cyclization was reported to be important to maximize tumour uptake of this peptide, but did not improve the overall pharmacokinetic profile.
Stabilization of the peptide requires further optimization to obtain a radioligand suitable for diagnostic and/or therapeutic applications. Stabilization could possibly also be achieved by dimerization of MG11. Sosabowski et al. synthesized MGD5, a DOTA-conjugated dimeric gastrin peptide [31]. The K d of the dimeric peptide was 0.69 nM as compared to 2.9 nM for the monomeric 111In-APH070 peptide. In addition, the internalization rate of 111In-MGD5 was nearly double that of 111In-APH070. SPECT/CT imaging and biodistribution studies have revealed that 111In-MGD5 has a clearly higher tumour uptake than 111In-APH070.
Recently, the biodistribution and in vitro characteristics of 12 CCK2R-targeting peptides have been investigated in a collaborative effort by various research groups [32–34]. These studies indicated that three peptides are optimal since they combined high tumour uptake with low kidney retention. Besides the above-mentioned cycloMG1 and dimeric MGD5 peptides, the linear minigastrin analogue PP-F-11 showed good in vivo characteristics. In this peptide the five N-terminal l-Glu residues of MG0 are replaced by six d-Glu residues [35], resulting in a kidney uptake only one-tenth that of MG0. Potentially the stability of these peptides could be further improved. Ocak et al. [34] found variable stability of the various gastrin peptides with serum half-lives ranging from 4.5 ± 0.1 h to 198 ± 0.1 h. In the urine of mice, only metabolized peptide fragments were detected, even at short times after injection for all peptides. MALDI-TOF MS revealed a major cleavage site in all gastrin derivatives between Asp and Phe-NH2 at the C-terminal end of the peptide.
Clinical studies with radiolabelled CCK2R-binding ligands
In the past few years, several studies in MTC patients have been performed with both gastrin-like [5, 22, 36–38] and CCK-like [39] peptides. The evaluation of 111In-DTPA-CCK8[Nle3,6] in patients was reported by Kwekkeboom et al. [39]. The results showed high background activity levels in the scintigraphic images, relatively low uptake in the strongly CCK receptor-positive stomach and a rapid degradation of 111In-DTPA-CCK8[Nle3,6] in the serum. Although confirmed MTC lesions could be visualized in two patients, small MTC lesions could not be detected.
Behr et al. reported a clinical pilot study with 111In-DTPA-MG0 in four MTC patients [5]. They found CCK2 receptor targeting in physiologically CCK2 receptor-expressing tissues (e.g. the stomach) as well as in metastatic MTC lesions. In 2002, Behr and Béhé reported a clinical study using 111In-DTPA-MG0 for both imaging (75 MTC patients) and therapy (8 MTC patients) [36, 40]. In the imaging study, 185–259 MBq of 111In-DTPA-MG0 was injected and whole-body scans were performed at several time-points after injection. They found that normal organ uptake was restricted to the stomach and the kidneys. In patients with known disease, all tumours known from conventional imaging modalities were visualized as early as 1 h after injection, although optimal scans were obtained at 24 h after injection. More importantly, in 29 out of the 32 MTC patients with occult disease, at least one lesion was visualized. Uptake in the liver and spleen was very low.
Gotthardt et al. reported a tumour detection rate of 87% by scintigraphy using 111In-DTPA-MG0 in a group of 26 MTC patients [37]. Most of the patients in that study, however, had known metastases and in the group of patients with occult disease, tumour lesions were found in only one. Recently, Fröberg et al. investigated and compared 99mTc-Demogastrin 2, evaluated earlier in vitro and in vivo by Nock et al. [22], with 111In-DOTA-CCK8 and 111In-DOTA-MG11 [38]. In an earlier study by this group [41], radiolabelling conditions were optimized and preclinical aspects of the compounds were investigated. 99mTc-Demogastrin 2 showed the best tumour visualization, which may have been due to better imaging properties of 99mTc compared to those of 111In. 99mTc-Demogastrin 2 visualized all known lesions in six MTC patients, whereas with the other two compounds several known lesions were missed. In addition, in four patients, new lesions in the neck, brain, bone and liver were discovered with 99mTc-Demogastrin 2. Both 111In-DOTA-CCK8 and 111In-DOTA-MG11 were shown to be less suitable for scintigraphy as the sensitivity as well as the uptake in visible lesions were limited and appeared to be insufficient for radionuclide therapy in these tumours. Moreover, the stability of 111In-DOTA-MG11 was poor. Although this peptide was reported to be stable in ex vivo human serum, in this study HPLC analysis of blood samples from patients showed that 10 min after administration, only 10% of the original peptide was still intact. 99mTc-Demogastrin 2 was more stable, illustrated by the fact that more than 60% of the radioligand was still intact at the same time-point. Therefore, it was concluded that 99mTc-Demogastrin 2 appears to be a promising diagnostic tool in patients with MTC.
GLP-1 peptides
GLP-1 is an intestinal hormone that stimulates insulin secretion through receptors expressed on islet cells. GLP-1 receptors are expressed mainly in the pancreas, stomach and brain. In addition, these receptors are also abundantly overexpressed on >90% of insulinomas at a mean density twice that of SSTR type 2 (SSTR2) [42, 43]. To investigate the feasibility of radiolabelled GLP-1 for the detection of insulinomas, tumour-targeting studies have been performed with radioiodinated GLP-1 and the GLP-1 receptor antagonist exendin-3 [44]. Studies were performed in rats bearing rat insulinoma RINm5F tumours. GLP-1(7–36)amide and exendin-3 were labelled with 125I. Although with both peptides the tumours could be visualized, the antagonist performed better. Radioiodinated GLP-1 was rapidly degraded in vivo (<10 min) , whereas the 125I-exendin-3 showed better stability in vivo. Therefore, it was concluded that GLP-1 antagonists, such as exendin-3, might be suitable tracers for scintigraphic imaging of insulinomas. In a second study, the GLP-1 analogue exendin-4 was synthesized with an additional Lys residue at position 40, allowing site-specific conjugation of DTPA. Biodistribution of 111In-labelled exendin-4 was studied in mice and rats. Specific uptake was observed in receptor-positive organs such as the stomach, pancreas, lungs and adrenals. High uptake was seen in the kidneys. Tumour targeting of 111In-DTPA-[Ahx-Lys40]exendin-4 was studied in transgenic Rip1Tag2 mice which spontaneously develop insulinomas. Due to the high GLP-1 receptor expression in these tumours, the tumour uptake was very high (287 ± 62%ID/g at 4 h after injection). Tumours as small as 1 mm could be detected by multipinhole SPECT imaging [45].
More recently, Brom et al. investigated the potential of PET imaging using DOTA-[Lys40]-exendin-3 [46]. In nude mice with subcutaneous rat insulinoma INS-1 tumours 68Ga-DOTA-[Lys40]-exendin-3 was compared to 111In-DTPA-[Lys40]-exendin-3. The IC50 values of both compounds were similar and were in the low nanomolar range. Remarkably, tumour uptake of 68Ga-DOTA-[Lys40]-exendin-3 was significantly lower than that of 111In-DTPA-[Lys40]-exendin-3 at 1 h after injection (25.1 ± 7.2%ID/g vs. 8.9 ± 3.1%ID/g, respectively). Despite this lower uptake, small subcutaneous tumours could be visualized by PET/CT imaging, and the authors considered that clinical studies should be conducted to investigate the potential of 68Ga-DOTA-[Lys40]-exendin-3 for insulinoma imaging in humans.
A similar study has been performed comparing 111In-DOTA[Ahx-Lys40]exendin-4, 68Ga-DOTA[Ahx-Lys40]exendin-4 and 99mTc-EDDA/HYNIC[Ahx-Lys40]exendin-4 in Rip1Tag2 mice [47]. The 99mTc-labelled compound showed a significantly lower tumour uptake than the 111In and 68Ga-labelled peptides. Dosimetry calculations revealed that 111In-DOTA[Ahx-Lys40]exendin-4 resulted in the highest effective dose (extrapolated to humans, 155 μSv/MBq), whereas the effective dose of 68Ga-DOTA[Ahx-Lys40]exendin-4 and 99mTc-EDDA/HYNIC [Ahx-Lys40]exendin-4 were much lower (32 μSv/MBq and 3.7 μSv/MBq, respectively).
Recently, a novel GLP-1 analogue, EM3106B, was labelled with 18F using N-2-(4-18F-fluorobenzamido)ethylmaleimide (18F-FBEM) [48]. This compound was synthesized in a 25% yield in 60 min. The IC50 value for the GLP-1 receptor was 1.07 ± 0.84 nM, which was similar to that of the unlabelled compound, but approximately fivefold better than the IC50 of GLP-1. In vivo studies in INS-1 tumour-bearing mice revealed high (28.5 ± 4.7%ID/g) and specific tumour uptake at 1 h after injection which could be clearly visualized by microPET imaging. Specific uptake was also noted in the pancreas, spleen, stomach and intestine.
