
Abstract
Purpose
The expression of human epidermal growth factor receptor-2 (HER2) receptors in cancers is correlated with a poor prognosis. If assessed in vivo, it could be used for selection of appropriate therapy for individual patients and for monitoring of the tumor response to targeted therapies. We have radiolabeled a HER2-binding Affibody molecule with fluorine-18 for in vivo monitoring of the HER2 expression by positron emission tomography (PET).
Materials and methods
The HER2-binding ZHER2:342-Cys Affibody molecule was conjugated with N-2-(4-[18F]fluorobenzamido)ethyl]maleimide ([18F]FBEM). The in vitro binding of the resulting radioconjugate was characterized by receptor saturation and competition assays. For in vivo studies, the radioconjugate was injected into the tail vein of mice bearing subcutaneous HER2-positive or HER2-negative tumors. Some of the mice were pre-treated with non-labeled ZHER2:342−Cys. The animals were sacrificed at different times post-injection, and the radioactivity in selected tissues was measured. PET images were obtained using an animal PET scanner.
Results
In vitro experiments indicated specific, high-affinity binding to HER2. PET imaging revealed a high accumulation of the radioactivity in the tumor as early as 20 min after injection, with a plateau being reached after 60 min. These results were confirmed by biodistribution studies demonstrating that, as early as 1 h post-injection, the tumor to blood concentration ratio was 7.5 and increased to 27 at 4 h. Pre-saturation of the receptors with unlabeled ZHER2:342-Cys lowered the accumulation of radioactivity in HER2-positive tumors to the levels observed in HER2-negative ones.
Conclusion
Our results suggest that the [18F]FBEM-ZHER2:342 radioconjugate can be used to assess HER2 expression in vivo.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Progress in tumor biology has resulted in characterization of specific cancer markers possessing prognostic and predictive value. Among the most studied markers is the human epidermal growth factor receptor-2 (HER2/neu/c-erbB-2), a member of the epidermal growth factor receptor family of tyrosine kinases (EGFR) [1, 2]. This 185-kDa transmembrane phosphoglycoprotein plays a key role in many cellular processes, including cell growth, differentiation, cell survival, and cell adhesion and migration. Whereas normal epithelial cells do not express HER2 protein (or express a very low level) on the cell surface, amplification of the HER2/neu gene and/or overexpression of the protein have been identified in 20-30% of invasive breast and lung cancers, as well as in ovarian carcinomas and B-cell acute lymphoblastic leukemia [3, 4]. The majority of the reports have linked the overexpression with shorter time to disease progression and decreased overall survival. The HER2-positive status is associated with resistance of cancers to hormone therapy and certain types of chemotherapy [5]. Therefore, the expression of HER2 receptors in cancers is correlated with a poor prognosis [6].
Because the HER2 expression levels in distant metastases may be quite distinct from levels in the primary tumor [7, 8], an assessment of the global expression of these receptors on cancer cells in vivo is essential for adequate diagnosis and selection of appropriate therapy.
Recently, significant advances have been made to develop probes for targeting antigens commonly overexpressed in human cancer cells. Several monoclonal antibodies and antibody fragments have already been approved for clinical use [9], including a recombinant humanized version of anti-HER2 monoclonal antibody trastuzumab (Herceptin), which has shown significant clinical efficacy in trials with HER2-overexpressing breast cancers [10]. The success of trastuzumab was followed by the introduction of other therapeutic antibodies targeting HER2 receptors, e.g., pertuzumab [11]. It has been reported that the outcome of these therapies in individual patients depends on the HER2 status of the treated tumors [12].
Radiolabeled trastuzumab [13-16] or F(ab′) fragments of trastuzumab labeled with In-111 and Ga-68 have been used for single-photon emission computed tomography (SPECT) and positron emission tomography (PET) imaging, respectively. However, antibodies are rather bulky proteins as compared with the molecular probes typically used for PET imaging [17, 18], which make the tumor penetration and clearance from the circulation rather slow, and this, in turn, considerably affects imaging studies. Furthermore, trastuzumab-based tracers cannot be used to monitor possible changes in HER2 expression caused by treatment with this antibody because of the competition of the imaging and therapeutic molecules for binding to these receptors.
A promising alternative to antibodies as targeting agents are Affibody molecules—highly stable proteins that are four times smaller than single-chain antibody fragments (scFv) and 20 times smaller than monoclonal antibodies [19]. The cysteine-free Affibody molecules provide a strong framework independent of disulfide bonds for its folding, and their small size enables penetration into solid tumors and rapid clearance from the bloodstream. Although the existing library of Affibody molecules can be used to select binders to different structures, in this work, we use the term in reference to the HER2-binding Affibody molecules. They constitute a highly suitable carrier for directing radioisotopes or toxins to HER2-positive tumor cells because of specific target binding and lack of specific non-receptor interactions, such as the Fc receptor binding displayed by some antibodies.
It has been shown that radiolabeled Affibody molecules display specific binding to HER2 receptors and rapid uptake in tumor xenografts [20, 21]. Furthermore, the binding is unaffected by pretreatment of the target tumor cells with trastuzumab [22], indicating that radiolabeled Affibody molecules and trastuzumab bind to distinct regions of the extracellular domain of HER2. This observation suggests that radiolabeled Affibody molecules can be used effectively for monitoring of receptor expression in patients undergoing treatment with trastuzumab.
