
Abstract
Secretion of proteins into the extracellular space has great advantages for the production of recombinant proteins. Type 1 secretion systems (T1SS) are attractive candidates to be optimized for biotechnological applications, as they have a relatively simple architecture compared to other classes of secretion systems. A paradigm of T1SS is the hemolysin A type 1 secretion system (HlyA T1SS) from Escherichia coli harboring only three membrane proteins, which makes the plasmid-based expression of the system easy. Although for decades the HlyA T1SS has been successfully applied for secretion of a long list of heterologous proteins from different origins as well as peptides, but its utility at commercial scales is still limited mainly due to low secretion titers of the system. To address this drawback, we engineered the inner membrane complex of the system, consisting of HlyB and HlyD proteins, following KnowVolution strategy. The applied KnowVolution campaign in this study provided a novel HlyB variant containing four substitutions (T36L/F216W/S290C/V421I) with up to 2.5-fold improved secretion for two hydrolases, a lipase and a cutinase.
Key points
• An improvement in protein secretion via the use of T1SS
• Reaching almost 400 mg/L of soluble lipase into the supernatant
• A step forward to making E. coli cells more competitive for applying as a secretion host
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Secretion of proteins is important for recombinant protein production as it can lead to protein production in high quality and yield, while at same time reducing the production costs (Kleiner-Grote et al. 2018). Protein secretion in Gram-negative bacteria, such as Escherichia coli, often results in low titers. This limitation is of particular concern and reduces the general utilities of biotechnological hosts, such as E. coli, as secreting hosts (Burdette et al. 2018; Kleiner-Grote et al. 2018).
To enhance the secretion efficiency of different classes of secretion systems, different approaches have been applied. For instance, these approaches include modification of secretion signal (Freudl 2018), transferring the secretion system to a more efficient secretion host (Eom et al. 2014), and modification of genetical elements such as enhancer fragment (Pourhassan et al. 2022a). In addition to these approaches, protein engineering has proved to be a powerful tool for tailoring protein properties (Wong et al. 2006). Until now, different secretion systems have been subjected to various engineering approaches, but these efforts have commonly had little success due to the complexity of the secretion systems (Burdette et al. 2018).
Among different secretion systems of Gram-negative bacteria, type 1 secretion systems (T1SS) have relatively simple architecture, and this makes them attractive targets to be optimized (Burdette et al. 2018). T1SS translocates proteins in an unfolded state across both membranes of Gram-negative bacteria in a single step. Subsequently, proteins fold in their functional state in the extracellular space (Holland et al. 2016).
The T1SS paradigm is the hemolysin A type 1 secretion system (HlyA T1SS). The HlyA T1SS machinery consists of three membrane proteins: the ABC transporter HlyB, the membrane fusion protein HlyD, and the outer membrane protein TolC. TolC is recruited upon an interaction between the substrate (HlyA) and the inner-membrane complex (HlyB and HlyD). Then, these membrane proteins form a tunnel-channel redirecting HlyA from the cytosol to the extracellular space (Holland et al. 2016; Pourhassan et al. 2021).
Recently, the single particle cryo-EM structure of HlyB/HlyD complex revealed that a trimer of HlyB dimers, each associated with two molecules of HlyD (per HlyB dimer), forms the inner membrane of the system complex (Zhao et al. 2022).
HlyB is an ATP-binding cassette (ABC) transporter that powers the secretion of HlyA by hydrolyzing ATP. HlyB has a domain organization consists of a transmembrane domain (TMD) composed of six transmembrane helices (TMH), a hydrophilic cytoplasmic nucleotide-binding domain (NBD), and an N-terminal cytosolic domain called the C39-peptidase like domain (CLD) (Lecher et al. 2012; Schmitt et al. 2003). In addition to its ATPase activity, NBD has a supplementary function. Specifically, it interacts with HlyA prior to secretion, which is proposed to represent the substrate recognition step (Benabdelhak et al. 2003; Pourhassan et al. 2022b). Based on a series of pull-down assays, Lecher et al. suggested that the CLD is involved in positioning HlyA into the secretion pathway, indicating a chaperone-like activity for this domain (Lecher et al. 2012).
HlyD, also refers as the periplasmic adaptor protein of the system, interacts with HlyB and TolC to stabilize and seal the tunnel-channel of HlyA T1SS (Alav et al. 2021; Holland et al. 2016). In terms of topology, HlyD has an N-terminal cytosolic domain (60 residues) that follows by a single transmembrane helix (20 residues), and a large periplasmic region (Schtilein et al. 1992). A crystal structure of a soluble fragment of HlyD (lacking some of both N-terminal and C-terminal residues) became available. Based on this structural information, a hexameric state of HlyD was modeled (Kim et al. 2016).
Within various protein engineering strategies, KnowVolution is a general applicable protein engineering strategy to improve protein properties (Cheng et al. 2015). KnowVolution, also refers as knowledge gaining directed evolution, combines directed evolution and computational analysis in order to maximize improvements with a minimal experimental effort. A KnowVolution campaign is divided into four phases: (I) identification; (II) determination; (III) computational analysis; (IV) recombination (Yang et al. 2015). KnowVolution has been reported for the successful engineering of, e.g., the ligase PigC (Brands et al. 2021), laccases (Novoa et al. 2019), aryl sulfotransferases (Islam et al. 2018), and polymer-binding peptides (Rübsam et al. 2018).
HlyA1 or HlyAc, a 23-kDa C-terminal fragment of HlyA containing the secretion signal plus three GG repeats, is a carrier for the secretion of heterologous proteins (Nicaud et al. 1986). While it has been successful in secreting various proteins, its potential as a universal secretion platform is limited by low secretion titers. Improving the efficiency of HlyA T1SS for recombinant proteins remains a challenge (Pourhassan et al. 2021).