Clinical studies with radiolabelled GLP-1R-targeting peptides
The feasibility of 111In-DTPA[Ahx-Lys40]exendin-4 for clinical use was first demonstrated in two patients with insulinoma [49]. After this proof of concept, a clinical study with an exendin peptide for insulinoma imaging was performed in six patients by the groups in Basel and Bern [50]. They used 111In-DOTA[Ahx-Lys40]exendin-4 in patients with proven endogenous hyperinsulinaemic hypoglycaemia. A peptide dose of 30 μg (82–97 MBq) was injected and SPECT/CT scans were recorded at 20 min, and 4 and 23 h after injection. In all six patients, a nadir in glucose levels occurred at 40 min after injection. In three patients, an exogenous glucose infusion was necessary. Blood sampling revealed a biexponential blood clearance with approximately 70% of the administered dose cleared in the alpha phase. The clearance occurred exclusively via the kidneys. In four of the six patients, the tumour could be clearly visualized at 4 h after injection, while in two patients the tumours could only be visualized in late scans (3–7 days), most likely due to their location close to the kidneys. The authors also showed that the in vivo determination of GLP-1R using 111In-DOTA[Ahx-Lys40]exendin-4 correlated with in vitro autoradiography and histology analysis of GLP-1R. Finally, it was demonstrated that the intraoperative use of a gamma probe 2–14 days after injection of 111In-DOTA[Ahx-Lys40]exendin-4 was highly beneficial for the in situ localization of tumours.
Recently, GLP-1R targeting with 111In-DTPA[Ahx-Lys40]exendin-4 was compared to SSTR2 targeting of 68Ga-DOTA-TATE PET/CT in patients with suspected insulinoma [51]. Of the 11 patients studied, GLP-1R targeting was positive in four, while SSTR2 targeting was positive in eight. In one patients both receptors were expressed. It was concluded that, in contrast to benign insulinomas, malignant insulinomas do not always express GLP-1R, but more often express SSTR2. They always express one of the two receptors.
CXCR4-binding peptides
Chemokines are small proteins (8–14 kDa) that chemoattract leucocytes by binding to cell surface receptors, chemokine receptors. In 1996, one of these receptors, CXCR4, was identified as a coreceptor for the entry of T-cell line-tropic HIV-1 [52]. Later, it was found that CXCR4 and its ligand, stromal cell-derived factor-1 (SDF-1), also play an important role in tumour metastasis [53]. Müller et al. [53] reported that CXCR4 is highly expressed in breast cancer and SDF-1 is highly expressed in organs representing the first destinations of metastasis. Moreover, they demonstrated that neutralization with anti-CXCR4 monoclonal antibody significantly inhibits the metastasis of breast cancer cells in mice. Similar results were obtained in other types of cancer (see reference [54] for review). Based on these findings, several groups exploited targeting of CXCR4 as a potential tool for the imaging of metastatic tumours.
The first study in which the development of an 111In-labelled CXCR4 targeting peptide was investigated was published in 2006 [54]. Based on the previously discovered T22 peptidic CXCR4 inhibitor a DTPA-conjugated labelled analogue was designed. This cyclic peptide, designated DTPA-Az-TZ14011 was stabilized by amidation at the C-terminus and N-terminally acetylated. The authors showed that 111In-DTPA-Tz-14011 had an IC50 for CXCR4 of 7.9 nM. An in vivo study in tumour-bearing mice demonstrated specific targeting in the tumour, with tumour-to-blood ratios increasing from 1.31 ± 0.14 at 1 h after injection to 5.65 ± 2.68 at 24 h. High and specific uptake was noted in the liver and spleen, which could be attributed to the high expression of CXCR4 mRNA in mice.
Based on this compound, a radiofluorinated peptide was developed [55]. The peptide was labelled with 18F using the well-established [18F]SFB method in a 50–60% decay-corrected yield. The IC50 value as determined in CXCR4-transfected CHO cells was 2.5 nM. Biodistribution in mice bearing CHO-CXCR4 tumours revealed moderate uptake in the tumour (2.3 ± 0.7%ID/g) which was not significantly higher than in the CXCR4-negative control tumour (1.7 ± 0.9%ID/g). Relatively high uptake was found in CXCR4-rich organs such as the spleen and bone marrow. Remarkably high blood levels were found, with the majority of the radiolabelled peptide (94.5%) in the red blood cell fraction. Association of the peptide with red blood cells in the circulation most likely resulted in a decreased availability of the peptide to bind to CXCR4 in the tumour.
More recently, a non-peptidic CXCR4 inhibitor was radiolabelled with 64Cu [56]. In this inhibitor, AMD3100, the two cyclam moieties are linked by a 1,4-phenylenebis(methylene) bridge. AMD3100 has been identified as a specific inhibitor of CXCR4. AMD3100 was originally developed as an inhibitor of HIV. It was also found to inhibit SDF-1-induced chemotaxis through CXCR4, suggesting that it might also be used to inhibit cancer metastasis. One of the cyclam rings was used to radiolabel the compound with 64Cu. Biodistribution was studied in non-tumour-bearing mice. High uptake was noted in the liver and kidneys and in CXCR4-expressing organs such as the spleen, lymph nodes and bone marrow. This compound was also tested in mice with human cancer xenografts [57]. Proof of principle of CXCR4 imaging with [64Cu]AMD3100 was shown in mice bearing CXCR4-transfected U87MG tumours. PET/CT imaging revealed high and specific uptake (35%ID/g at 90 min after injection) in the tumour as well as significant uptake in the liver, kidneys and bladder. Similar results were found in a CXCR4-expressing orthotopic MDA-MB-231 breast cancer model. Most importantly, lung metastases derived from the same breast cancer tumour could be visualized by PET/CT imaging. Weiss et al. reported similar results with [64Cu]AMD3100 in mice with transfected tumours in the liver and lungs [58]. Although promising, the bicyclam AMD3100 has a relatively low affinity (approximately 650 nM). In search of agents that are amenable to structural modification, the same group investigated the monocyclam analogue AMD3465 to image CXCR4 expression. Compared with the previous compound, AMD3465 has higher affinity, and reduced size and charge [59]. Despite the improved affinity and kinetics, [64Cu]AMD3465 also shows considerable liver uptake. Some of this liver uptake might be attributed to possible transchelation of 64Cu from [64Cu]AMD3465 to proteins such as superoxide dismutase and ceruloplasmin. Clinical studies with these peptides have not been published.
Gastrin-releasing peptide receptor-targeting peptides
BN is a 14-amino-acid C-terminally amidated peptide of amphibian origin with the sequence Glu1-Gln2-Arg3-Leu4-Gly5-Asn6-Gln7-Trp8-Ala9-Val10-Gly11-His12-Leu13-Met14-NH2 (see Table 1). Gastrin-releasing peptide (GRP) is its 27-amino-acid human counterpart which shares the seven C-terminal amino acids with BN. There are three BN mammalian receptor subtypes, namely BB1 (formerly designated as the neuromedin B receptor), BB2 (formerly designated as the GRP receptor, GRPR) and BB3 (the orphan receptor, for which the native ligand has yet to be identified). BN and GRP both bind with high affinity to GRPR, the subtype found to be most frequently expressed in tumours [60]. This receptor is overexpressed in some common tumour types such as carcinomas of the prostate and breast as well as small-cell lung cancer, renal cell carcinoma and gastrointestinal stromal tumours (GIST) [60–62]. For this reason, much of the research effort has focused on high-affinity ligands of GRPR. These ligands are generally analogues of full-length BN(1–14) or are truncated analogues based on the C-terminal amino acid sequence (BN(7–14)) which confers receptor binding affinity.
Preclinical studies with radiolabelled BN analogues
A recurrent theme in the development of 99mTc-labelled BN analogues is their tendency to be somewhat lipophilic and to clear via the hepatobiliary pathway (thus hindering imaging in the abdominal region). Baidoo et al. described the synthesis of 99mTc diaminodithiol conjugates of Lys3-BN [63]. Subsequent work by this group involved modifications of this compound to reduce the lipophilicity of these analogues to allow scintigraphy of the abdominal region, introducing DTPA into the sequence as a hydrophilic pharmacokinetic modifier [64, 65]. The group of Volkert has used P2S2 [66, 67] and N3S BN(7–14) conjugates [68] using different carbon chain spacers between the Tc-binding moiety and the peptide. Smith et al. showed that a spacer of three to eight carbon atoms could be used without compromising the agonist binding affinity of the 99mTc-N3S-X-BN(7–14)NH2 constructs [68]. Van de Wiele et al. demonstrated the clinical utility of these compounds using a 99mTc-N3S-Gly-5-Ava-BN(7–14) developed by Resolution Pharmaceuticals, Canada (99mTc-RP527) (see Table 1) [69], but again hepatobiliary excretion of this compound made imaging in the abdominal areas problematic [70]. Nock et al. investigated a series of compounds, one based on a GRPR antagonist as well as four other GRPR agonists. Demobesin 1 is based on the potent antagonist [D-Phe6, Leu-NHEt13,des-Met14]BN(6–14) [71], to which they attached an open chain tetraamine (N4) chelator for labelling with 99mTc. Although this compound showed minimal internalization in prostate cancer cells, it demonstrated very high and persistent uptake in PC-3 human xenografts in nude mice (15.61 ± 1.19%ID/g at 4 h) [72]. Using the tetraamine chelator, four further agonist compounds were developed: [N 04 ,Pro1,Tyr4]BN and its Nle14-substituted analogue (Demobesin 3 and 4) as well as two truncated BN analogues, [(N4-Bzdig)0]BN(7–14) and its Nle14-substituted analogue, using a benzylaminodiglycolic acid spacer between the chelator and the receptor-binding sequence (Demobesin 5 and 6) [73]. All these compounds showed high receptor affinity (IC50 <0.06 nM in competition binding assays) and rapid internalization (about 75 % within 30 min) in PC-3 tumour cells. In vivo, Demobesin 3 and 4 demonstrated high tumour uptake (9–11%ID/g at 1 h, 7–9%ID/g at 4 h after injection) with rapid clearance from nontarget tissues. These compounds proved to be much less lipophilic than the truncated BN analogues and are excreted mainly via the kidneys. The two truncated analogues, Demobesin 5 and 6, showed lower tumour uptake and largely hepatobiliary clearance. Due to the higher tumour uptake and favourable excretion route, clinical trials have been started on the Demobesin 4 compound. A 99mTc-EDDA/HYNIC-[Lys3]BN compound has been developed by Ferro-Flores et al. [74]. Although this compound shows predominantly renal excretion, it has relatively low uptake in PC-3 tumours in vivo.