PET imaging has been routinely used in the neurosciences for in situ quantification of receptor expression [23, 24]. Delforge et al. [25] used PET for in vivo quantification of cardiac β-adrenergic receptors and was able to obtain their density maps. Recent studies showed that PET might also serve as a useful imaging modality to measure growth factor receptor expression on tumor cells [26]. The global assessment of receptor expression by PET will help prevent false-negative results because of the heterogeneity of tumors that cannot be fully represented by biopsy probes. This approach will permit the non-invasive identification of patients that are suitable for targeted therapy designed against HER2-positive tumors and will provide information about the immediate response to therapeutic interventions, which can then be adjusted for individual patients based on the actual status of those receptors. 18F is routinely applied in clinical oncology in the form of fluorodeoxyglucose (FDG), a Food and Drug Administration-approved glucose analog. Therefore, it is widely used and readily available.
This study is the first to describe the synthesis as well as in vitro and in vivo characterization of a novel HER2-specific [18F]FBEM-ZHER2:342-Affibody molecule radioconjugate that may allow the application of PET imaging to assess in vivo HER2 expression.
Materials and methods
General
Unless otherwise specified, all reagents were analytical grade and were obtained from commercial sources. The ZHER2:342-Cys Affibody molecules were kindly provided by our Cooperative Research and Development Agreement (CRADA) partner in Sweden (Affibody AB; http://www.affibody.com). 18F radionuclide was obtained from the NIH/CC cyclotron facility from proton irradiation of O-18-enriched water. Trastuzumab (Herceptin) was purchased from Genentech, San Francisco, CA, USA, as a lyophilized white powder and reconstituted with the supplied solvent. Pentamethylbenzyl (4-trimethylammonium) benzoate trifluoromethanesulfonate was prepared as described previously [27].
Synthesis of N-(2-(4-[18F]fluorobenzamido)ethyl)maleimide ([18F]FBEM)
[18F]Fluorobenzoic acid was prepared as previously described using a manual synthesis method [28]. Briefly, pentamethylbenzyl (4-trimethylammonium) benzoate trifluoromethanesulfonate (3 mg, 6 µmol) was heated for 10 min in a sealed V-vial with [18F]fluoride (550 to 2,220 MBq) in the presence of Kryptofix (2.2.2; 6 µmol) and K2CO3 (3 µmol) in CH3CN (0.1 ml). The intermediate was treated with trifluoroacetic acid (TFA) to provide 4-[18F]fluorobenzoic acid. The 4-[18F]fluorobenzoic acid was treated with N-(2-aminoethyl)maleimide (1.52 mg, 6 µmol), diethyl cyanophosphonate (1.3 mg, 8 µmol), and N,N-diisopropylethyl amine (10 ml). The resulting solution was heated at 70°C for 5 min. This solution was diluted with 150 µl water and injected onto an high-performance liquid chromatography (HPLC) column [Luna C-18 (Phenomenex), 10 × 250 mm, 20% CH3CN, 80% water, 5 ml/min]. The product eluting at about 17 min was collected, diluted to about 20 ml with water and trapped on a BondElut C-18 (500 mg). The product was eluted from the column with CH2Cl2; the organic layer was carefully separated from the small amount of water and carefully evaporated into the bottom of a 1.5-ml Eppendorf tube.
Conjugation of N-2-(4-[18F]fluorobenzamido)ethyl]maleimide with ZHER2:342−Cys ([18F]FBEM-ZHER2:342-Affibody)
The isolated [18F]FBEM (111-370 MBq) was dissolved in 10 µl of CH3CN. In the meantime, stock Affibody molecule ZHER2:342-Cys (100 µg in 100 µl phosphate-buffered saline, PBS; 12 nmol) was treated with 10 µl of 1 M dithiothreitol (DTT) for 40 min at 37°C. The solution was then eluted in 0.25 ml fractions of 0.1 M NaOAc, 0.1% ascorbic acid, pH 7, from a NAP-5 column (GE Healthcare Bio-Science AB, Uppsala, Sweden; cut-off, 5 kDa). Fraction 4 (0.25 ml) from this column was added to the solution of [18F]FBEM. This solution was incubated for 15-30 min at room temperature and loaded onto a NAP-5 column. The NAP-5 column was eluted with 250 µl portions of PBS. The most concentrated fraction containing the radiolabeled protein (fraction 4) was collected and used for the biological experiments. The radiochemical yield of 18F-labeled Affibody conjugate using this un-optimized procedure was approximately 5% based on starting 18F-fluoride and uncorrected for decay. The total procedure requires about 2 h.
Protein concentration of the labeled preparation was determined by measuring UV absorbance at 280 nm (NanoDrop Technologies, Wilmington, DE, USA) using the conversion 1 AU = 0.99 mg/ml.
HPLC analysis of [18F]FBEM-ZHER2:342-Affibody
Analysis of the protein product was conducted using a Vydac protein C-4 column (4.5 × 150 mm). Gradient elution was employed (5-75% of 0.1% TFA in acetonitrile versus 0.1% TFA in water over 15 min). [18F]FBEM-ZHER2:342-Affibody molecules were eluted at about 8 min.
Cell lines
Human parental breast (SKBR-3, MCF7) and ovarian (SKOV-3) cancer cell lines that express different levels of HER2, as well as a HER2-negative human glioblastoma cell line (U251), were purchased from the American Type Culture Collection (Rockville, MD, USA). The breast cancer cell line, stably transfected with HER2 (MCF7/HER2-18), was kindly provided by Dr. John W. Park, University of California, San Francisco, CA, USA. The cells were cultured in DMEM/F12 medium (SKBR-3, SKOV-3, U251) or in DMEM (MCF7, MCF7/HER2-18) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (GIBCO, Grand Island, NY, USA) and penicillin/streptomycin (100 U/ml of each). Cells were grown as a monolayer at 37°C in a humidified atmosphere containing 5% CO2. In the case of MCF7/HER2 transfectants, the culture medium also contained 400 µg/ml Geneticin, G418 (GIBCO, Grand Island, NY, USA).