To optimize the secretion efficiency of the system, we recently worked on the HlyA enhancer fragment and have achieved great success (Pourhassan et al. 2022a), which promoted us to a further improvement of the system. In the current study, we report a two-round based KnowVolution campaigns on the inner membrane complex of HlyA T1SS (HlyB and HlyD) to achieve further improved HlyA T1SS secretion. The KnowVolution on HlyB yielded a variant with four beneficial substitutions that increased the secretion efficiency of lipase for 2.5-folds. In addition, the general applicability of the approach was also investigated with a cutinase as a second model system.
Materials and methods
Materials
All chemicals used in this study were purchased from Sigma-Aldrich, Roche Diagnostics GmbH, and Applichem GmbH if not stated otherwise. Enzymes were purchased from New England Biolabs (England), Promega GmbH (Germany), and SeSam-Biotech GmbH (Germany). All oligonucleotides were purchased from either Eurofins MWG Operon or GeneScript. The commercial available kits NucleoSpin plasmid miniprep kit and PCR clean-up kit were purchased from Macherey Nagel (Düren, Germany). Gibson assembly master mix was purchased from New England Biolabs (England). DNA sequencing was performed by Microsynth Seqlab (Göttingen, Germany).
Generation of hlyB and hlyD casting error-prone PCR mutant library
For cloning and library generation, E. coli BL21-Gold (DE3) was used. Oligonucleotides used in this work are listed in Tables S1 and S2. Plasmid pK184_EF(Best)-lipA-hlyA1-Ter-hlyBD (Pourhassan et al. 2022a) was used as the backbone vector for the library generation. Note that the gene encoding the TolC protein is endogenous, and the genes encoding HlyB and HlyD are plasmid-based. The abbreviations in the name of the plasmid stand for: EF for enhancer fragment, lipA for lipase gene from Serratia marcescens, hly for hemolysin, and Ter for a terminator region. The lac promoter of plasmid pK184 is inducible with isopropyl β-D-1-thiogalactopyranoside (IPTG). This plasmid contains a kanamycin resistance gene.
Casting error-prone PCR (cepPCR) was used for generation of random mutagenesis libraries for HlyB and HlyD according to the published cepPCR protocol (Yang et al. 2017). Accordingly, the hlyB gene from E. coli UTI89, except of the region encoding for the NBD, was divided into six fragments, listed in Table S1.
The hlyD gene from E. coli UTI89 was divided into six fragments, listed in Table S1. For each fragment, a primer set, forward and reverse, was designed and employed for amplification of the fragment via epPCR.
The parameters and reaction conditions of cepPCR (50 µL) were as follows: 1X ThermoPol buffer, 0.2 mM dNTP mix, 400 pM of each primer, 10 U polymutarase polymerase, 60 ng DNA template, 0.1–0.9 mM MnCl2. The PCR was performed as follows: 94 °C for 2 min (1 cycle), 94 °C for 30 s, primer melting temperature (Tm) for 30 s, 68 °C for 1 min (for 30 cycles), and 68 °C for 10 min (1 cycle). To obtain mutations of the first amino acids of each fragment, the forward primers annealed upstream of reverse primer of the previous fragment. PCR products were checked on agarose gel and purified with the PCR clean-up kit according to the instructions of the manufacturer.
For cloning of the individual mutant fragments into plasmid pK184_EF(Best)-lipA-hlyA1-Ter-hlyBD, either the megawhop PCR or Gibson assembly was used, with the following details.
For each individual fragment, nine mutant libraries applying different MnCl2 concentrations (0.1 to 0.9 mM) were generated. The inactive ratio of libraries were determined by prescreening on tributyrin agar plates (trypton 10 g/L, yeast extract 5 g/L, NaCl 10 g/L, agar 15 g/L, gum arabic 1.5 g/L, tributyrin 15 g/L, kanamycin 50 μg/mL). The condition of library generation for each fragment was adjusted to obtain libraries with an inactive ratio of 40–50%. To construct the entire vector, the amplified fragment containing mutations was used as either megaprimer for megawhop PCR or insert for Gibson assembly.
The megawhop PCR was performed based on the published protocol (Miyazaki 2011). The megawhop PCR products were digested with DpnI (20 U, 37 °C, overnight) and purified using the PCR clean-up kit.
For Gibson assembly cloning, the vector template lacking the related fragment was amplified using a high-fidelity polymerase, Q5 polymerase. The PCR mixture (50 μL) contained 50 ng of template plasmid, 1 × Q5 standard reaction buffer, 0.2 mM dNTP mix, 200 pM of each primer, and 1 U Q5 DNA polymerase. Subsequently, the epPCR product of the related fragments and the PCR product of the backbone vector were digested with DpnI enzyme (overnight, at 37 °C) to remove any undigested wildtype vector. The PCR products were purified using a PCR clean-up kit. The whole plasmid construction of the epPCR product and the linear plasmid was performed via Gibson assembly according to the instructions of the manufacturer.
Subsequently, 2 μL of the assembled product of Gibson assembly reaction or the megawhop PCR product was transformed into chemically competent cells of E. coli BL21-Gold (DE3). The transformed cells were cultured directly on tributyrin LB agar plates containing kanamycin 50 μg/mL. Ten single cell clones of each library were picked and sent for sequencing. For each fragment, single active clones were picked up and cultured in 96-well plates (PS-F-bottom, Sarstedt, Germany).
Site-saturation mutation of potential beneficial positions
Site-saturation mutagenesis of potential beneficial positions was performed by two-step PCR based on the published protocol (Wang and Malcolm 1999). The primers having degenerative nucleotides used for site-saturation mutagenesis are listed in Table S2. The PCR reaction was performed in two steps. In the first step, two separate reactions in separate tubes were performed; one tube containing the forward primer and the another one containing the reverse primer. In the second step, both tubes were mixed and amplification was continued.