99mTc-BN derivatives have also been prepared via the [99mTc(H2O)3(CO)3]+ precursor. La Bella et al. demonstrated high binding affinity for the GRP receptor, and rapid internalization into PC-3 cells, of their [99mTc(CO)3-Nα-histidinylacetate]BN(7–14) compound; however, tumour uptake was low and no blocking was seen. Since uptake into the pancreas was high and specific, the authors speculated that this was due to weak vascularization of the tumour xenografts. This group developed another tricarbonyl technetium BN conjugate: [99mTc(I)-PADA-AVA]BN(7–14), which demonstrated hepatobiliary clearance in vivo and relatively low (but specific) tumour uptake [75]. Stabilization of the peptide sequence and insertion of hydrophilic spacers to counteract the lipophilicity of the [99mTc(CO)3((NαHis)Ac)] chelate led to the development of [99mTc(CO)3((NαHis)Ac)]-X-(Cha13,Nle14) BN(7–14) (where Cha is cyclohexylalanine) where the most hydrophilic spacer Lys(sha)-βAla–βAla- (where sha is shikimic acid) gave the highest tumour uptake 3.2%ID/g at 1.5 h in PC-3 tumours in mice as well as the highest tumour-to-nontumour ratios compared with compounds without spacers or stabilized peptide sequence [76–78]. This group further investigated the effect of glycation and charge on lipophilicity, tumour uptake and tumour-to-nontarget tissue ratios [79, 80]. Smith et al. also used tricarbonyl 99mTc complexes such as [99mTc(X)(CO)3-Dpr-SSS-BN(7–14)NH2] (where X is H2O or P(CH2OH)3 and DPr is diaminopropionic acid), and demonstrated specific uptake in PC-3 tumour xenografts of 3.7 ± 0.9%ID/g at 1 h after injection (better than that of the 99mTc-N3S conjugate in the same animal model) [68, 81]. More recently, this group has found tumour uptakes of these derivatives in T47-D breast cancer xenografts of up to 3.7 ± 1.8%ID/g at 1 h [82], and also found that insertion of PEG5 or PEG8 into the spacer had no significant effect on tumour retention. On the other hand, Dapp et al. found that the incorporation of PEG5 into their ((NαHis)Ac)-X-(Cha13,Nle14)BN(7–14) derivative had a positive effect on pharmacokinetics [83].
In parallel with this work on 99mTc-labelled BN analogues, other workers have been developing radiolabelled BN analogues using chelators such as DTPA and DOTA for radiolabelling with trivalent radiometals such as 111In and 67Ga for SPECT and 68Ga and 86Y for PET imaging, as well as 177Lu for therapy. The Rotterdam group investigated a number of 111In-radiolabelled DTPA conjugates of BN (both agonists and antagonists), selecting an agonist [111In-DTPA-Pro1,Tyr4]BN (termed 111In-MP2248) based on its level of uptake into GRPR-positive cells and tissues [84, 85]. Subsequently, this group developed a DOTA analogue [111In-DOTA-Pro1,Tyr4]BN (termed MP2346) which displayed better target-to-blood ratios and uptake in GRPR-expressing tissues than the DTPA analogue [86]. Further improvements in GRPR affinity were seen by shortening the sequence and inserting non-natural amino acids to develop MP2653, [111In-DTPA-ACMpip5,Tha6,βAla11,Tha13,Nle14]BN(5–14), where ACMpip is the cationic spacer 4-aminocarboxymethyl-piperidine and Tha is β-(2-thienyl) alanine. This resulted in higher uptake in PC-3 xenografts than seen with [111In-DTPA-Pro1,Tyr4]BN DTPA [87].
Similar to their work on 99mTc-labelled BN analogues [68], Hoffman et al. investigated the effect of spacer length using 111In-DOTA-labelled peptides of the structure: [DOTA-X]BN(7–14), where X represents a spacer containing 0, 3, 5, 8 or 11 carbon atoms. They found that the [111In-DOTA-8-Aoc]BN(7–14) analogue had higher affinity in vitro in PC-3 cells as well as the highest uptake in normal pancreas in vivo. Uptake in PC-3 tumours was 3.63 ± 1.1%ID/g at 1 h [88].
A different approach was taken by Zhang et al. [89] who developed DTPA- and DOTA-γ-aminobutyric acid-[D-Tyr6,β-Ala11,Thi13,Nle14]BN(6–14) derivatives (BZH1 and BZH2) that target all three human receptors (i.e. GRPR, NMB-R and BB3) based on their concomitant expression in some tumour types [60]. However, relatively low serum stability was seen for [111In]BZH1 and [111In]BZH2 and this was coupled with relatively fast tumour washout [89, 90]. 67Ga-DOTA-PEG2-[D-Tyr6,β-Ala11,Thi13,Nle14]BN(6–14) was also evaluated in AR42J tumour-bearing mice (5.26 ± 1.3%ID/g in tumour) [91]. Zhang et al. went on to evaluate 67Ga-DOTA-PEG4-BN(7–14) (DOTA-PESIN) which proved to have higher and more prolonged tumour uptake than the pan-BN analogues in PC-3 tumour-bearing nude mice (8.77 ± 1.88%ID/g at 4 h) [90].
As is the case when there are several groups working in the field, it becomes difficult to compare studies carried out in different centres using xenografts arising from cells at differing passage numbers in different mouse models. Still, the Rotterdam group has compared their full-length DOTA-BN analogue MP2346 and their truncated DTPA-BN analogue MP2653 with three of the most promising compounds from other groups (as judged by uptake in prostate cancer tumours) under standardized conditions: [111In]DOTA-PESIN, [111In]AMBA (DOTA-glycyl-4-aminobenzoyl BN(7–14)) [92] and the antagonist [99mTc]Demobesin-1. [99mTc]Demobesin-1 performed the best as a potential imaging agent followed by [111In]AMBA and [111In]DOTA-PESIN, both of which could be used for imaging or PRRT [93]. Clinical studies of [99mTc]Demobesin-1 are currently being carried out in Rotterdam.
The unparalleled tumour targeting ability of the 99mTc-Demobesin-1 antagonist has led others to work on radiolabelling antagonists with trivalent metals. These are usually des-Met14 analogues of BN with modifications at Leu13 which affect their affinity for GRPR. For example, the 111In-labelled DOTA-aminohexanoyl-[D-Phe6,Leu-NHCH2CH2CH 133 ,des-Met14]BN(6–14) (Bomproamide) developed by Abd-Elgaliel et al. [94] showed rapid and high uptake in PC-3 tumours in SCID mice (6.90 ± 1.06%ID/g) whilst internalization into PC-3 cells in vivo was only 14% (compared with 25% for Demobesin-1 which has a C-terminal Leu-NHCH2CH 133 ). Mansi et al. developed an antagonist, 111In-RM1, for comparison with the potent agonist 111In-AMBA. These two compounds have the same chelator and linker and differ only in that the antagonist is a des-Met14 analogue with an inserted statyl group at BN(13) [95]. Despite lower GRPR affinity (K d of 8.5 ± 2.7 nM for the natIn-RM1 antagonist vs. 0.6 ± 0.3 nM for the agonist), 111In-RM1 showed higher tumour uptake than 111In-AMBA (13.4 ± 0.8%ID/g vs. 3.69 ± 0.74%ID/g at 4 h) with better tumour-to-normal tissue ratios. This group went on to develop a further antagonist molecule with a positively charged linker (which they postulated would increase affinity), i.e. DOTA-4-amino-1-carboxymethyl-piperidine-D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2 (RM2) [96]. Although the affinity of natIn-RM2 was higher (K d of 2.9 ± 0.4 nM) than that of natIn-RM1, the only significant improvement in vivo was due to lower liver uptake, leading to higher tumour-to-liver ratio. In addition to higher tumour uptake than the agonist BN derivatives and slower washout from tumours, the antagonists overall have shown much faster relative clearance from the pancreas and other abdominal GRPR receptor-expressing organs [94–96], and thus have more favourable tumour-to-normal tissue ratios.
Due to the ready availability of the 68Ga positron emitter (t 1/2 = 68 min) through the 68Ge/68Ga generator system, a number of DOTA-conjugated BN derivatives have been evaluated preclinically as 68Ga-PET diagnostic imaging agents in tumour models at 1 h [90, 91, 95–98]. The antagonist compounds in particular show very high tumour-to-normal tissue ratios at early time-points [95, 96], which is advantageous for imaging with 68Ga. Recently, the prostate tumour-targeting properties of 68Ga-labelled DOTA-G-4-aminobenzoyl BN(7–14) (AMBA) (a BN agonist) and 18F-methylcholine (18F-FCH) have been directly compared in nude mice bearing VCaP human tumour xenografts, and 68Ga-AMBA peptide targeting was found to be superior to the metabolism-based 18F-FCH targeting [97].