In vitro cell-binding assays
The in vitro binding characteristics of the [18F]FBEM-ZHER2:342-Affibody molecules were assessed using saturation and displacement cell-binding assays. The day before the experiments, cells were seeded in six-well plates at concentration of 2.5 × 105 cells per ml. For receptor saturation analysis, the cells were incubated (4°C, 2 h) with increasing concentrations (3.3-420 nM) of [18F]FBEM-ZHER2:342-Affibody alone or with additional 100-fold excess of non-labeled Affibody molecules. Then, the incubation medium was collected, and the cells were washed with cold PBS and trypsinized, and the cell-associated radioactivity was measured using a γ-counter (1480 Wizard 3, Automatic Gamma Counter, Perkin-Elmer, Waltham, MA, USA). The total non-specific and specific binding were plotted against the total molar concentration of added [18F]FBEM-ZHER2:342-Affibody, and the data were analyzed by non-linear regression using GraphPad Prism (GraphPad Software, San Diego, CA, USA)
For the displacement assay, the cells were incubated with 216 nM of [18F]FBEM-ZHER2:342-Affibody and increasing concentrations (1.2 nM-120 µM) of non-labeled Affibody molecules. The rest of the experimental procedures were the same as those used for the saturation studies.
The specificity of binding was also checked for cell lines that express different numbers of HER2 receptors. The experiments were carried out at the same way as for saturation analysis; however, in this case, only one concentration of [18F]FBEM-ZHER2-Affibody was chosen. In addition, some wells were preincubated (30 min) with 100-fold excess amount of unlabeled Affibody or trastuzumab (1 µg/ml).
Tumor model
Five- to 7-week-old female athymic nude mice were injected subcutaneously with 1 × 107 of SKOV-3 or with 106 of U251 cells suspended in Matrigel (BD Bioscience, San Jose, CA, USA). Tumors were placed on the hind leg or on the back. These locations were chosen to optimize the PET images by placing the tumor well away from the radioactivity in major body structures and reduce background radioactivity. U251 and SKOV-3 tumors (50-250 mg) developed after 2 and 4-5 weeks, respectively. All animal studies were conducted in accordance with the principles and procedures outlined in the National Institute of Health Guide for the Care and Use of Animals on approved studies from the National Institutes of Health Institutional Animal Care and Use Committee.
Biodistribution studies
Athymic nude mice bearing SKOV-3 tumors were injected with 1.8-2.2 MBq (1.6-2 µg, 100 µl) of [18F]FBEM-ZHER2-Affibody into the tail vein. Groups of three to six mice were sacrificed, and their organs dissected 1, 2, 3, 4, and 6 h post-injection. Then, blood, tumor, and major organs were collected and weighed. The radioactivity in the tissues was measured along with a standard of the injected dose using γ-counter. The results were calculated as percentage injected dose per gram of tissue (% ID/g).
To study the blood kinetics and the stability of [18F]FBEM-ZHER2:342-Affibody in vivo, the blood was collected at 1-6 h post-injection in vials containing 30 µl heparin. The samples were weighed, and the radioactivity was measured by γ-counter. Then, the blood was centrifuged (10 min at 10,000×g) and fractions of plasma (200-300 µl) were separated on NAP-5 columns. The mean concentrations of radioactivity in the blood (% ID/g) were plotted against time after injection, and the curve was fitted by single exponential decay. Similar procedure was used to analyze high molecular weight (HMW)/low molecular weight (LMW) ratio of radioactivity in the urine collected 60 min post-injection.
To confirm the specificity of binding in vivo, one group of animals bearing HER2-negative U251 tumors was injected with the [18F]FBEM-ZHER2:342-Affibody and another group bearing HER2-positive SKOV-3 tumors was pre-injected with 400 µg of non-labeled Affibody 45 min before the [18F]FBEM-ZHER2:342-Affibody was injected. Two hours later, all animals were sacrificed, and their organs analyzed as described above.
PET studies
Mice were anesthetized using isoflurane/O2 (1.5-5% v/v) and injected with approximately 3.7-4.4 MBq (3.4-4 µg, 100 µl) of the [18F]FBEM-ZHER2:342-Affibody via the tail vein. PET scans were performed using the Advanced Technology Laboratory Animal Scanner (ATLAS) PET scanner [29]. Whole-body (1, 2, 3, 4, and 6 h; four bed positions, each 15 min) and dynamic data acquisition (1 frame, 10 min, up to 12 frames) were started about 2 min after radiotracer injection and recorded with a 100-700 keV energy window. The images were reconstructed by a two-dimensional ordered-subsets expectation maximum (2D-OSEM) algorithm, and no correction was applied for attenuation or scatter. For each scan, regions of interest were drawn over the tumor, normal tissue, and major organs. The maximum radioactivity accumulation within the tumor or organs was obtained from mean pixel values within the multiple region of interest (ROI). The results were calculated as a percentage injected dose per gram (% ID/g) and standard uptake value (SUV). At the end of the study, an 18F source of known activity was imaged to obtain MBq of 18F per counts per second for the imaging system (calibration factor). Then, every ROI (counts per second per cubic centimeter) was multiplied by this factor and divided by injected activity.