The PCR mixture (50 μL) of the first step contained 50 ng backbone plasmid, 1X Q5 standard reaction buffer, 0.2 mM dNTP mix, 200 pM of either forward or reverse primer, and 1 U Q5 DNA polymerase. The PCR was performed as follows: 98 °C for 2 min (1 cycle); 98 °C for 20 s, Tm for 20 s, 72 °C for 8 min (for 3 cycles), and 72 °C for 10 min (1 cycle). After mixing two tubes, another PCR reaction was performed as follows: 98 °C for 2 min (1 cycle); 98 °C for 20 s, primer melting temperature (Tm) for 20 s, 72 °C for 8 min (for 15 cycles), and 72 °C for 10 min (1 cycle). The PCR products were checked on the agarose gel, and subsequently were subjected to DpnI digestion (20 U, 37 °C, overnight). DNA purification of the PCR products was performed using PCR purification kit. Purified PCR products were transformed directly into chemically competent E. coli BL21-Gold (DE3) for expression and screening.
Recombination of compatible substitutions experimentally
The recombination library of the HlyB was generated using an overlap extension PCR method based on the published protocol (Hilgarth and Lanigan 2020). Two oligonucleotides of the hlyB gene with 6 and 7 substitutions were synthesized by GenScript. Both synthesized oligonucleotides, the hlyB wildtype gene, plus the plasmids with improved positions were used as templates for the overlap extension PCR. Subsequently, the HlyB overlap extension library was cloned into the pK184 plasmid using Gibson assembly method and transformed into E. coli BL21-Gold (DE3) chemical competent cells.
Cultivation and expression of libraries in 96-well plates
For each library, single active clones were picked and placed in 150 µL LBkan medium in 96-well plates (PS-F-bottom, Sarstedt, Germany) along with four wildtype clones and four empty vector clones as positive and negative controls, respectively. Active clones were identified by generating halo on the tributyrin-agar plates. The cultures were incubated overnight (24 h, 37 °C, 900 rpm, and 80% humidity) in a 96-well MTP shaker (SI505 MTP shaker incubator, Avantor). Then, 50 μL of glycerol 50% (v/v) were added to each wells and the plates, named as master plates, were stored at − 80 °C for further experiments. For expression, each master plate was sub-cultured into a new 96-well MTP plate containing 150 µL LBkan medium and cultivated (24 h, 37 °C, 900 rpm, and 80% humidity). The cultures were used to inoculate new 96-well MTP plates containing 150 µL LBkan medium and cultivated for 3 to 4 h (37 °C, 900 rpm, and 80% humidity). Subsequently, main cultures were induced with 1 mM of IPTG and 5 mM CaCl2. The cultures were further cultivated for 16 h (37 °C, 900 rpm, and 80% humidity) in 96-well MTP shaker incubator. Afterwards, the MTPs were centrifuged (4 °C, 20 min, 2900 g) and supernatants were harvested and used for the further investigation.
Colorimetric screening system
The screening of the libraries with the para-nitrophenyl butyrate (pNPB) assay was performed as previously reported in 96-well MTPs (Pourhassan et al. 2022a). Screening of the recombinant library via lipase colorimetric assay proved to be a challenging task as the amount of lipase in the supernatant of clones was high and reached the saturation level of the assay. To solve this issue, the supernatant was further diluted in the assay reaction buffer (20-fold dilution) and the incubation time before inducing the cultures was reduced to 3 h.
Computational method to analyze stability
The structure of HlyB was modeled based on the structure of PCAT1 (PDB: 4RY2) (Lin et al. 2015) with TopModel tool (Mulnaes et al. 2020). The homology model of HlyB was used for the computational analysis. For visualization, the HlyB /HlyD structure (PDB: 7SGR) driven from Cryo-EM from Zhao et al. work was used (Zhao et al. 2022).
The ΔΔGfold (ΔΔGfold = ΔΔGfold, Sub − ΔΔGfold, WT) values of individual substitutions were computed using FoldX method version 3b5.1 (Guerois et al. 2002), in YASARA structure version as described previously (Cui et al. 2020; Van Durme et al. 2011). Internal-atomic distance between 12 beneficial positions of the HlyB wildtype measured by YASARA (Table S3). Default FoldX parameters were used (temperature 298 K; ionic strength 0.05 m; pH 7) to generate the substitutions. Mutate residues commands of FoldX were used to compute the ΔΔGfold values of substitutions (Table S4). PyMOL molecular graphics system, version 1.8.6.0 enhanced for Mac OS X (DeLano 2002) was used for visualization.
Results
Overall, two KnowVolution campaigns were employed on the HlyA T1SS of E. coli inner membrane complex, which resulted in an increased secretion. First, we describe the results of the four phases of the KnowVolution campaign on HlyB. Encouraged by these results, we also conducted a KnowVolution campaign on HlyD to further improve the secretion efficiency.
KnowVolution of HlyB toward increasing secretion efficiency
To identify amino acid positions of the HlyB transporter that increase the secretion efficiency of HlyA T1SS, a KnowVolution campaign, in 4 phases was conducted (Fig. 1).