The development of BN analogues for diagnostic imaging using 64Cu PET has been dominated by the need to develop chelators that are stable to in vivo transchelation of the Cu(II) to liver and blood proteins, and this has recently been the subject of an editorial by Hoffman and Smith [99]. Although both DOTA and TETA (1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetraacetic acid) were used as chelators for radiolabelling BN analogues with 64Cu [100–103], high background due to transchelation was seen, leading to the development of more stable analogues mainly based on 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA) [104–106] and cross bridged derivatives of TETA, i.e. CB-TE2A [107]. Smith and coworkers have developed [64Cu-NO2A-8-Aoc]BN(7–14) and compared it with [64Cu-TE2A-8-Aoc]BN(7–14) and [64Cu-DO2A-8-Aoc]BN(7–14) [99, 104]. The NO2A analogue was found to be superior for imaging in PC-3 tumour-bearing SCID mice. However, when the same group compared a range of 64Cu-NO2A-X-BN(7–14) derivatives in which X represented different linkers, 64Cu-NO2A-AMBA-BN(7–14) (where AMBA is para-aminobenzoic acid) had higher PC-3 tumour uptake (6.05 ± 1.15%ID/g at 1 h) and more rapid clearance from nontarget tissues than [64Cu-NO2A-8-Aoc]BN(7–14), possibly due to the more hydrophilic nature of the linker [108]. Following on from their work on 64Cu-DOTA-BN(7–14) peptides containing different amino acid linkers [109], Lears et al. have recently used Sar-Ar (1-N-(4-aminobenzyl)-3,6,10,13,16,19-hexaazabicyclo[6.6.6]-eicosane-1,8-diamine) [110] as a stable chelator for Cu(II) [111]. They conjugated Sar-Ar to BN using succinic acid (SA), 8-aminooctanoic acid (Aoc), glycine and serine to form linkers. While the tumour uptake of 64Cu-SarAr-SA-Aoc-BN(7–14) and 64Cu-SarAr-SA-Aoc-GSG-BN(7–14) in PC-3 tumours in mice was high (13.0 ± 0.9%ID/g and 8.5 ± 0.8%ID/g at 1 h, respectively) and comparable with that seen for the best of the radiolabelled BN agonists, there was no improvement in tumour-to-normal tissue ratios due to slower clearance from the normal tissues than other 64Cu-BN analogues [111]. The clearance from blood, liver, tumour and pancreas was faster for the analogue with the more hydrophilic GSG-containing linker, and it may be that investigation of more hydrophilic linkers would improve this molecule. To date there have yet to be any studies published on the use of 64Cu-labelled BN antagonists.
A few 18F-BN derivatives (both agonists and antagonists) have been developed and evaluated [103, 112–119]. Zhang et al. radiofluorinated a full-length ([Lys3]BN) and a truncated derivative Aca-BN(7–14) (where Aca is aminocaproic acid) using N-succinimidyl-4-18F-fluorobenzoate (18F-SFB) reacted at the Lys or the Aca amino groups, respectively. They found the [18F-FB-Lys3]BN derivative to be superior to the Aca analogue in terms of affinity, PC-3 tumour uptake (5.94 ± 0.78%ID/g at 60 min) and pharmacokinetics [112]. This group went on to develop 18F-BBN-RGD heterodimers for targeting coexpressed GRPR and αvβ3 integrin in prostate cancer cells [103, 114, 115, 119]. Their 18F-FB-PEG3-Glu-RGD-BN molecule showed 4.00 ± 0.08%ID/g in PC-3 tumours with more rapid clearance from the kidney than a derivative without PEG3 in the linking moiety [114].
In a collaboration between the Centre for Radiopharmaceutical Sciences of ETH, PSI and USZ, Switzerland and Bayer Schering Pharma, Hohne et al. developed a one-step 18F-labelling process which involved the incorporation of a lipophilic silyl labelling moiety into an antagonist BN structure (similar to the 111In/68Ga-labelled RM1 and RM2 above) to give [18F]2-(4-(di-tert-butylfluorosilyl)phenyl)acetyl-Arg-Ava-Gln-Trp-Ala-Val-NMeGly-His-Sta-Leu-NH2 [113], where Ava is 5-aminopentanoic acid. However this molecule showed low tumour uptake and substantial hepatobiliary clearance and the group focused on developing derivatives with a less-lipophilic benzonitrile one-step labelling moiety and inserting polar spacers between the labelling moiety and the binding sequence. They found that the negatively charged derivative, 3-cyano-4-18F-fluoro-benzoyl-Ala(SO3H)-Ava-Gln-Trp-Ala-Val-NMeGly-His-Sta-Leu-NH2 had much higher uptake in PC-3 tumours (4.88 ± 0.36%ID/g) than the corresponding positively charged analogue (3-cyano-4-18F-fluoro-benzoyl-Arg-Ava-Gln-Trp-Ala-Val-NMeGly-His-Sta-Leu-NH2) [116]. To further increase the hydrophilicity of this compound, an additional Ala(SO3H) moiety was inserted into the spacer, and this compound (termed 18F-BAY-86-4367) showed subnanomolar GRPR binding affinity, behaving as an antagonist. High tumour uptake (6.19 ± 2.49%ID/g in PC-3 xenografts) was seen with fast, predominantly renal clearance (about 70%) and better tumour-targeting properties than either 18F-FCH or 18F-FDG [117].
Clinical studies with radiolabelled BN analogues
The first clinical studies on radiolabelled BN derivatives were carried out by van de Wiele et al. using 99mTc-N3S-Gly-5-Ava-BN(7–14) (99mTc-RP527) [69]. Planar and SPECT images were acquired and tumour uptake was seen in four of six breast cancers (as well as low diffuse uptake in normal breast) and one of four androgen-resistant bone-metastasized prostate carcinomas with good tumour-to-normal tissue ratios [69]. Primarily renal, and to a lesser extent, hepatobiliary excretion of this compound was seen [70]. The rights to RP527 were acquired by Bracco Diagnostics who went on to develop 177Lu-AMBA for PRRT [92]. The latter compound has been the subject of a phase I clinical trial which has thus far only been published in abstract form [120]. SPECT imaging with 177Lu-AMBA in seven patients with hormone-refractory prostate cancer showed lesions in five of the seven patients. High pancreatic uptake was also seen. Scopinaro et al. reported the detection of breast cancer using dynamic and static planar imaging of cys-(6-amino-n-hexanoic acid)-BN(2–14) (termed [Leu13]BN) [121] modified on its N-terminus to directly bind 99mTc [122]. In five patients with primary breast cancer, five of five primary tumours were visualized at 3 h with 99mTc[Leu13]BN, as well as axillary involvement in two of the five patients. Again diffuse uptake in normal breast was seen, but tumour-to-normal breast tissue ratios were better than those seen with 99mTc-sestamibi [121]. In addition, dynamic planar and SPECT images were acquired in ten patients with a primary prostate tumour (two benign adenomas). All eight patients with prostate cancer were positive and pelvic lymph node involvement was correctly detected in three patients [123]. Neither patient with benign adenoma showed uptake. In a dynamic, planar and SPECT study of colorectal cancer in 13 patients carried out by the same group, 11 cancers were detected using 9mTc[Leu13]BN [124]. However, little information has been given about the mode of excretion or uptake of this compound in normal tissues. To address the question as to whether or not GRPR are present in human pancreas, four patients underwent planar and dynamic scintigraphy with the agonist 111In-MP2248 ([111In-DTPA-Pro1,Tyr4]BN) and the antagonist 99mTc-Demobesin 1. Clear pancreatic uptake was seen for both compounds suggesting the presence of GRPR [125].
Clinical studies of a pan-BN derivative termed 68Ga-DOTABOM, GABA-[D-Tyr6,β-Alal11,Thi13,Nle14]BN(6–14) [126], or BZH2, have been published in abstract form and summarized by Hofmann et al. [127] and Maecke et al. [128]. PET scans in 11 patients with prostate cancer were performed. Primary tumours were visible in all patients as well as lymph node metastases in three patients. Pancreatic uptake was seen in four patients and clearance was predominantly renal with >75%ID recovered in the urine at 60 min. A similar pan-BN compound, DOTA PEG2-[D-Tyr6,β-Ala11,Thi13,Nle14]BN(6–14), 68Ga-BZH3, was compared with 18F-FDG in 17 patients with GIST [129]. Of the 17 patients, 7 were positive for uptake of 68Ga-BZH3 (8/30 lesions) whereas 14 were positive for 18F-FDG (25/30 lesions). One recurrent GIST in the stomach showed positive 68Ga-BZH3 uptake but was negative for 18F-FDG. Enhanced uptake of 68Ga-BZH3 was seen in the pancreas of all patients. The median tumour SUV values were 3.3 and 7.9 for 68Ga-BZH3 and 18F-FDG, respectively. The authors concluded that 68Ga-BZH3 may be diagnostically useful in a subset of patients with GIST in whom viable tumour is suspected but 18F-FDG is negative. The same authors have recently evaluated 68Ga-BZH3 and 18F-FDG dynamic PET in patients with recurrent gliomas in comparison with grading. Of 15 patients, 10 showed enhanced 68Ga-BZH3 uptake visually while 6 showed enhanced 18F-FDG metabolism visually. They concluded that 68Ga-BZH3 may be helpful in differentiating between low- and high-grade recurrent gliomas.