Results
Conjugation and labeling
Affibody molecule with a C-terminal cysteine provided functionality for site-specific labeling using maleimide chemistry. We prepared N-(2-(4-[18F]fluorobenzamido)ethyl)maleimide [18F]FBEM using a similar method (Fig. 1) to that utilized to prepare [18F]fluoropaclitaxel [28]. An alternative synthesis of [18F]FBEM has been previously reported [26]. 4-[18F]Fluorobenzoic acid was obtained by reaction of pentamethylbenzyl 4-trimethylaniliniumbenzoate trifluoromethanesulfonate with [18F]fluoride followed by cleavage of the ester function with TFA. 2-Aminoethylmaleimide was coupled to [18F]fluorobenzoic acid using diethyl cyanophosphonate as the coupling reagent. [18F]FBEM was obtained in 22.0 ± 4.7% radiochemical yield (n = 44, uncorrected). The [18F]FBEM was purified by HPLC and, because of the complexity of the overall synthesis, was not commonly evaluated for purity. However, in the limited number of radiochemical purity assays of the isolated [18F]FBEM, we consistently observed values greater than 95%.

Synthetic route used for labeling of the ZHER2:342-Cys Affibody molecule with 18F. DECP Diethyl cyanophosphonate, DIPEA N,N-diisoproplyethylamine
Because ZHER2:342-cys Affibody molecules form disulfide on storage, treatment with DTT and removal of excess DTT using a NAP-5 column was utilized. The reduced protein was incubated with [18F]FBEM for 15-30 min. The labeled protein was isolated by elution through a NAP-5 column with PBS and obtained in 6.5 ± 2.2% radiochemical yield (based on starting [18F]fluoride, n = 26, uncorrected). HPLC demonstrates the presence of a single radiolabeled species. Protein staining of the polyacrylamide gel electrophoresis showed a mixture of monomeric and dimeric proteins; however, greater than 90% of the radioactivity migrated with the 8-kDa protein band. The specific radioactivity of the resulting radioconjugate was in the range 1-2.3 MBq/μg at the end of radiochemical synthesis.
Binding specificity in vitro
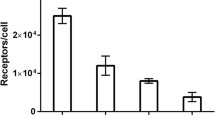
Competition for binding between [18F]FBEM-ZHER2:342-Affibody and non-radioactive Affibody molecules demonstrate that [18F]FBEM-ZHER2:342-Affibody can be displaced by increasing amounts of unlabeled molecules (Fig. 2a). This provides evidence for receptor-mediated binding to HER2-expressing cells. Saturation analysis shows a single class of high-affinity binding sites that had a mean equilibrium dissociation constant (K D) of 15 nM for the SKOV-3 cell line. Figure 2b shows the total non-specific and specific-binding plotted against the total molar concentration of added [18F]FBEM-ZHER2:342-Affibody. Similar studies were performed with the SKBR-3 cell line, which demonstrated that the [18F]FBEM-ZHER2:342-Affibody conjugate bound with similar affinity (data not shown). Specificity of binding was also tested using cell lines expressing different numbers of HER2 receptors. As shown in Fig. 3, binding to HER2-negative cells was comparable to that observed in experiments in which addition of 100-fold excess of unlabeled Affibody molecules almost completely blocked the HER2 receptors for [18F]FBEM-ZHER2:342-Affibody binding. We also tested whether pre-incubation with trastuzumab interferes with binding of [18F]FBEM-ZHER2:342-Affibody. Radiolabeled Affibody molecules and trastuzumab bound non-competitively, which confirms that [18F]FBEM-ZHER2:342-Affibody and trastuzumab bind to different epitopes on the extracellular domain of HER2.

Characterization of HER2-specific binding. a Representative results of competition-binding assay using SKOV-3 cells (log of concentration of competitor compound versus percent of the maximum specific binding of radiolabeled Affibody molecule). b Representative results of saturation assay using SKOV-3 cells (concentration of radiolabeled Affibody molecule versus concentration bound: NSB non-specific binding obtained by saturation of the receptors with 100-fold excess of non-labeled Affibody, TB total binding, and SB specific binding)

The binding of [18F]FBEM-ZHER2:342-Affibody molecules to cells with different levels of HER2 expression [cell line versus normalized cell-associated radioactivity (%) and the effect of pre-incubation with either Affibody molecules or trastuzumab on the binding, n = 3]. The standard errors were in the range 0.1 to 3.1%
Biodistribution studies
The results of the biodistribution studies are summarized in Tables 1 and 2. Among the organs evaluated, the most prominent [18F]FBEM-ZHER2:342-Affibody uptake was found in the kidneys, bone, and tumor. However, the radioactivity in the kidneys decreased from 14% ID/g to 1.5% ID/g at, respectively, 1 and 4 h post-injection because of excretion of the tracer to the urinary bladder. All other tested organs exhibited very low levels of tracer uptake over the entire time course, which resulted in a significantly high tumor-to-background tissue radioactivity accumulation ratio (Table 2). It is noteworthy that the uptake of radioactivity 2 h post-injection was higher in the tumor than in any other organ. This level remained steady until the 6-h time point.
