
Abstract
Epstein–Barr virus (EBV) is an oncogenic virus that is closely associated with several malignant and lymphoproliferative diseases. Studies have shown that the typical characteristic of EBV-associated diseases is aberrant methylation of viral DNA and the host genome. EBV gene methylation helps EBV escape from immune monitoring and persist in host cells. EBV controls viral gene promoter methylation by hijacking host epigenetic machinery to regulate the expression of viral genes. EBV proteins also interact with host epigenetic regulatory factors to mediate the methylation of the host’s important tumour suppressor gene promoters, thereby participating in the occurrence of tumorigenesis. Since epigenetic modifications, including DNA methylation, are reversible in nature, drugs that target DNA methylation can be developed for epigenetic therapy against EBV-associated tumours. Various methylation modes in the host and EBV genomes may also be of diagnostic and prognostic value. This review summarizes the regulatory roles of DNA methylation on the promotor of EBV gene and host genome in EBV-associated diseases, proposes the application prospect of DNA methylation in early clinical diagnosis and treatment, and provides insight into methylation-based strategies against EBV-associated diseases.
Key points
• Methylation of both the host and EBV genomes plays an important role in EBV-associated diseases.
• The functions of methylation of the host and EBV genomes in the occurrence and development of EBV-associated diseases are diverse.
• Methylation may be a therapeutic target or biomarker in EBV-associated diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epstein–Barr virus (EBV) was originally discovered by Michael Epstein and Yvonne Barr in a cultured lymphoma cell line from African Burkitt’s lymphoma (BL) patients through electron microscopy in 1964, and it was the first identified human tumor virus (Epstein et al. 1964). EBV belongs to the γ-herpesvirus family and is also known as human herpes virus type 4. The DNA genome of EBV is estimated to be 172 kb, and it encodes more than 90 genes (Odumade et al. 2011). Humans are susceptible to developing EBV infections, and EBV can establish lifelong latent infection in more than 90% of the adult population (Kwok and Chiang 2016). EBV is mainly transmitted through saliva and transfusions. The primary infection with EBV usually occurs by contact with saliva from infected individuals, and then, the virus enters epithelial cells of the oropharynx, where the virus is amplified through lytic replication and subsequently infects B cells and oropharyngeal epithelial cells (Bu et al. 2019). EBV shows obvious tropism to B lymphocytes and transforms them into continuously proliferating lymphoblastoid cell lines (LCLs). Ultimately, latent infection is established with the expression of viral latent genes and without the production of infectious virus (Hutt-Fletcher 2007).
Most EBV primary infections occur during childhood and adolescence and are mostly asymptomatic, whereas some patients infected with EBV manifest infectious mononucleosis (IM) (Bu et al. 2019), which is benign and self-limited. The course ranges from 2 to 3 weeks, and the majority of patients recover well; however, a few individuals manifest with chronic recurrent IM-like symptoms over a long period of time, accompanied by an unusual pattern of antibodies against EBV and high EBV DNA load in the peripheral blood, which is referred as chronic active EBV infection (CAEBV). The pathogenesis of CAEBV has not yet been clarified, and the prognosis is unsatisfactory and often results in high mortality rates. Unfortunately, there is still no definite therapy for CAEBV (Cohen et al. 2011). In addition, EBV, as an important oncogenic virus, is causally associated with various malignancies (Zhang et al. 2020), including epithelioid malignancies such as nasopharyngeal carcinoma (NPC), EBV-associated gastric carcinoma (EBVaGC), breast cancer (BC) and lymphoid malignancies such as BL, Hodgkin’s lymphoma (HL), and T/natural killer (NK) cell lymphoma (Ai and Xie 2018; Tsao et al. 2017). Although EBV has been identified and studied for over 50 years, its complex mechanisms have not been fully elucidated. At present, plenty of evidence shows that aberrant epigenetics are involved in EBV-associated diseases.
Epigenetic modifications are heritable and affect gene expression without altering DNA sequences (Jones and Baylin 2007; Sharma et al. 2010). Epigenetic modifications include but may not be limited to DNA methylation, chromatin remodeling, nucleosome positioning, and histone modifications, with DNA methylation being the most commonly studied epigenetic modification in humans (Leong and Lung 2021). DNA methylation is generated by the activity of DNA methyltransferases (DNMTs), which enzymatically transfer a methyl group from S-adenosyl methionine (SAM) to cytosine, mainly occurring at the fifth carbon atom of cytosine (5mC) in mammalian DNA (Jair et al. 2006). DNA methylation influences gene expression patterns without altering the corresponding DNA sequence, and it is a reversible and heritable epigenetic modification that is important in gene regulation and organism development. There are three major DNMTs for DNA methylation. DNMT1 is mainly responsible for the maintenance of DNA methylation by “copying” the cytosine-phosphate-guanine (CpG) methylation pattern from the parental strand onto the daughter strand during DNA replication. DNMT3a and DNMT3b generally perform de novo methylation of either unmethylated or hemimethylated DNA (Moore et al. 2013). DNA methylation occurs primarily at cytosines (C) in CpG-enriched regions called CpG islands (CGIs) (Belleau et al. 2018). CGIs are thought to be predominantly located in the promoter region of genes, and approximately 50% of the human genome in the 5′ end of the gene promoter region contains CGIs. In normal cells, CGIs located in the housekeeping gene promoter are protected from methylation and allow for transcription of the downstream gene, whereas those located in genomic sequences other than the promoter CpG island tend to be methylated. Approximately 70–80% of CpG sites in the mammalian cells are methylated, but both the CpG sites and their degrees of methylation are unevenly distributed in the genome (Esteller 2007; Leong and Lung 2021). However, aberrant DNA hypermethylation of CGIs in promoters (promoter hypermethylation) usually occurs in many cancers. The three main mechanisms of promoter hypermethylation are as follows. (i) Direct effect: DNA methylation can directly interfere with the binding of transcription factors to their recognition sites. (ii) Indirect effect: the methylation of gene regulatory sequence in the 5′ end binds to the methylated CpG sequence-binding proteins and prevents the transcription factors from forming transcriptional complexes with the gene. (iii) CpG methylation: moderately changes the structural properties of DNA and thereby affects transcription through regulation of the chromatin structure (Zhang et al. 2021).