The antagonist BN derivative, 18F-BAY-4367 has recently been compared to 18F-FEC (18F-ethyl-choline), one of the diagnostic PET imaging agents for recurrent prostate cancer in Europe. Both recurrent and primary prostate cancers (five patients each) were imaged. 18F-BAY-4367 showed uptake in one of the five patients with recurrent disease, compared to positive uptake in four of the five using 18F-FEC. Of the five patients with primary disease, three were positive for 18F-BAY-4367, while 18F-FEC was able to delineate malignant lesions in all four of the patients imaged. 18F-BAY-4367 was safe and well tolerated. The authors concluded that 18F-BAY-4367 is not reliable for PET imaging of recurrent disease, but is able to visualize primary tumours in a subset of patients [130]. This conclusion is consistent with the recent finding that GRPR are overexpressed in primary prostate cancer but that this overexpression drops in high-grade or advanced disease [131]. It is clear that the androgen regulation of GRPR in prostate cancer is a factor that needs to be further understood to fully utilize these GRPR-targeting radiopharmaceuticals [132].
Conclusion
A wide series of radiolabelled peptide analogues targeting receptors specifically expressed on tumours are under investigation. This is mainly due to the success achieved with somatostatin analogues, increasing knowledge about receptor expression and advances in the radiochemistry and peptide chemistry. The majority of these new peptides have been evaluated only in animal models, aiming to optimize in vivo stability, target affinity and target-to-nontarget ratios. Clinical studies have generally been performed in small numbers of patients, mainly to investigate the feasibility of these radiolabelled peptides.
References
Ivy AC, Oldberg E. A hormone reaction for gallbladder contraction and evacuation. Am J Physiol. 1928;86:599–613.
Vanderhaeghen JJ, Signeau JC, Gepts W. New peptide in vertebrate CNS reacting with antigastrin antibodies. Nature. 1975;257:604–5.
Noble F, Wank SA, Crawley JN, Bradwejn J, Seroogy KB, Hamon M, et al. International Union of Pharmacology. XXI. Structure, distribution, and functions of cholecystokinin receptors. Pharmacol Rev. 1999;51:745–81.
Noble F, Roques BP. CCK-B receptor: chemistry, molecular biology, biochemistry and pharmacology. Prog Neurobiol. 1999;58:349–79.
Behr TM, Jenner N, Béhé M, Angerstein C, Gratz S, Raue F, et al. Radiolabeled peptides for targeting cholecystokinin-B/gastrin receptor-expressing tumors. J Nucl Med. 1999;40:1029–44.
Wank SA, Pisegna JR, de Weerth A. Cholecystokinin receptor family. Molecular cloning, structure, and functional expression in rat, guinea pig, and human. Ann N Y Acad Sci. 1994;713:49–66.
Sankaran H, Goldfine ID, Deveney CW, Wong KY, Williams JA. Binding of cholecystokinin to high-affinity receptors on isolated rat pancreatic acini. J Biol Chem. 1980;255:1849–53.
Innis RB, Snyder SH. Distinct cholecystokinin receptors in brain and pancreas. Proc Natl Acad Sci USA. 1980;77:6917–21.
Hellmich MR, Rui XL, Hellmich HL, Fleming RY, Evers BM, Townsend Jr CM. Human colorectal cancers express a constitutively active cholecystokinin-B/gastrin receptor that stimulates cell growth. J Biol Chem. 2000;275:32122–8.
Reubi JC, Waser B. Unexpected high incidence of cholecystokinin-B/gastrin receptors in human medullary thyroid carcinomas. Int J Cancer. 1996;67:644–7.
Reubi JC, Schaer JC, Waser B. Cholecystokinin(CCK)-A and CCK-B/gastrin receptors in human tumors. Cancer Res. 1997;57:1377–86.
Smith JP, Verderame MF, McLaughlin P, Martenis M, Ballard E, Zagon IS. Characterization of the CCK-C (cancer) receptor in human pancreatic cancer. Int J Mol Med. 2002;10:689–94.
Korner M, Waser B, Reubi JC, Miller LJ. CCK(2) receptor splice variant with intron 4 retention in human gastrointestinal and lung tumours. J Cell Mol Med. 2010;14:933–43.
Behr TM, Jenner N, Radetzky S, Béhé M, Gratz S, Yucekent S, et al. Targeting of cholecystokinin-B/gastrin receptors in vivo: preclinical and initial clinical evaluation of the diagnostic and therapeutic potential of radiolabelled gastrin. Eur J Nucl Med. 1998;25:424–30.
Reubi JC, Waser B, Schaer JC, Laederach U, Erion J, Srinivasan A, et al. Unsulfated DTPA- and DOTA-CCk analogs as specific high-affinity ligands for CCK-B receptor-expressing human and rat tissues in vitro and in vivo. Eur J Nucl Med. 1998;25:481–90.
de Jong M, Bakker WH, Bernard BF, Valkema R, Kwekkeboom DJ, Reubi JC, et al. Preclinical and initial clinical evaluation of 111In-labeled nonsulfated CCK8 analog: a peptide for CCK-B receptor-targeted scintigraphy and radionuclide therapy. J Nucl Med. 1999;40:2081–7.
Laverman P, Béhé M, Oyen WJ, Willems PH, Corstens FH, Behr TM, et al. Two technetium-99m-labeled cholecystokinin-8 (CCK8) peptides for scintigraphic imaging of CCK receptors. Bioconjug Chem. 2004;15:561–8.
Laverman P, Roosenburg S, Gotthardt M, Park JS, Oyen WJG, de Jong M, et al. Targeting of a CCK2 receptor splice variant with In-111-labelled cholecystokinin-8 (CCK8) and In-111-labelled minigastrin. Eur J Nucl Med Mol Imaging. 2008;35:386–92.
Aloj L, Caraco C, Panico M, Zannetti A, Del Vecchio S, Tesauro D, et al. In vitro and in vivo evaluation of In-111-DTPAGlu-G-CCK8 for cholecystokinin-B receptor imaging. J Nucl Med. 2004;45:485–94.
Aloj L, Panico M, Caraco C, Del Vecchio S, Arra C, Affuso A, et al. In vitro and in vivo characterization of indium-111 and technetium-99m labeled CCK-8 derivatives for CCK-B receptor imaging. Cancer Biother Radiopharm. 2004;19:93–8.
Béhé M, Becker W, Gotthardt M, Angerstein C, Behr TM. Improved kinetic stability of DTPA-dGlu as compared with conventional monofunctional DTPA in chelating indium and yttrium: preclinical and initial clinical evaluation of radiometal labelled minigastrin derivatives. Eur J Nucl Med Mol Imaging. 2003;30:1140–6.
Nock BA, Maina T, Béhé M, Nikolopoulou A, Gotthardt M, Schmitt JS, et al. CCK-2/gastrin receptor-targeted tumor imaging with (99m)Tc-labeled minigastrin analogs. J Nucl Med. 2005;46:1727–36.
Béhé M, Kluge G, Becker W, Gotthardt M, Behr TM. Use of polyglutamic acids to reduce uptake of radiometal-labeled minigastrin in the kidneys. J Nucl Med. 2005;46:1012–5.
Mather SJ, McKenzie AJ, Sosabowski JK, Morris TM, Ellison D, Watson SA. Selection of radiolabeled gastrin analogs for peptide receptor-targeted radionuclide therapy. J Nucl Med. 2007;48:615–22.
Roosenburg S, Laverman P, Joosten L, Eek A, Oyen WJ, De JM, et al. Stabilized (111)in-labeled sCCK8 analogues for targeting CCK2-receptor positive tumors: synthesis and evaluation. Bioconjug Chem. 2010;21:663–70.
Good S, Walter MA, Waser B, Wang X, Müller-Brand J, Béhé MP, et al. Macrocyclic chelator-coupled gastrin-based radiopharmaceuticals for targeting of gastrin receptor-expressing tumours. Eur J Nucl Med Mol Imaging. 2008;35:1868–77.
Dijkgraaf I, Kruijtzer JAW, Frielink C, Soede AC, Hilbers HW, Oyen WJG, et al. Synthesis and biological evaluation of potent alpha(v)beta(3)-integrin receptor antagonists. Nucl Med Biol. 2006;33:953–61.
Haubner R. alpha(v)beta(3)-integrin imaging: a new approach to characterise angiogenesis? Eur J Nucl Med Mol Imaging. 2006;33:S54–63.
von Guggenberg E, Sallegger W, Helbok A, Ocak M, King R, Mather SJ, et al. Cyclic minigastrin analogues for gastrin receptor scintigraphy with technetium-99m: preclinical evaluation. J Med Chem. 2009;52:4786–93.
von Guggenberg E, Rangger C, Sosabowski J, Laverman P, Reubi JC, Virgolini IJ, et al. Preclinical evaluation of radiolabeled DOTA-derivatized cyclic minigastrin analogs for targeting cholecystokinin receptor expressing malignancies. Mol Imaging Biol. 2011. doi:10.1007/s11307-011-0506-2.