The specificity of binding in vivo was evaluated in two independent experiments. In each case, mice were sacrificed 2 h post-injection, and the radioactivity in the blood and major organs was measured. As expected, blocking the HER2 receptors in mice bearing HER2-positive SKOV-3 tumors with an excess of unlabeled Affibody resulted in a significant decrease of radioactivity in the tumor, as only tumors expressed high numbers of HER2 receptors. In the blood and the rest of organs examined, there was no significant change because of pre-treatment with non-labeled molecules (Fig. 4a). Receptor-mediated binding of radiotracer was confirmed by successful blocking with non-labeled Affibody molecules.

a The [18F]FBEM-ZHER2:432-Affibody uptake 2 h post i.v. injection in athymic nude mice bearing either HER2-positive SKOV-3 cells after pretreatment with or without non-radioactive Affibody or HER2-negative U251 tumors (tissue type versus % ID/g tissue). Each bar represents an average ± SD from n = 3-6. b Blood kinetics of [18F]FBEM-ZHER2:342-Affibody. The squares represent % ID/g in the blood with an exponential curve fit. Insert Average HMW fractions of the plasma-associated radioactivity. Each point represents mean ± SD (three to four mice)
The in vivo HER2-binding specificity was also tested using a group of animals that were bearing HER2-negative U251 tumors. The tumor-associated radioactivity in this group was the same as that observed in those animals pretreated with an excess of non-labeled Affibody (Fig. 4a). This confirms that the [18F]FBEM-ZHER2:342-Affibody accumulation is HER2-dependent rather than a non-specific trapping of proteins because of variations in the vascularization of the tumor tissue.
Pharmacokinetic studies
The mean radioactivity expressed as % ID/g in the blood over time for the group of three to six mice after intravenous administration of [18F]FBEM-ZHER2:342-Affibody is shown in Fig. 4b. The clearance from the blood system was rapid. The radioactivity concentration was 2.7 ± 0.06, 1.31 ± 0.25, and 0.28 ± 0.07% ID/g at 30, 60, and 120 min post-injection, respectively (n = 3-6). The fitting of the kinetic curve with exponential decay indicated that the elimination half-life of the tracer in blood is 36 min. Mouse plasma was also evaluated for metabolic stability of [18F]FBEM-ZHER2:342-Affibody. To distinguish radiolabeled Affibody (high molecular weight) from small molecule metabolites (low molecular weight), plasma samples were eluted through NAP-5 columns (5 kDa cut-off) with PBS. Reference analysis of the [18F]FBEM-ZHER2:342-Affibody on the NAP5 column revealed 67% in the HMW fraction. The majority of radioactivity from plasma samples was associated with the HMW fractions, indicating that the [18F]FBEM-ZHER2:342-Affibody was present in the blood at late time points (table insert, Fig. 4b). Relatively low level of the radioactivity in the HMW fraction (<20%) of the urine obtained at 60 min post-injection suggests that most of the radioactivity present in the urine cannot be attributable to radiolabeled protein.
PET imaging
Representative whole-body decay-corrected coronal images were obtained from mice bearing SKOV-3 tumors at different time points after tracer injection (Fig. 5). These images confirmed that the [18F]FBEM-ZHER2:342-Affibody molecules accumulate in the tumor. Tumor uptake was significantly above the background as early as 20 min post-[18F]FBEM-ZHER2:342-Affibody injection. The liver and kidney radioactivity was high only during the first hour because of non-specific accumulation but then gradually dropped over time. The prominent radioactivity accumulation in the kidneys and urinary bladder suggests that the agent was primarily cleared through the renal pathway.

An image of a mouse bearing SKOV-3 tumor located on the left shoulder and decay-corrected whole-body PET images of the same animal obtained 1, 2, 3, and 4 h after i.v. injection of [18F]FBEM-ZHER2-342-Affibody
Dynamic PET scans were recorded up to 2 h post-injection. Representative coronal images were selected at different time points (Fig. 6, upper panels). Time-activity curves showed high tumor activity accumulation (SUV = 2.4) as early as 20 min post-injection (Fig. 6, lower panel), which reached a plateau by 40 min (SUV = 3.15, 3.32, 3.43, 60, 90, and 120 min, respectively). Whereas the kidney’s uptake was much higher than that of tumor only during the first hour, very rapid clearance was observed at later time points. The ideal time point for monitoring tumors appears to be 1 h after injection of tracer. At this time point tumor uptake reached the maximum of accumulation according to high-contrast PET images. The uptake observed in major organs also correlated well with biodistribution studies (data not presented).

Upper panels Transverse and coronal slices recorded during a dynamic study (acquisition time, 10 min each frame). Tumor is indicated by the arrows. Lower panel Quantitative results of the image analysis
Discussion
An accurate assessment of HER2 expression is crucial for the selection of patients that may benefit from HER2-targeted therapies. Currently, ex vivo techniques, such as immunohistochemistry and fluorescence in situ hybridization, are used to estimate HER2 expression before recommending HER2-tageted molecular therapies, of which the most common is treatment with trastuzumab. However, these screening approaches have significant limitations. New methods are needed to allow for non-invasive or minimally invasive evaluation of HER2 expression in vivo that can be repeated in individual patients and can provide an assessment of global HER2 expression in both primary tumors and distant metastasis. It is critical that these methods are also capable of monitoring efficacy in response to targeted therapy as measured by receptor levels on tumors. Thus, the high resolution, sensitivity, and quantitative nature of PET imaging offer an ideal alternative for assessing HER2 expression status. Radiotracers suitable for non-invasive imaging of HER2 in vivo have been previously reported. Robinson et al. developed and labeled with 124I a divalent antibody (C6.5) fragment that effectively functions in the imaging of HER2-positive tumors with PET. Moreover, they validated a method for using a clinical PET/computed tomography scanner to quantify tumor uptake [17]. The p185HER2 in xenografts could be distinguished by microPET imaging using a different size and format of anti-p185HER2 antibody fragments [30]. Smith-Jones et al. [16] used an F(ab′) fragment of trastuzumab labeled with 111In and 68Ga for SPECT and PET, respectively. However, the targeting agent they used cannot be applied to monitor response to trastuzumab, which is the current standard treatment for HER2-positive breast cancer, because the probe will compete for the same binding sites on HER2 receptors as non-labeled trastuzumab. Therefore, a decrease in signal will not correlate with decreased expression.