An overview of the HlyB KnowVolution campaign aimed at increasing the secretion efficiency of the HlyA T1SS. In phase I, the hlyB gene underwent random mutagenesis, resulting in the identification of 12 potential beneficial positions. In phase II, all 12 positions were subjected to site-saturation mutagenesis. In phase III, the computer-assisted recombination (CompassR) strategy was used to select substitutions for recombination based on the HlyB homology model. Finally, in phase IV, recombination was performed resulting in a variant harboring four substitutions (T36L, F216W, S290C, V421I). This variant showed a 2.5-fold improvement in secretion efficiency compared to the wildtype
Phase I (identification)
In the first phase of KnowVolution campaign on HlyB, beneficial positions of the hlyB gene were identified through a random mutagenesis approach known as casting error-prone PCR (cepPCR) according to the published protocol (Yang et al. 2017). In cepPCR approach, the gene of interest is divided into smaller fragments, and subsequently, fragments were subjected to epPCR individually. To generate a library on HlyB using cepPCR approach, the gene of hlyB was divided into six fragments (F1B: 275 bp, F2B: 251 bp, F3B: 283 bp, F4B: 256 bp, F5B: 279 bp, F6B: 291 bp). The region of hlyB gene encoding the NBD was excluded. Excluding the region of the hlyB gene that encodes the NBD was expected to reduce the number of inactive clones in the resulting library, as this region contains highly conserved motifs that are essential for transporter function.
Random mutagenesis libraries of those six fragments were generated using a low-fidelity DNA polymerase in buffer conditions with different MnCl2 concentrations (ranging from 0.1 to 0.9 mM). The mutation frequency was adjusted by altering the MnCl2 concentration to obtain libraries with an inactive ratio of 40–50%. The inactive ratio of libraries was determined via prescreening clones on tributyrin agar plates, as active clones were able to generate halos because of the secreted lipase-HlyA1, which was used as model substrate. The mutation load of libraries was in the range of 3.6 to 5.2 mutations per fragment (on average almost 15 mutations per kbp).
In the first phase, ~ 4576 clones were screened in a 96-well MTP format using the para-nitrophenyl butyrate (pNPB) assay to identify E. coli clones that secrete the lipase with higher titers. The promising variants showing higher secretion level in compared to wildtype variant were rescreened four times via pNPB assay to eliminate the false positive clones. In this phase, the selected promising variants showed increased secretion efficiency in a range of 1.2- to 1.6-fold compared to the parental type (wild type).
Despite generating and screening of a larger library for the fifth fragment, we did not identify any beneficial position. Upon aligning the sequence of the promising variants, it was observed that twelve positions were repeatedly mutated, which were considered as “potential beneficial positions.” These positions were T36, S71, V108, L164, F216, E226, S272, S290, I407, V421, F434, and N465.
Phase II (determination)
To obtain the full diversity of those 12 identified positions, individual site-saturation mutagenesis were performed on each position to identify variants with increased secretion levels. A total of 96 variants were screened for each position to ensure that all possible amino acids were represented. From each individual library, clones with higher secretion level in compared to wildtype variant were isolated. To be sure that isolated variant are not false positive, they rescreened four time via the pNPB assay. After the rescreening, those confirmed promising variants were sent for sequencing. Table 1 summarizes the substitutions that exhibited higher secretion efficiency compared to the wildtype.
Phase III (selection)
No three-dimensional structure of HlyB was available during the experimental phase of the study. Therefore, we created a homology model based on the crystal structure of the ABC transporter PCAT1 (Lin et al. 2015) to perform the calculations. However, a single particle cryo-EM structure of HlyB/HlyD was recently published (Zhao et al. 2022). This structure (PDB: 7SGR) used for visualization of the identified beneficial positions (Figs. 2 and 3).

An illustration of the potential beneficial positions in the three-dimensional structure of HlyB wildtype (PDB: 7SGR). HlyB dimer is in cyan and green. a I49 of HlyD is among the residues involved in the interaction with the positively charged region of the CLD. b F434 of HlyB is located in close proximity of the amphipathic helix of HlyD. The amphipathic helix is oriented parallel to the membrane. c Residue S71 is located in the protomer-protomer interface. The figure was generated using the PyMol software (www.pymol.org)

An illustration of the potential beneficial positions in the three-dimensional structure of the HlyB wildtype (PDB: 7SGR). a Carton representation of HlyB dimer (cyan and green) associated with two HlyD molecules (light brown and red), with the yellow surface mapping the HlyA-interaction region on the CLD (Lecher et al. 2012). The magenta color highlights residue 108, which is located within this interaction region. b An illustration of the identified beneficial positions determined in the current study as black dots. The positions suggested by Holland et al. involved in oligomer formation are underlined (Holland et al. 2016), and the positions at the opening of HlyB suggested by Zhao et al. (2022) are highlighted by an asterisk. The figure was generated using PyMol software (www.pymol.org)
To investigate whether the identified beneficial positions exhibit cooperative effects, we explored the recombination of these residues. However, recombining multiple beneficial positions is a major challenge in protein engineering, as it often leads to variants with reduced or even no activity after recombining three or four substitutions (Liebeton et al. 2000). To address this challenge, we employed a computer-assisted recombination (CompassR) strategy, which has been previously validated for improving the performance of the BSLA lipase (Cui et al. 2020). The CompassR strategy relies on the calculation of the relative free energy of folding (ΔΔGfold) of different substitutions and enables the in silico selection of substitutions that can be combined for improved protein function. Table 2 summarizes the calculated ΔΔGfold values for HlyB substitutions. Based on the CompassR selection criteria (ΔΔGfold < 0.36), we identified nine beneficial substitutions (Table 1) that were recombined in phase IV.
Phase IV (recombination)
Recombination between selected substitutions from phase III was accomplished through an overlap extension PCR according to a published protocol (Hilgarth and Lanigan 2020). The resulting library was estimated to have 1500 different combinations. Variants showing higher secretion levels of lipase were isolated and rescreened four times. The screening of the recombinant library yielded a variant, termed as 15-H7, with an improvement of around 2.5-folds in the secretion of lipase-HlyA1 (Fig. 4a). This variant contained 4 substitutions: T36L, F216W, S290C, and V421I.