Sosabowski JK, Matzow T, Foster JM, Finucane C, Ellison D, Watson SA, et al. Targeting of CCK-2 receptor-expressing tumors using a radiolabeled divalent gastrin peptide. J Nucl Med. 2009;50:2082–9.
Laverman P, Joosten L, Eek A, Roosenburg S, Peitl PK, Maina T, et al. Comparative biodistribution of 12 (111)In-labelled gastrin/CCK2 receptor-targeting peptides. Eur J Nucl Med Mol Imaging. 2011;38:1410–6.
Aloj L, Aurilio M, Rinaldi V, D'ambrosio L, Tesauro D, Peitl PK, et al. Comparison of the binding and internalization properties of 12 DOTA-coupled and (111)In-labelled CCK2/gastrin receptor binding peptides: a collaborative project under COST Action BM0607. Eur J Nucl Med Mol Imaging. 2011;38:1417–25.
Ocak M, Helbok A, Rangger C, Peitl PK, Nock BA, Morelli G, et al. Comparison of biological stability and metabolism of CCK2 receptor targeting peptides, a collaborative project under COST BM0607. Eur J Nucl Med Mol Imaging. 2011;38:1426–35.
Kolenc-Peitl P, Mansi R, Tamma M, Gmeiner-Stopar T, Sollner-Dolenc M, Waser B, et al. Highly improved metabolic stability and pharmacokinetics of indium-111-DOTA-gastrin conjugates for targeting of the gastrin receptor. J Med Chem. 2011;54:2602–9.
Béhé M, Behr TM. Cholecystokinin-13 (CCK-B)/gastrin receptor targeting peptides for staging and therapy of medullary thyroid cancer and other CCK-B receptor expressing malignancies. Biopolymers. 2002;66:399–418.
Gotthardt M, Béhé MP, Beuter D, Battmann A, Bauhofer A, Schurrat T, et al. Improved tumour detection by gastrin receptor scintigraphy in patients with metastasised medullary thyroid carcinoma. Eur J Nucl Med Mol Imaging. 2006;33:1273–9.
Fröberg AC, de Jong M, Nock BA, Breeman WAP, Erion JL, Maina T, et al. Comparison of three radiolabelled peptide analogues for CCK-2 receptor scintigraphy in medullary thyroid carcinoma. Eur J Nucl Med Mol Imaging. 2009;36:1265–72.
Kwekkeboom DJ, Bakker WH, Kooij PP, Erion J, Srinivasan A, De JM, et al. Cholecystokinin receptor imaging using an octapeptide DTPA-CCK analogue in patients with medullary thyroid carcinoma. Eur J Nucl Med. 2000;27:1312–7.
Behr TM, Béhé MP. Cholecystokinin-B/gastrin receptor-targeting peptides for staging and therapy of medullary thyroid cancer and other cholecystokinin-B receptor-expressing malignancies. Semin Nucl Med. 2002;32:97–109.
Breeman WAP, Fröberg AC, De Blois E, van Gameren A, Melis M, de Jong M, et al. Optimised labeling, preclinical and initial clinical. Aspects of CCK-2 receptor-targeting with 3 radiolabeled peptides. Nucl Med Biol. 2008;35:839–49.
Reubi JC, Waser B. Concomitant expression of several peptide receptors in neuroendocrine tumours: molecular basis for in vivo multireceptor tumour targeting. Eur J Nucl Med Mol Imaging. 2003;30:781–93.
Korner M, Stockli M, Waser B, Reubi JC. GLP-1 receptor expression in human tumors and human normal tissues: potential for in vivo targeting. J Nucl Med. 2007;48:736–43.
Gotthardt M, Fischer M, Naeher I, Holz JB, Jungclas H, Fritsch HW, et al. Use of the incretin hormone glucagon-like peptide-1 (GLP-1) for the detection of insulinomas: initial experimental results. Eur J Nucl Med Mol Imaging. 2002;29:597–606.
Wild D, Béhé M, Wicki A, Storch D, Waser B, Gotthardt M, et al. [Lys(40) (Ahx-DTPA-In-111)NH2]exendin-4, a very promising ligand for glucagon-like peptide-1 (GLP-1) receptor targeting. J Nucl Med. 2006;47:2025–33.
Brom M, Oyen WJG, Joosten L, Gotthardt M, Boerman OC. (68)Ga-labelled exendin-3, a new agent for the detection of insulinomas with PET. Eur J Nucl Med Mol Imaging. 2010;37:1345–55.
Wild D, Wicki A, Mansi R, Béhé M, Keil B, Bernhardt P, et al. Exendin-4-based radiopharmaceuticals for glucagonlike peptide-1 receptor PET/CT and SPECT/CT. J Nucl Med. 2010;51:1059–67.
Gao H, Niu G, Yang M, Quan Q, Ma Y, Murage EN, et al. PET of insulinoma using (18)F-FBEM-EM3106B, a new GLP-1 analogue. Mol Pharm. 2011;8:1775–82.
Wild D, Macke H, Christ E, Gloor B, Reubi JC. Glucagon-like peptide 1-receptor scans to localize occult insulinomas. N Engl J Med. 2008;359:766–8.
Christ E, Wild D, Forrer F, Brandle M, Sahli R, Clerici T, et al. Glucagon-like peptide-1 receptor imaging for localization of insulinomas. J Clin Endocrinol Metab. 2009;94:4398–405.
Wild D, Christ E, Caplin ME, Kurzawinski TR, Forrer F, Brandle M, et al. Glucagon-like peptide-1 versus somatostatin receptor targeting reveals 2 distinct forms of malignant insulinomas. J Nucl Med. 2011;52:1073–8.
Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–7.
Müller A, Homey B, Soto H, Ge NF, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6.
Hanaoka H, Mukai T, Tamamura H, Mori T, Ishino S, Ogawa K, et al. Development of a In-111-labeled peptide derivative targeting a chemokine receptor, CXCR4, for imaging tumors. Nucl Med Biol. 2006;33:489–94.
Jacobson O, Weiss ID, Kiesewetter DO, Farber JM, Chen X. PET of tumor CXCR4 expression with 4-18F-T140. J Nucl Med. 2010;51:1796–804.
Jacobson O, Weiss ID, Szajek L, Farber JM, Kiesewetter DO. 64Cu-AMD3100 – a novel imaging agent for targeting chemokine receptor CXCR4. Bioorg Med Chem. 2009;17:1486–93.
Nimmagadda S, Pullambhatla M, Stone K, Green G, Bhujwalla ZM, Pomper MG. Molecular imaging of CXCR4 receptor expression in human cancer xenografts with [64Cu]AMD3100 positron emission tomography. Cancer Res. 2010;70:3935–44.
Weiss ID, Jacobson O, Kiesewetter DO, Jacobus JP, Szajek LP, Chen X, et al. Positron emission tomography imaging of tumors expressing the human chemokine receptor CXCR4 in mice with the use of (64)Cu-AMD3100. Mol Imaging Biol. 2011. doi:10.1007/s11307-010-0466-y.
De Silva RA, Peyre K, Pullambhatla M, Fox JJ, Pomper MG, Nimmagadda S. Imaging CXCR4 expression in human cancer xenografts: evaluation of monocyclam 64Cu-AMD3465. J Nucl Med. 2011;52:986–93.
Reubi JC, Wenger S, Schmuckli-Maurer J, Schaer JC, Gugger M. Bombesin receptor subtypes in human cancers: detection with the universal radioligand (125)I-[D-TYR(6), beta-ALA(11), PHE(13), NLE(14)] bombesin(6-14). Clin Cancer Res. 2002;8:1139–46.
Reubi JC, Korner M, Waser B, Mazzucchelli L, Guillou L. High expression of peptide receptors as a novel target in gastrointestinal stromal tumours. Eur J Nucl Med Mol Imaging. 2004;31:803–10.
Reubi JC. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr Rev. 2003;24:389–427.
Baidoo KE, Lin KS, Zhan Y, Finley P, Scheffel U, Wagner Jr HN. Design, synthesis, and initial evaluation of high-affinity technetium bombesin analogues. Bioconjug Chem. 1998;9:218–25.
Lin KS, Luu A, Baidoo KE, Hashemzadeh-Gargari H, Chen MK, Brenneman K, et al. A new high affinity technetium-99m-bombesin analogue with low abdominal accumulation. Bioconjug Chem. 2005;16:43–50.
Lin KS, Luu A, Baidoo KE, Hashemzadeh-Gargari H, Chen MK, Pili R, et al. A new high affinity technetium analogue of bombesin containing DTPA as a pharmacokinetic modifier. Bioconjug Chem. 2004;15:1416–23.
Gali H, Hoffman TJ, Sieckman GL, Owen NK, Katti KV, Volkert WA. Synthesis, characterization, and labeling with 99mTc/188Re of peptide conjugates containing a dithia-bisphosphine chelating agent. Bioconjug Chem. 2001;12:354–63.
Karra SR, Schibli R, Gali H, Katti KV, Hoffman TJ, Higginbotham C, et al. 99mTc-labeling and in vivo studies of a bombesin analogue with a novel water-soluble dithiadiphosphine-based bifunctional chelating agent. Bioconjug Chem. 1999;10:254–60.