The Affibody molecules described by Orlova et al. [31] and Steffen et al. [32] offer a trastuzumab-compatible alternative for HER2 imaging. Using 125I-labeled ZHER2:342-Affibody molecules, tumor-specific accumulation of radioactivity was demonstrated, and a high-contrast visualization of HER2-positive xenografts in mice was obtained 6 h post-injection.
More recently, promising results using the synthetic Affibody molecule 111In-DOTA-ZHER22:342-pep2 (DOTA, 1,4,7,10-tetraazacyclododecane-N,N′,N″,N″′-tetraacetic acid) for SPECT imaging were also reported by the same group [22]. In this study, biodistribution analysis revealed very high accumulation of the new tracer in tumor only 1 h post-injection (23% ID/g). Consequently, high-contrast images as soon as 1 h post-injection using a Gamma Camera could be obtained. The images obtained after treatment with 17-AAG (heat-shock protein inhibitor) showed significant changes in the observed signal. The same DOTA-modified constructs can also be labeled with a positron emitter, 68Ga. Considering the very efficient and straightforward labeling procedure and the relatively easy access to 68Ga from commercially available generators, this approach presents an attractive alternative for PET imaging. However, we are not aware of any publication describing use of such conjugates.
Mume et al. [33] used maleimide chemistry for site-specific radiboromination of (ZHER2:4)2-Cys. The resulting conjugate was shown to bind specifically to the HER2-expressing cell-line, SKOV-3, and the biodistribution studies confirmed its ability to accumulate in SKOV-3 xenografts. This preferential tumor binding combined with fast clearance from normal organs resulted in high tumor-to-normal tissue ratios at 4 h post-injection.
Cysteine-based chelators were incorporated by Tran et al. [34] to ZHER2:342 for site specific labeling with 99mTc. Again, the Affibody molecule-based radioconjugates, in this case 99mTc-CGG-ZHER2:342, were shown to accumulate specifically in SKOV-3 xenografts and, because of rapid clearance from the blood and other organs (except kidneys), allowed clear images of tumors by Gamma-camera at 6 h post-injection.
In this work, we have further demonstrated the feasibility of using Affibody molecules to monitor HER2 expression. By labeling ZHER2:342-Cys Affibody molecule with 18F, we have created a new tracer that will allow the application of a well-established PET methodology to quantify HER2 expression in vivo. The radionuclide chosen for the imaging agent offers several distinct advantages: (1) broad availability, (2) high positron yield (almost 100%), (3) a half-life (T 1/2 = 109.7 min) that closely matches the relatively short biological lifetime of Affibody molecules.
The cysteine residue on the C-terminal of ZHER2:342-Cys allowed for the application of a well-defined labeling chemistry using [18F]fluorine-labeled maleimide. Cai et al. [18] described the radiosynthesis of N-(2-(4-[18F]fluorobenzamido)ethyl)maleimide ([18F]FBEM) and applied this molecule to the radiolabeling of an arginine-glycine-aspartic acid peptide for imaging of αvβ3 integrin expression. We developed an alternative synthesis of [18F]FBEM that employed the radiosynthesis of [18F]fluorobenzoic acid and then coupling to aminoethylmaleimide using diethylcyanophosphate. This procedure was based on our previous experience in the radiosynthesis of fluoropaclitaxel [28]. The preparation of the [18F]FBEM was completed in a two-pot, three-step reaction sequence and provided sufficient product for coupling to the reduced ZHER2:342-Cys Affibody molecule. The terminal cysteine caused the stock ZHER2-Cys Affibody molecules to exist as a mixture of monomeric and dimeric forms. Initially, we attempted to reduce the disulfide bonds with Tris(2-carboxyethyl)phosphine (TCEP), but we observed that the TCEP reacted with the limited amount of [18F]FBEM and precluded successful conjugation. We then adapted the procedure of Mume et al. [33] that used DTT to reduce the protein and then removed the excess DTT using a NAP-5 column before conjugation. These conditions have proven quite reproducible for the preparation of conjugated Affibody molecules.
The affinity observed when the radiotracer was bound to HER2 receptors on SKOV-3 and SKBR-3 cells in vitro was comparable to that of typical antibodies used clinically for imaging and therapy (e.g., the reported affinity of trastuzumab to HER2 is 5 nM) [35]. Our studies of the radiotracer binding to tumor cells expressing different levels of HER2 showed that the total cell-associated radioactivity was (1) correlated with the level of receptor expression, (2) reduced by pre-treatment of the cells with non-labeled Affibody molecules, and (3) was not affected by pre-treatment with trastuzumab. Consequently, our radioconjugate might be used to assess the level of receptor expression and possibly to monitor the changes of its expression in tumors also in patients treated with trastuzumab.