Secretion of lipase-HlyA1 through the HlyA T1SS. a SDS-PAGE of the unconcentrated supernatant of wildtype and the 15-H7 variant secreting lipase-HlyA1. b Semi-quantification of SDS-PAGE gel via the ImageJ software (Abràmoff et al. 2004). Ratio of the improvements of 15-H7 variant compared to the wildtype. One-way ANOVA (p < 0.05) indicated a significance differences between the time points. The error bars are based on three independent replicates with the error bars reporting S.D. The molecular weight of the marker proteins (M) is given on the left (kDa). xh, x hours after induction
Comparing the secretion level of HlyB wildtype and 15-H7 variant
A test secretion experiment was conducted in 100-mL Erlenmeyer flasks to compare the secretion levels of the 15-H7 variant (the best variant obtained from the HlyB KnowVolution campaign) and the wildtype. The unconcentrated supernatant of both cultures was analyzed by SDS-PAGE (Fig. 4). Prior to loading on the SDS-PAGE, the samples were normalized based on OD600 of the cultures. A semi-quantification of the SDS-PAGE gel using the ImageJ software (Collins 2007) confirmed approximately a 2.5-fold increase in the secretion efficiency of the 15-H7 variant of compared to the wildtype at 1 h after induction. However, the improvement decreased to 1.5-fold after the second hour due to aggregate formation, as evident from the formation of white foam in the supernatant of the 15-H7 culture (Fig. 4b). The semi-quantified SDS-PAGE signals were statistically investigated via analysis of variance (ANOVA) and it indicated a significant differences between the different time points.
In addition, the supernatant samples collected every hour after induction were used to perform the pNPB assay to determine lipase activity. The absorbance intensities of samples were measured at 410 nm. The pNPB assay results confirmed the SDS-PAGE analysis and showed that the secretion level of 15-H7 was higher than the wildtype (Fig. 5a, b). The improvement in secretion was higher in the early hours after induction, with the amount of soluble enzyme in the 15-H7 culture being 2.5-fold higher than in the wildtype during the first hour after induction. However, the amount of secreted Lipase-HlyA1 in the 15-H7 culture reached a concentration that promoted aggregation, visible as white particles.

Enhanced secretion of lipaase-HlyA1 by the 15-H7 variant. a Activity of the lipase-HlyA1 in the supernatant of the culture dedicated to either the “15-H7” or wildtype HlyB using the pNPB assay. b Ratio of improved lipase activity of the wildtype and the improved variants using the pNPB assay. The error bars are based on three independent replicates with the error bars reporting S.D
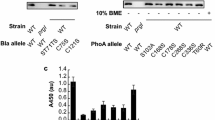
The level of expression of HlyB (Fig. 6a) and HlyD (Fig. 6b) was analyzed by western blotting for E. coli cells having either the wildtype or the 15-H7 variants. This analysis performed in three biological replicates and confirmed that HlyB and HlyD were expressed at comparable levels in both variants. Thus, the improved secretion efficiency of the 15-H7 variant cannot be attributed to a higher expression of the HlyA secretion system apparatus.

Western blot analysis of E. coli cells secreting lipaase-HlyA1 through the HlyA T1SS. a Western blot of HlyB and b western blot of HlyD using polyclonal antibodies against HlyB and HlyD, respectively. The blots indicate that both membrane proteins were expressed for both variants at comparable levels. The molecular weight (kDa) of the marker proteins (M) is given on the left; xh: cell pellets of culture, where x denotes the number of hours after induction
To determine the amount of secreted lipase-HlyA1 in the 15-H7 culture, a serial dilution of purified HlyA with known concentration was prepared and a calibration curve was generated. The concentration of secreted lipase-HlyA1 in the supernatant was quantified to approximately 400 mg/L. The amount of lipase-HlyA1 (wet weight) collected in the white particles was estimated to be 5 mg from 25 mL culture (200 mg/L).
Improved secretion of cutinase-HlyA1 through HlyA T1SS
We were eager to see if the 15-H7 HlyB variant could promote the secretion of a different target protein rather than lipase. To do this, a fusion gene encoding the cutinase protein from Fusarium solani f.sp. pisi was cloned instead of the lipase gene in the plasmid harboring the 15-H7 HlyB variant. The test expression was conducted for wildtype and 15-H7 variant, and the supernatant of the cultures was analyzed via western blotting. This results showed that the secretion of cutinase-HlyA1 in the presence of the 15-H7 HlyB variant was 2.5-fold higher than the wildtype one. This observation demonstrated that the novel HlyB is capable of enhancing the secretion of another target protein, cutinase-HlyA1 (Figure S1).
Application of KnowVolution to HlyD for increased secretion efficiency
Following the success in improving HlyB secretion efficiency, we applied the KnowVolution strategy to HlyD in an attempt to further enhance the secretion efficiency of the HlyA T1SS system. The KnowVolution strategy for directed evolution of HlyD was the same as already explained for HlyB.
In the identification step (phase I), we identified three potential beneficial positions located in the cytoplasmic domain of HlyD: M6, I49, V69. However, despite screening site-saturation libraries for these positions, no promising variant was found. We attempted to regenerate the mutation libraries on fragments 1 and 2 to cover more beneficial variants, but still no positions were determined.
Discussion
Secretion, as a strategy of recombinant protein production, could offer several advantages such as increased stability and solubility of the target protein, as well as a significant reduction in the production costs due to simplified downstream processes (Burdette et al. 2018; Kleiner-Grote et al. 2018).