Smith CJ, Gali H, Sieckman GL, Higginbotham C, Volkert WA, Hoffman TJ. Radiochemical investigations of (99m)Tc-N(3)S-X-BBN[7-14]NH(2): an in vitro/in vivo structure-activity relationship study where X = 0-, 3-, 5-, 8-, and 11-carbon tethering moieties. Bioconjug Chem. 2003;14:93–102.
van de Wiele C, Dumont F, Vanden Broecke R, Oosterlinck W, Cocquyt V, Serreyn R, et al. Technetium-99m RP527, a GRP analogue for visualisation of GRP receptor-expressing malignancies: a feasibility study. Eur J Nucl Med. 2000;27:1694–9.
van de Wiele C, Dumont F, Dierckx RA, Peers SH, Thornback JR, Slegers G, et al. Biodistribution and dosimetry of (99m)Tc-RP527, a gastrin-releasing peptide (GRP) agonist for the visualization of GRP receptor-expressing malignancies. J Nucl Med. 2001;42:1722–7.
Wang LH, Coy DH, Taylor JE, Jiang NY, Moreau JP, Huang SC, et al. des-Met carboxyl-terminally modified analogues of bombesin function as potent bombesin receptor antagonists, partial agonists, or agonists. J Biol Chem. 1990;265:15695–703.
Nock B, Nikolopoulou A, Chiotellis E, Loudos G, Maintas D, Reubi JC, et al. [99mTc]Demobesin 1, a novel potent bombesin analogue for GRP receptor-targeted tumour imaging. Eur J Nucl Med Mol Imaging. 2003;30:247–58.
Nock BA, Nikolopoulou A, Galanis A, Cordopatis P, Waser B, Reubi JC, et al. Potent bombesin-like peptides for GRP-receptor targeting of tumors with 99mTc: a preclinical study. J Med Chem. 2005;48:100–10.
Ferro-Flores G, de Murphy CA, Rodriguez-Cortes J, Pedraza-Lopez M, Ramirez-Iglesias MT. Preparation and evaluation of 99mTc-EDDA/HYNIC-[Lys 3]-bombesin for imaging gastrin-releasing peptide receptor-positive tumours. Nucl Med Commun. 2006;27:371–6.
La Bella R, Garcia-Garayoa E, Langer M, Blauenstein P, Beck-Sickinger AG, Schubiger PA. In vitro and in vivo evaluation of a 99mTc(I)-labeled bombesin analogue for imaging of gastrin releasing peptide receptor-positive tumors. Nucl Med Biol. 2002;29:553–60.
Garcia Garayoa E, Ruegg D, Blauenstein P, Zwimpfer M, Khan IU, Maes V, et al. Chemical and biological characterization of new Re(CO)3/[99mTc](CO)3 bombesin analogues. Nucl Med Biol. 2007;34:17–28.
Garcia Garayoa E, Schweinsberg C, Maes V, Ruegg D, Blanc A, Blauenstein P, et al. New [99mTc]bombesin analogues with improved biodistribution for targeting gastrin releasing-peptide receptor-positive tumors. Q J Nucl Med Mol Imaging. 2007;51:42–50.
Brans L, Maes V, Garcia-Garayoa E, Schweinsberg C, Daepp S, Blauenstein P, et al. Glycation methods for bombesin analogs containing the (NalphaHis)Ac chelator for 99mTc(CO)3 radiolabeling. Chem Biol Drug Des. 2008;72:496–506.
Garcia Garayoa E, Schweinsberg C, Maes V, Brans L, Blauenstein P, Tourwe DA, et al. Influence of the molecular charge on the biodistribution of bombesin analogues labeled with the [99mTc(CO)3]-core. Bioconjug Chem. 2008;19:2409–16.
Schweinsberg C, Maes V, Brans L, Blauenstein P, Tourwe DA, Schubiger PA, et al. Novel glycated [99mTc(CO)3]-labeled bombesin analogues for improved targeting of gastrin-releasing peptide receptor-positive tumors. Bioconjug Chem. 2008;19:2432–9.
Smith CJ, Sieckman GL, Owen NK, Hayes DL, Mazuru DG, Kannan R, et al. Radiochemical investigations of gastrin-releasing peptide receptor-specific [(99m)Tc(X)(CO)3-Dpr-Ser-Ser-Ser-Gln-Trp-Ala-Val-Gly-His-Leu-Met-(NH2)] in PC-3, tumor-bearing, rodent models: syntheses, radiolabeling, and in vitro/in vivo studies where Dpr = 2,3-diaminopropionic acid and X = H2O or P(CH2OH)3. Cancer Res. 2003;63:4082–8.
Retzloff LB, Heinzke L, Figureoa SD, Sublett SV, Ma L, Sieckman GL, et al. Evaluation of [99mTc-(CO)3-X-Y-bombesin(7-14)NH2] conjugates for targeting gastrin-releasing peptide receptors overexpressed on breast carcinoma. Anticancer Res. 2010;30:19–30.
Dapp S, Garayoa EG, Maes V, Brans L, Tourwe DA, Müller C, et al. PEGylation of (99m)Tc-labeled bombesin analogues improves their pharmacokinetic properties. Nucl Med Biol. 2011;38:997–1009.
Breeman WA, Hofland LJ, de Jong M, Bernard BF, Srinivasan A, Kwekkeboom DJ, et al. Evaluation of radiolabelled bombesin analogues for receptor-targeted scintigraphy and radiotherapy. Int J Cancer. 1999;81:658–65.
Breeman WA, de Jong M, Bernard BF, Kwekkeboom DJ, Srinivasan A, van der Pluijm ME, et al. Pre-clinical evaluation of [(111)In-DTPA-Pro(1), Tyr(4)]bombesin, a new radioligand for bombesin-receptor scintigraphy. Int J Cancer. 1999;83:657–63.
Breeman WA, de Jong M, Erion JL, Bugaj JE, Srinivasan A, Bernard BF, et al. Preclinical comparison of (111)In-labeled DTPA- or DOTA-bombesin analogs for receptor-targeted scintigraphy and radionuclide therapy. J Nucl Med. 2002;43:1650–6.
de Visser M, Bernard HF, Erion JL, Schmidt MA, Srinivasan A, Waser B, et al. Novel 111In-labelled bombesin analogues for molecular imaging of prostate tumours. Eur J Nucl Med Mol Imaging. 2007;34:1228–38.
Hoffman TJ, Gali H, Smith CJ, Sieckman GL, Hayes DL, Owen NK, et al. Novel series of 111In-labeled bombesin analogs as potential radiopharmaceuticals for specific targeting of gastrin-releasing peptide receptors expressed on human prostate cancer cells. J Nucl Med. 2003;44:823–31.
Zhang H, Chen J, Waldherr C, Hinni K, Waser B, Reubi JC, et al. Synthesis and evaluation of bombesin derivatives on the basis of pan-bombesin peptides labeled with indium-111, lutetium-177, and yttrium-90 for targeting bombesin receptor-expressing tumors. Cancer Res. 2004;64:6707–15.
Zhang H, Schuhmacher J, Waser B, Wild D, Eisenhut M, Reubi JC, et al. DOTA-PESIN, a DOTA-conjugated bombesin derivative designed for the imaging and targeted radionuclide treatment of bombesin receptor-positive tumours. Eur J Nucl Med Mol Imaging. 2007;34:1198–208.
Schuhmacher J, Zhang H, Doll J, Macke HR, Matys R, Hauser H, et al. GRP receptor-targeted PET of a rat pancreas carcinoma xenograft in nude mice with a 68Ga-labeled bombesin(6-14) analog. J Nucl Med. 2005;46:691–9.
Lantry LE, Cappelletti E, Maddalena ME, Fox JS, Feng W, Chen J, et al. 177Lu-AMBA: synthesis and characterization of a selective 177Lu-labeled GRP-R agonist for systemic radiotherapy of prostate cancer. J Nucl Med. 2006;47:1144–52.
Schroeder RP, Müller C, Reneman S, Melis ML, Breeman WA, De Blois E, et al. A standardised study to compare prostate cancer targeting efficacy of five radiolabelled bombesin analogues. Eur J Nucl Med Mol Imaging. 2010;37:1386–96.
Abd-Elgaliel WR, Gallazzi F, Garrison JC, Rold TL, Sieckman GL, Figueroa SD, et al. Design, synthesis, and biological evaluation of an antagonist-bombesin analogue as targeting vector. Bioconjug Chem. 2008;19:2040–8.
Mansi R, Wang X, Forrer F, Kneifel S, Tamma ML, Waser B, et al. Evaluation of a 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid-conjugated bombesin-based radioantagonist for the labeling with single-photon emission computed tomography, positron emission tomography, and therapeutic radionuclides. Clin Cancer Res. 2009;15:5240–9.
Mansi R, Wang X, Forrer F, Waser B, Cescato R, Graham K, et al. Development of a potent DOTA-conjugated bombesin antagonist for targeting GRPr-positive tumours. Eur J Nucl Med Mol Imaging. 2011;38:97–107.
Schroeder RP, van Weerden WM, Krenning EP, Bangma CH, Berndsen S, Grievink-de Ligt CH, et al. Gastrin-releasing peptide receptor-based targeting using bombesin analogues is superior to metabolism-based targeting using choline for in vivo imaging of human prostate cancer xenografts. Eur J Nucl Med Mol Imaging. 2011;38:1257–66.
Liu Z, Niu G, Wang F, Chen X. (68)Ga-labeled NOTA-RGD-BBN peptide for dual integrin and GRPR-targeted tumor imaging. Eur J Nucl Med Mol Imaging. 2009;36:1483–94.