The results of our in vivo studies were very promising. The calculated elimination half-life was about 36 min, which definitely facilitated the imaging studies, whereas clearance of antibody fragments is in the range of a few hours [36], and trastuzumab requires about 25 days [37]. High accumulation of the radioactivity in HER2-positive tumors as compared with the background was observed as early as 20 min after injection as shown by PET imaging reached the plateau after 40 min and remained at that level for the duration of the experiments. For imaging purposes, this ratio at such a short time post-injection is much better than the previously published data with other HER2-targeting agents that demonstrated much weaker tumor accumulation and worse signal-to-background ratio [17, 31]. Four hours later, the ratio increased to 69, which resulted in further improvement of the contrast between tumor and the surrounding tissues, as shown by the whole body scans (Fig. 6, upper panels). By that time, only the bowel and the urinary bladder indicated any accumulation of radioactivity. These results were confirmed by the biodistribution studies indicating that 1 h post-injection, the concentration of radioactivity (% ID/g) in tumor was 7.5 times higher than that in the blood. Two hours after injection, the tumor uptake was higher than the uptake in any other organs, and levels remained steady over the course of the study. Pretreatment with non-radiolabeled Affibody molecules decreased the tumor accumulation to the levels observed in HER2-negative tumors, which was comparable to that found in the normal organs, with the exception of the kidneys, where the uptake was significantly higher. However, the radioactivity in the kidneys decreased rapidly because of excretion of the tracer to the urinary bladder. This rapid clearance of radioactivity from the kidneys distinguishes our radioconjugate from other Affibody-based radiotracers that (or their radioactive degradation products) tend to accumulate in the kidneys. On the other hand, [18F]FBEM-ZHER2:342 produced a higher level of radioactivity in the bone as a result of possible dehaloganation. Nevertheless, according to extrapolation of pre-clinical biodistribution data using the Olinda simulation package [38], several PET scans per year could be carried out using the typical dose of 185 MBq without exceeding the radiation doses to the patient that are allowed for routine diagnostic purposes.
Conclusion
Our results suggest that the [18F]FBEM-ZHER2-Affibody radioconjugate described herein can be used to assess HER2 expression in vivo by PET imaging and, therefore, might be used to monitor possible changes of receptor expression in response to therapeutic interventions. The combination of a radionuclide commonly used in the clinic and an Affibody molecule characterized by a relatively low molecular weight (compared to antibodies or single chain antibody fragments), high affinity to the target receptors, and fast clearance from the blood and normal organs makes it an optimal candidate for clinical applications.
References
Ross JS, Fletcher JA, Bloom KJ, et al. HER-2/neu testing in breast cancer. Am J Clin Pathol 2003;120:S53-71.
Cooke T, Reeves J, Lanigan A, Stanton P. HER2 as a prognostic and predictive marker for breast cancer. Ann Oncol 2001;12(Suppl 1):S23-8.
Menard S, Pupa SM, Campiglio M, Tagliabue E. Biologic and therapeutic role of HER2 in cancer. Oncogene 2003;22:6570-8.
Meric-Bernstam F, Hung MC. Advances in targeting human epidermal growth factor receptor-2 signaling for cancer therapy. Clin Cancer Res 2006;12:6326-30.
Burstein HJ. The distinctive nature of HER2-positive breast cancers. N Engl J Med 2005;353:1652-4.
Dowsett M, Cooke T, Ellis I, et al. Assessment of HER2 status in breast cancer: why, when and how? Eur J Cancer 2000;36:170-6.
Zidan J, Dashkovsky I, Stayerman C, Basher W, Cozacov C, Hadary A. Comparison of HER-2 overexpression in primary breast cancer and metastatic sites and its effect on biological targeting therapy of metastatic disease. Br J Cancer 2005;93:552-6.
Carlsson J, Nordgren H, Sjostrom J, et al. HER2 expression in breast cancer primary tumours and corresponding metastases. Original data and literature review. Br J Cancer 2004;90:2344-8.
Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov 2006;5:147-59.
Rueckert S, Ruehl I, Kahlert S, Konecny G, Untch M. A monoclonal antibody as an effective therapeutic agent in breast cancer: trastuzumab. Expert opinion on biological therapy 2005;5:853-66.
Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann Oncol 2007;18:977-84.
Pegram MD, Konecny GE, O’Callaghan C, Beryt M, Pietras R, Slamon DJ. Rational combinations of trastuzumab with chemotherapeutic drugs used in the treatment of breast cancer. J Natl Cancer Inst 2004;96:739-49.
Milenic DE, Garmestani K, Brady ED, et al. Targeting of HER2 antigen for the treatment of disseminated peritoneal disease. Clin Cancer Res 2004;10:7834-41.
Kobayashi H, Shirakawa K, Kawamoto S, et al. Rapid accumulation and internalization of radiolabeled herceptin in an inflammatory breast cancer xenograft with vasculogenic mimicry predicted by the contrast-enhanced dynamic MRI with the macromolecular contrast agent G6-(1B4M-Gd)(256). Cancer Res 2002;62:860-6.
Lub-de Hooge MN, Kosterink JG, Perik PJ, et al. Preclinical characterisation of 111In-DTPA-trastuzumab. Br J Pharmacol 2004;143:99-106.
Smith-Jones PM, Solit DB, Akhurst T, Afroze F, Rosen N, Larson SM. Imaging the pharmacodynamics of HER2 degradation in response to Hsp90 inhibitors. Nat Biotechnol 2004;22:701-6.
Robinson MK, Doss M, Shaller C, et al. Quantitative immuno-positron emission tomography imaging of HER2-positive tumor xenografts with an iodine-124 labeled anti-HER2 diabody. Cancer Res 2005;65:1471-8.