Note almost one-third of all FDA-approved recombinant proteins are produced by E. coli (Puetz and Wurm 2019; Sharma and Chaudhuri 2017), but to our knowledge, there is no example of usage of E. coli as a secretion host in industrial scales. Despite the benefits of using E. coli in large-scale protein production, the low secretion levels have posed a challenge. To address this issue, researchers have attempted to engineer different secretion systems of E. coli, but with little success so far (Burdette et al. 2018; Pourhassan et al. 2021).
T1SS have a relatively simple architecture among other secretion systems of Gram-negative bacteria (Costa et al. 2015), making this class of secretion systems attractive targets for engineering efforts. The best achieved secretion titer for T1SS in different Gram-negative bacteria has been reported for metalloprotease secreted by TliDEF, a T1SS from Pseudomonas fluorescens, with a yield of 789 mg/mL (Burdette et al. 2018; Pourhassan et al. 2021).
Previously, we reported on a successful improvement of the secretion efficiency of the HlyA T1SS through a KnowVolution approach on the HlyA enhancer fragment, as well as including a terminator region. Our previous work resulted in a secretion level of approximately 180 mg/L for soluble lipase-HlyA1. Additionally, the white particles in a small culture volume of only 25 mL had a high protein content estimated to be above 1.5 mg of lipase-HlyA1 (Pourhassan et al. 2022a).
So far two case studies attempted to apply directed evolution for engineering of the inner membrane proteins of the Hly secretion system, but both reported studies were restricted to traditional protein evolution methods, epPCR (Eom et al. 2005; Sugamata and Shiba 2005). In the current study, we applied for the first time a combination of directed evolution and computational assisted strategy, CompassR, to explore the protein sequence of the inner membrane proteins of the Hly secretion system more effectively. It has already reported that applying cepPCR for random library generation increases the chance of identifying the beneficial positions of a gene of interest (Yang et al. 2017).
In the current work, we applied KnowVolution strategy on the inner membrane complex of the system to further optimize secretion of target proteins, resulting in significant improvements in secretion efficiency. Our T1SS system achieved now more than 400 mg/L soluble lipase plus around 5 mg in form of aggregated enzyme in a culture volume of only 25 mL through a KnowVolution strategy.
We acknowledge that the optimized HlyA T1SS in the current study is still away from being a potential platform in industrial scales for different substrates, but the obtained improvement indeed indicates that putting more efforts on the HlyA system is worth as there might be still rooms for improvement in efficiency of the HlyA system. It might also be beneficial using modern machine learning technologies to explore the sequence space of different system components either individually or in the context of each other. Latter one might be achieved by the provided practical supports from a deeper understanding of the HlyA T1SS secretion mechanism in the future.
In contrast to previous KnowVolution campaigns that used epPCR or SeSAM (Ji et al. 2020) for diversity generation, we employed a cepPCR method (Yang et al. 2017) that enables us to adjust mutation frequencies to small peptide stretches. This approach led to the identification of several beneficial amino acid positions within the cepPCR library for HlyB, with 12 positions distributed in the CLD and TMD. Furthermore, the CompassR strategy (Cui et al. 2020) was performed in the phase III of the HlyB KnowVolution campaign. The best variant after all four phases contained four mutations: T36L/F216W/S290C/V421I.
During the KnowVolution campaign, three positions (T36, S71, and V108) were identified located in the CLD, which is the domain that interacts with HlyA before secretion (Lecher et al. 2012). Recently, Zhao et al. showed that an interaction between HlyD and the CLD is essential for secretion. They also defined the main interface between the three protomers, which is a projection between the cytosolic domain of HlyD, residues 35–50, with a positively charged surface of the CLD. Interestingly, one of the positions we identified as potentially beneficial for HlyD, I49, is located within this region of HlyD. In addition, another identified possible beneficial of HlyD, V69, is involved in making contact with TM1/2 of HlyB. However, site saturation of these positions did not result in any improved variants.
One potential position of HlyB, S71, is located in the inter-protomer interface in close proximity to the residues involved in the HlyD/CLD interaction. In the work by Zhao et al., the S71C mutation was tested and led to the conclusion that oligomerization between HlyB and HlyD occurs in the native environment, which is a prerequisite for secretion (Zhao et al. 2022).
Previous reports have indicated that the central part of the CLD interaction site is composed of hydrophobic residues, and upon interaction with substrate, chemical shifts were observed in the first α-helix and β-sheets number 3 to 5 of the CLD (Lecher et al. 2012). Notably, the identified potential beneficial position V108 is located within the β-sheet 5 of the CLD.
Experiments conducted by Holland laboratory using cross-linking reported that two HlyB mutants, E256K and S279L, were unable to form oligomers with HlyD (Holland et al. 2016). In the work by Zhao et al., residues R256, F323, and A327 were identified as being located at the lateral opening of the protomer involved in HlyA translocation. Furthermore, they identified several positions of HlyB that were in close proximity to the substrate eGFP-HlyA1, including positions 256, 263, 319, 323, 327, and 422 (Zhao et al. 2022).
Our suggested possible beneficial positions of HlyB, including F216, E226, S272, and S290, appear to be in the vicinity of the substrate interaction region of the TMD of HlyB. Especially, V421, which was identified as a beneficial positions in the current study, is located within the translocation pathway (Zhao et al. 2022).
Interestingly, the potential beneficial position I407 is located in the periplasmic loop P3 which is between TMHs 5 and 6. This loop comprising 8 residues in HlyB is conserved in a wide variety of ABC transporters. A site-saturation mutagenesis study on the residues of this loop demonstrated that at least four residues of this loop are essential for secretion, as for instance, I401T and D404G mutants showed only 20% of hemolytic activity and a double mutant, S402P and D404K, showed no hemolytic activity at all (Holland et al. 2016).