Hoffman TJ, Smith CJ. True radiotracers: Cu-64 targeting vectors based upon bombesin peptide. Nucl Med Biol. 2009;36:579–85.
Rogers BE, Bigott HM, McCarthy DW, la Manna D, Kim J, Sharp TL, et al. MicroPET imaging of a gastrin-releasing peptide receptor-positive tumor in a mouse model of human prostate cancer using a 64Cu-labeled bombesin analogue. Bioconjug Chem. 2003;14:756–63.
Chen X, Park R, Hou Y, Tohme M, Shahinian AH, Bading JR, et al. MicroPET and autoradiographic imaging of GRP receptor expression with 64Cu-DOTA-[Lys3]bombesin in human prostate adenocarcinoma xenografts. J Nucl Med. 2004;45:1390–7.
Rogers BE, Manna DD, Safavy A. In vitro and in vivo evaluation of a 64Cu-labeled polyethylene glycol-bombesin conjugate. Cancer Biother Radiopharm. 2004;19:25–34.
Yan Y, Chen K, Yang M, Sun X, Liu S, Chen X. A new 18F-labeled BBN-RGD peptide heterodimer with a symmetric linker for prostate cancer imaging. Amino Acids. 2010;41:439–47.
Prasanphanich AF, Nanda PK, Rold TL, Ma L, Lewis MR, Garrison JC, et al. [64Cu-NOTA-8-Aoc-BBN(7-14)NH2] targeting vector for positron-emission tomography imaging of gastrin-releasing peptide receptor-expressing tissues. Proc Natl Acad Sci USA. 2007;104:12462–7.
Gasser G, Tjioe L, Graham B, Belousoff MJ, Juran S, Walther M, et al. Synthesis, copper(II) complexation, (64)Cu-labeling, and bioconjugation of a new bis(2-pyridylmethyl) derivative of 1,4,7-triazacyclononane. Bioconjug Chem. 2008;19:719–30.
Prasanphanich AF, Retzloff L, Lane SR, Nanda PK, Sieckman GL, Rold TL, et al. In vitro and in vivo analysis of [(64)Cu-NO2A-8-Aoc-BBN(7-14)NH(2)]: a site-directed radiopharmaceutical for positron-emission tomography imaging of T-47D human breast cancer tumors. Nucl Med Biol. 2009;36:171–81.
Garrison JC, Rold TL, Sieckman GL, Figueroa SD, Volkert WA, Jurisson SS, et al. In vivo evaluation and small-animal PET/CT of a prostate cancer mouse model using 64Cu bombesin analogs: side-by-side comparison of the CB-TE2A and DOTA chelation systems. J Nucl Med. 2007;48:1327–37.
Lane SR, Nanda P, Rold TL, Sieckman GL, Figueroa SD, Hoffman TJ, et al. Optimization, biological evaluation and microPET imaging of copper-64-labeled bombesin agonists, [64Cu-NO2A-(X)-BBN(7-14)NH2], in a prostate tumor xenografted mouse model. Nucl Med Biol. 2010;37:751–61.
Parry JJ, Kelly TS, Andrews R, Rogers BE. In vitro and in vivo evaluation of 64Cu-labeled DOTA-linker-bombesin(7-14) analogues containing different amino acid linker moieties. Bioconjug Chem. 2007;18:1110–7.
Di Bartolo NM, Sargeson AM, Donlevy TM, Smith SV. Synthesis of a new cage ligand, SarAr, and its complexation with selected transition metal ions for potential use in radioimaging. J Chem Soc Dalton Trans. 2001;15:2303–9.
Lears KA, Ferdani R, Liang K, Zheleznyak A, Andrews R, Sherman CD, et al. In vitro and in vivo evaluation of 64Cu-labeled SarAr-bombesin analogs in gastrin-releasing peptide receptor-expressing prostate cancer. J Nucl Med. 2011;52:470–7.
Zhang X, Cai W, Cao F, Schreibmann E, Wu Y, Wu JC, et al. 18F-labeled bombesin analogs for targeting GRP receptor-expressing prostate cancer. J Nucl Med. 2006;47:492–501.
Hohne A, Mu L, Honer M, Schubiger PA, Ametamey SM, Graham K, et al. Synthesis, 18F-labeling, and in vitro and in vivo studies of bombesin peptides modified with silicon-based building blocks. Bioconjug Chem. 2008;19:1871–9.
Li ZB, Wu Z, Chen K, Ryu EK, Chen X. 18F-labeled BBN-RGD heterodimer for prostate cancer imaging. J Nucl Med. 2008;49:453–61.
Liu Z, Yan Y, Chin FT, Wang F, Chen X. Dual integrin and gastrin-releasing peptide receptor targeted tumor imaging using 18F-labeled PEGylated RGD-bombesin heterodimer 18F-FB-PEG3-Glu-RGD-BBN. J Med Chem. 2009;52:425–32.
Mu L, Honer M, Becaud J, Martic M, Schubiger PA, Ametamey SM, et al. In vitro and in vivo characterization of novel 18F-labeled bombesin analogues for targeting GRPR-positive tumors. Bioconjug Chem. 2010;21:1864–71.
Honer M, Mu L, Stellfeld T, Graham K, Martic M, Fischer CR, et al. 18F-labeled bombesin analog for specific and effective targeting of prostate tumors expressing gastrin-releasing peptide receptors. J Nucl Med. 2011;52:270–8.
Yang M, Gao H, Zhou Y, Ma Y, Quan Q, Lang L, et al. F-18-labeled GRPR agonists and antagonists: a comparative study in prostate cancer imaging. Theranostics. 2011;1:220–9.
Liu Z, Yan Y, Liu S, Wang F, Chen X. (18)F, (64)Cu, and (68)Ga labeled RGD-bombesin heterodimeric peptides for PET imaging of breast cancer. Bioconjug Chem. 2009;20:1016–25.
Bodei L, Ferrari M, Nunn A, Llull J, Cremonesi M, Martano L, et al. 177Lu-AMBA bombesin analogue in hormone refractory prostate cancer patients: a phase I escalation study with single-cycle administrations. Eur J Nucl Med Mol Imaging. 2007;34:S221.
Scopinaro F, Varvarigou AD, Ussof W, De Vincentis G, Sourlingas TG, Evangelatos GP, et al. Technetium labeled bombesin-like peptide: preliminary report on breast cancer uptake in patients. Cancer Biother Radiopharm. 2002;17:327–35.
Varvarigou AD, Scopinaro F, Leondiadis L, Corleto V, Schillaci O, De Vincentis G, et al. Synthesis, chemical, radiochemical and radiobiological evaluation of a new 99mTc-labelled bombesin-like peptide. Cancer Biother Radiopharm. 2002;17:317–26.
Scopinaro F, De Vincentis G, Varvarigou AD, Laurenti C, Iori F, Remediani S, et al. 99mTc-bombesin detects prostate cancer and invasion of pelvic lymph nodes. Eur J Nucl Med Mol Imaging. 2003;30:1378–82.
Scopinaro F, De Vincentis G, Corazziari E, Massa R, Osti M, Pallotta N, et al. Detection of colon cancer with 99mTc-labeled bombesin derivative (99mTc-leu13-BN1). Cancer Biother Radiopharm. 2004;19:245–52.
Fröberg A, Visser M, Maina T, Erion J, de Swart J, de Jong M, et al. Are GRP-receptors present in the human pancreas? J Nucl Med. 2006;47 Suppl 1:429P.
Meyer GJ, Macke H, Schuhmacher J, Knapp WH, Hofmann M. 68Ga-labelled DOTA-derivatised peptide ligands. Eur J Nucl Med Mol Imaging. 2004;31:1097–104.
Hofmann M, Machtens S, Stief C, Maecke H, Boerner AR, Knapp WH. Feasibility of Ga-68-DOTABOM PET in prostate carcinoma patients. Eur J Nucl Med Mol Imaging. 2004;31:S253.
Maecke HR, Hofmann M, Haberkorn U. (68)Ga-labeled peptides in tumor imaging. J Nucl Med. 2005;46 Suppl 1:172S–8S.
Dimitrakopoulou-Strauss A, Hohenberger P, Haberkorn U, Macke HR, Eisenhut M, Strauss LG. 68Ga-labeled bombesin studies in patients with gastrointestinal stromal tumors: comparison with 18F-FDG. J Nucl Med. 2007;48:1245–50.
Schaefer N, Valencia R, Borkowski S, Geissler E, Kristiansen G, Rohde B, et al. Comparison of BAY 86-4367, a new F-18 labeled bombesin analog, with F-18-ethyl-choline in recurrent and primary prostate cancer patients. J Nucl Med. 2011;52 Suppl 1:40.
Beer M, Montani M, Gerhardt J, Wild PJ, Hany TF, Hermanns T, et al. Profiling gastrin-releasing peptide receptor in prostate tissues: clinical implications and molecular correlates. Prostate. 2011. doi:10.1002/pros.21434
Schroeder RP, van Weerden WM, Bangma C, Krenning EP, de Jong M. Peptide receptor imaging of prostate cancer with radiolabelled bombesin analogues. Methods. 2009;48:200–4.
Conflicts of interest
None.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Laverman, P., Sosabowski, J.K., Boerman, O.C. et al. Radiolabelled peptides for oncological diagnosis. Eur J Nucl Med Mol Imaging 39 (Suppl 1), 78–92 (2012). https://doi.org/10.1007/s00259-011-2014-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-011-2014-7