Cai W, Olafsen T, Zhang X, et al. PET imaging of colorectal cancer in xenograft-bearing mice by use of an 18F-labeled T84.66 anti-carcinoembryonic antigen diabody. J Nucl Med 2007;48:304-10.
Tolmachev V, Orlova A, Nilsson FY, Feldwisch J, Wennborg A, Abrahmsen L. Affibody molecules: potential for in vivo imaging of molecular targets for cancer therapy. Expert Opin Biol Ther 2007;7:555-68.
Engfeldt T, Orlova A, Tran T, et al. Imaging of HER2-expressing tumours using a synthetic Affibody molecule containing the (99m)Tc-chelating mercaptoacetyl-glycyl-glycyl-glycyl (MAG3) sequence. Eur J Nucl Med Mol Imaging 2007;34:722-33.
Tolmachev V, Nilsson FY, Widstrom C, et al. 111In-benzyl-DTPA-ZHER2:342, an affibody-based conjugate for in vivo imaging of HER2 expression in malignant tumors. J Nucl Med 2006;47:846-53.
Orlova A, Tolmachev V, Pehrson R, et al. Synthetic affibody molecules: a novel class of affinity ligands for molecular imaging of HER2-expressing malignant tumors. Cancer Res 2007;67:2178-86.
Tai YF, Piccini P. Applications of positron emission tomography (PET) in neurology. J Neurol Neurosurg Psychiatry 2004;75:669-76.
Innis RB, Cunningham VJ, Delforge J, et al. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab 2007;27:1533-9.
Delforge J, Mesangeau D, Dolle F, et al. In vivo quantification and parametric images of the cardiac beta-adrenergic receptor density. J Nucl Med 2002;43:215-26.
Cai W, Chen K, Mohamedali KA, et al. PET of vascular endothelial growth factor receptor expression. J Nucl Med 2006;47:2048-56.
Lang L, Jagoda E, Schmall B, et al. Development of fluorine-18-labeled 5-HT1A antagonists. J Med Chem 1999;42:1576-86.
Kiesewetter DO, Jagoda EM, Kao CH, et al. Fluoro-, bromo-, and iodopaclitaxel derivatives: synthesis and biological evaluation. Nucl Med Biol 2003;30:11-24.
Seidel J. Resolution uniformity and sensitivity of the NIH atlas small animal PET scanner: comparison to simulated LSO scanners without depth-of-interaction capability. IEEE Trans Nucl Sci 2003;50:1347-57.
Olafsen T, Kenanova VE, Sundaresan G, et al. Optimizing radiolabeled engineered anti-p185HER2 antibody fragments for in vivo imaging. Cancer Res 2005;65:5907-16.
Orlova A, Magnusson M, Eriksson TL, et al. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res 2006;66:4339-48.
Steffen AC, Orlova A, Wikman M, et al. Affibody-mediated tumour targeting of HER-2 expressing xenografts in mice. Eur J Nucl Med Mol Imaging 2006;33:631-8.
Mume E, Orlova A, Larsson B, et al. Evaluation of ((4-hydroxyphenyl)ethyl)maleimide for site-specific radiobromination of anti-HER2 affibody. Bioconjug Chem 2005;16:1547-55.
Tran T, Engfeldt T, Orlova A, et al. In vivo evaluation of cysteine-based chelators for attachment of 99mTc to tumor-targeting Affibody molecules. Bioconjug Chem 2007;18:549-48.
De Lorenzo C, Cozzolino R, Carpentieri A, Pucci P, Laccetti P, D’Alessio G. Biological properties of a human compact anti-ErbB2 antibody. Carcinogenesis 2005;26:1890-5.
Olafsen T, Tan GJ, Cheung CW, et al. Characterization of engineered anti-p185HER-2 (scFv-CH3)2 antibody fragments (minibodies) for tumor targeting. Protein Eng Des Sel 2004;17:315-23.
Cobleigh M, Frame D. Is trastuzumab every three weeks ready for prime time? J Clin Oncol 2003;21:3900-01.
Stabin MG, Sparks RB, Crowe E. OLINDA/EXM: the second-generation personal computer software for internal dose assessment in nuclear medicine. J Nucl Med 2005;46:1023-7.
Acknowledgment
The authors appreciate the support of Affibody AB and the Swedish pioneers of the Affibody molecule-related research: Jörgen Carlsson, Fredrick Y. Nilsson, Stefan Ståhl, and Vladimir Tolmachev. We owe special thanks to one of the creators of ATLAS, Jürgen Seidel, for his introduction to and continuous help with this unique imaging system. We also appreciate constructive comments from Kevin Camphausen and David Goldstein, as well as the technical assistance of Alesia Holly. This research was supported in part by the Center for Cancer Research, an Intramural Research Program of the National Cancer Institute, and was funded in part with Federal funds from the National Cancer Institute, National Institutes of Health under contracts N01-CO-12400 and N01-CO-12401, and by Breast Cancer Research Stamp proceeds awarded through competitive peer review. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services nor does mention of trade names, commercial products, or organization imply endorsement by the US Government.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kramer-Marek, G., Kiesewetter, D.O., Martiniova, L. et al. [18F]FBEM-ZHER2:342-Affibody molecule—a new molecular tracer for in vivo monitoring of HER2 expression by positron emission tomography. Eur J Nucl Med Mol Imaging 35, 1008–1018 (2008). https://doi.org/10.1007/s00259-007-0658-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-007-0658-0