Structural analysis shows that our beneficial position, F434, is located exactly at a location in which the amphipathic helix of HlyD (residues 9–24) contacts the elbow helix of HlyB (Zhao et al. 2022).
The KnowVolution campaign in HlyD did not identify any single residue for improving the secretion efficiency of HlyA T1SS. This is consistent with two previous directed evolution studies on HlyA T1SS, which also did not identify any improved mutants harboring HlyD mutations (Low et al. 2010; Sugamata and Shiba 2005). One possible explanation for this is that only 60 residues of HlyD are located in the cytosol, limiting its direct contact with the substrate to these residues. The rest of the protein is either located in the membrane (20 residues) or in the periplasmic space (approximately 400 residues). Another possibility is that HlyD is already optimized by evolution and is in its best functional state.
The applied evolutionary approach of this study can be generally applied to other section systems to improve the secretion performance of bacterial secretion systems. An improved secretion system opens up an avenue for the usage of Gram-negative bacteria such as E. coli as a secreting host for industrial protein production.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Abràmoff MD, Magalhães PJ, Ram SJ (2004) Image processing with ImageJ. Biophoton Internatl 11(7):36–42
Alav I, Kobylka J, Kuth MS, Pos KM, Picard M, Blair JM, Bavro VN (2021) Structure, assembly, and function of tripartite efflux and type 1 secretion systems in Gram-negative bacteria. Chem Rev 121(9):5479–5596
Benabdelhak H, Kiontke S, Horn C, Ernst R, Blight MA, Holland IB, Schmitt L (2003) A specific interaction between the NBD of the ABC-transporter HlyB and a C-terminal fragment of its transport substrate haemolysin A. J Mol Biol 327(5):1169–1179
Brands S, Brass HU, Klein AS, Sikkens JG, Davari MD, Pietruszka J, Ruff AJ, Schwaneberg U (2021) KnowVolution of prodigiosin ligase PigC towards condensation of short-chain prodiginines. Catal Sci Technol 11(8):2805–2815
Burdette LA, Leach SA, Wong HT, Tullman-Ercek D (2018) Developing Gram-negative bacteria for the secretion of heterologous proteins. Microb Cell Fact 17(1):196
Cheng F, Zhu L, Schwaneberg U (2015) Directed evolution 2.0: improving and deciphering enzyme properties. Chem Commun 51(48):9760–9772
Collins TJ (2007) ImageJ for microscopy. Biotechniques 43(S1):S25–S30
Costa TR, Felisberto-Rodrigues C, Meir A, Prevost MS, Redzej A, Trokter M, Waksman G (2015) Secretion systems in Gram-negative bacteria: structural and mechanistic insights. Nat Rev Microbiol 13(6):343–359
Cui H, Cao H, Cai H, Jaeger KE, Davari MD, Schwaneberg U (2020) Computer-assisted recombination (CompassR) teaches us how to recombine beneficial substitutions from directed evolution campaigns. Chem–A Eur J 26(3):643–649
DeLano WL (2002) Pymol: an open-source molecular graphics tool. CCP4 Newsl Protein Crystallogr 40(1):82–92
Eom GT, Song JK, Ahn JH, Seo YS, Rhee JS (2005) Enhancement of the efficiency of secretion of heterologous lipase in Escherichia coli by directed evolution of the ABC transporter system. Appl Environ Microbiol 71(7):3468–3474
Eom GT, Lee SH, Oh YH, Choi JE, Park SJ, Song JK (2014) Efficient extracellular production of type I secretion pathway-dependent Pseudomonas fluorescens lipase in recombinant Escherichia coli by heterologous ABC protein exporters. Biotechnol Lett 36(10):2037–2042
Freudl R (2018) Signal peptides for recombinant protein secretion in bacterial expression systems. Microb Cell Fact 17(1):52
Guerois R, Nielsen JE, Serrano L (2002) Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol 320(2):369–387
Hilgarth RS, Lanigan TM (2020) Optimization of overlap extension PCR for efficient transgene construction. MethodsX 7:100759
Holland IB, Peherstorfer S, Kanonenberg K, Lenders M, Reimann S, Schmitt L (2016) Type I protein secretion-deceptively simple yet with a wide range of mechanistic variability across the family. EcoSal Plus 7(1). https://doi.org/10.1128/ecosalplus.ESP-0019-2015
Islam S, Laaf D, Infanzón B, Pelantová H, Davari MD, Jakob F, Křen V, Elling L, Schwaneberg U (2018) KnowVolution campaign of an aryl sulfotransferase increases activity toward cellobiose. Chem Eur J 24(64):17117–17124
Ji Y, Islam S, Cui H, Dhoke GV, Davari MD, Mertens AM, Schwaneberg U (2020) Loop engineering of aryl sulfotransferase B for improving catalytic performance in regioselective sulfation. Catal Sci Technol 10(8):2369–2377
Kim J-S, Song S, Lee M, Lee S, Lee K, Ha N-C (2016) Crystal structure of a soluble fragment of the membrane fusion protein HlyD in a type I secretion system of Gram-negative bacteria. Structure 24(3):477–485
Kleiner-Grote GR, Risse JM, Friehs K (2018) Secretion of recombinant proteins from Escherichia coli. Eng Life Sci 18(8):532–550
Lecher J, Schwarz CK, Stoldt M, Smits SH, Willbold D, Schmitt L (2012) An RTX transporter tethers its unfolded substrate during secretion via a unique N-terminal domain. Structure 20(10):1778–1787
Liebeton K, Zonta A, Schimossek K, Nardini M, Lang D, Dijkstra BW, Reetz MT, Jaeger K-E (2000) Directed evolution of an enantioselective lipase. Chem Biol 7(9):709–718
Lin DY-w, Huang S, Chen J (2015) Crystal structures of a polypeptide processing and secretion transporter. Nature 523(7561):425–430
Low KO, Mahadi NM, Rahim RA, Rabu A, Bakar FDA, Murad AMA, Illias RM (2010) Enhanced secretory production of hemolysin-mediated cyclodextrin glucanotransferase in Escherichia coli by random mutagenesis of the ABC transporter system. J Biotechnol 150(4):453–459
Miyazaki K (2011) MEGAWHOP cloning: a method of creating random mutagenesis libraries via megaprimer PCR of whole plasmids. Meth Enzymol 498:399–406 (Elsevier)
Mulnaes D, Porta N, Clemens R, Apanasenko I, Reiners J, Gremer L, Neudecker P, Smits SH, Gohlke H (2020) TopModel: template-based protein structure prediction at low sequence identity using top-down consensus and deep neural networks. J Chem Theory Comput 16(3):1953–1967
Nicaud J-M, Mackman N, Gray L, Holland I (1986) The C-terminal, 23 kDa peptide of Escherichi coli haemolysin 2001 contains all the information necessary for its secretion by the haemolysin (Hly) export machinery. FEBS Lett 204(2):331–335
Novoa C, Dhoke GV, Mate DM, Martínez R, Haarmann T, Schreiter M, Eidner J, Schwerdtfeger R, Lorenz P, Davari MD (2019) KnowVolution of a fungal laccase toward alkaline pH. ChemBioChem 20(11):1458–1466
Pourhassan ZN, Smits SH, Ahn JH, Schmitt L (2021) Biotechnological applications of type 1 secretion systems. Biotechnol Adv 53:107864
Pourhassan ZN, Cui H, Khosa S, Davari MD, Jaeger KE, Smits SH, Schwaneberg U, Schmitt L (2022a) Optimized hemolysin type 1 secretion system in Escherichia coli by directed evolution of the Hly enhancer fragment and including a terminator region. ChemBioChem 23(6):e202100702
Pourhassan ZN, Hachani E, Spitz O, Smits SH, Schmitt L (2022b) Investigations on the substrate binding sites of hemolysin B, an ABC transporter, of a type 1 secretion system. Front Microbiol 13:1055032
Puetz J, Wurm FM (2019) Recombinant proteins for industrial versus pharmaceutical purposes: a review of process and pricing. Processes 7(8):476
Rübsam K, Davari MD, Jakob F, Schwaneberg U (2018) KnowVolution of the polymer-binding peptide LCI for improved polypropylene binding. Polymers 10(4):423
Schmitt L, Benabdelhak H, Blight MA, Holland IB, Stubbs MT (2003) Crystal structure of the nucleotide-binding domain of the ABC-transporter haemolysin B: identification of a variable region within ABC helical domains. J Mol Biol 330(2):333–342
Schtilein R, Gentschev I, Mollenkopf H-J, Goebel W (1992) A topological model for the haemolysin translocator protein HlyD. Mol Gen Genet 234:155–163
Sharma A, Chaudhuri TK (2017) Revisiting Escherichia coli as microbial factory for enhanced production of human serum albumin. Microb Cell Fact 16(1):1–19
Sugamata Y, Shiba T (2005) Improved secretory production of recombinant proteins by random mutagenesis of hlyB, an alpha-hemolysin transporter from Escherichia coli. Appl Environ Microbiol 71(2):656–662
Van Durme J, Delgado J, Stricher F, Serrano L, Schymkowitz J, Rousseau F (2011) A graphical interface for the FoldX forcefield. Bioinformatics 27(12):1711–1712
Wang W, Malcolm BA (1999) Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChangeTM site-directed mutagenesis. Biotechniques 26(4):680–682
Wong TS, Zhurina D, Schwaneberg U (2006) The diversity challenge in directed protein evolution. Comb Chem High Throughput Screen 9(4):271–288
Yang J, Zhao H-L, Ran L-Y, Li C-Y, Zhang X-Y, Su H-N, Shi M, Zhou B-C, Chen X-L, Zhang Y-Z (2015) Mechanistic Insights into elastin degradation by pseudolysin, the major virulence factor of the opportunistic pathogen Pseudomonas aeruginosa. Sci Rep 5(1):9936. https://doi.org/10.1038/srep09936
Yang J, Ruff AJ, Arlt M, Schwaneberg U (2017) Casting epPCR (cepPCR): a simple random mutagenesis method to generate high quality mutant libraries. Biotechnol Bioeng 114(9):1921–1927
Zhao H, Lee J, Chen J (2022) The hemolysin A secretion system is a multi-engine pump containing three ABC transporters. Cell 185(18):3329-3340. e13
Acknowledgements
We thank all current and former members of the Institute of Biochemistry for support and fruitful discussions. We dedicate this manuscript to Karl-Erich Jaeger for his pioneering contributions in the field of molecular enzyme technology.
Funding
Open Access funding enabled and organized by Projekt DEAL. This research was funded by CLIB Competence Center Biotechnology (CKB) funded by the European Regional Development Fund ERDF (34.EFRE-0300096) to Z.P. and L.S.
Author information
Authors and Affiliations
Contributions
ZP, HC, and NM: performing experiments; ZP: writing the draft; ZP, MD, SS, and HC: conceptualization and revising the draft for the content; LS, US: supervision, conceptualization, and revising the draft for the content. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pourhassan, Z.N., Cui, H., Muckhoff, N. et al. A step forward to the optimized HlyA type 1 secretion system through directed evolution. Appl Microbiol Biotechnol 107, 5131–5143 (2023). https://doi.org/10.1007/s00253-023-12653-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-023-12653-7