When the virus persists during latent infection, most of the CGIs of early virus gene promoters are methylated, and the expression of virus genes is inhibited and maintains latent infection. For example, the promoter of the transcription initiation region of the bovine leukaemia virus is methylated under the action of the host DNMT3b, thereby directly inhibiting the binding of transcription factors, blocking viral transcription and maintaining virus latency (Pierard et al. 2010). The viral proteins VP26 and VP5 of herpes simplex virus 1 and the latent-related nuclear antigen encoded by Kaposi sarcoma-associated herpesvirus all bind to host DNMT3a to promote DNA methylation of their viral gene promoters (Rowles et al. 2015). Similarly, the X protein encoded by hepatitis B virus (HBV) can also upregulate host DNMT gene expression and promote the methylation of viral DNA and can lead to HBV latency in hepatocytes. In addition, the transcription activator Tax encoded by HBV downregulates the expression of DNMTs, resulting in virus reactivation (Vivekanandan et al. 2010). Methylation participates in genomic defense that resists viral infection and protects the human genome from a large number of repeated malignant effects. However, accumulating evidence has suggested that some viruses, including EBV, can selectively methylate and silence certain regions of their own genomes, especially when the viral gene expression of this region in the host cell is not required for the survival, establishment, and maintenance of the latent infection (Weber et al. 2019).
To address the importance of aberrant methylation in EBV-associated diseases, we summarized recent advances in EBV genome methylation characteristics during EBV latency states and lytic reactivation. The epigenetic profiles of DNA methylation have been extensively studied in several EBV-associated diseases, including EBVaGC, NPC, HL, and BL, and the mechanisms contributing to the unique epigenetic signatures have also been comprehensively summarized and compared. Recent knowledge on EBV methylation suggests its potential as a therapeutic target or biomarker in EBV-associated diseases.
DNA methylation of the EBV genome
Once EBV infects host cells, it starts to induce a latent or lytic infection with diverse expressed genes (Ma et al. 2011). According to the latent genes expressed by EBV in host cells, EBV infection can be classified into four distinct latent infection statuses (0, I, II, and III) (Chiu and Sugden 2016). Only two noncoding RNAs called EBV-encoded RNA 1 (EBER1) and EBER2 are expressed at latency 0. For example, EBV can latently infect memory B cells of healthy carriers. EBV infection in latency I expresses three latent genes in the host cell, EBV nuclear antigen 1 (EBNA1), EBERs, and BamH I-A rightward transcripts (BARTs); this includes BL and plasmablastic lymphoma (Navari et al. 2015). The EBV in latency II expresses four latent genes, EBNA1, EBERs, BARTs, and latent membrane protein (LMPs). For example, HL, CAEBV, peripheral T cell lymphoma, diffuse large B cell lymphoma, NPC, and EBVaGC are classified as type II latency diseases (Sakamoto et al. 2017; Tsao et al. 2017). Hosts with type III latency, for example, patients with IM or immunocompromised individuals and immortalized LCLs, express all latent genes, including six EBNAs (EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNA-LP), EBERs, LMP1, LMP2A, and LMP2B (Li et al. 2020b). Under certain conditions, such as the body having decreased immune function or certain physiochemical factors, EBV can be reactivated, and a large number of viral structural and regulatory genes can be expressed. The lytic replication cycle is characterized by the ordered and sequential expression of viral immediate-early (BZLF1 and BRLF1), early (BNLF1, BHRF1, BMRF1, and BARF1), and late (gp350, gp85, and gp110) genes, which are regulated primarily at the level of transcription (McKenzie and El-Guindy 2015). Latent genes are selectively expressed based on the different latent phases of EBV in the infected cells. The strategy of selective expression of latent genes of EBV contributes to evading immune surveillance and establishing a lifelong persistent infection in the host (Ok et al. 2015).
Promoters encoding the EBNAs include the BamH I W promoter (Wp), BamH I C promoter (Cp), and BamH I Q promoter (Qp). Wp and Cp are the common transcription promoters of EBNAs that can initiate the transcriptional synthesis of all six EBNA mRNAs. However, Qp transcribes only EBNA1 mRNA and is not involved in the expression of other EBNAs. The transcriptional promoter of LMP (LMPp) is located in the BamH I N region, which is transactivated by EBNA2, and EBNA2 induces the expression of LMPs (Kartika et al. 2020). The EBV lytic cycle is initiated by the activation of two promoters, the BZLF1 promoter (Zp) and BRLF1 promoter (Rp), that direct the expression of the immediate-early genes Zta and Rta, which in turn activate the downstream lytic genes individually or coordinately (Jin et al. 2010; Tonoyan et al. 2020).
The effects of epigenetic modifications, especially DNA methylation, are involved in the whole process of EBV infection and promote the development of EBV-associated diseases. Some studies have reported that the methylation status of the EBV promoter is related to the activation of the latent promoter. The EBV genome is unmethylated but becomes heavily methylated during the latent stage of the virus cycle in infected cells (Germi et al. 2016). How EBV promoters obtain methylated cytosines is not yet clear; once established, this methylation pattern is maintained in proliferating and latently infected cells (Kalla et al. 2012). In addition to Qp, there is a good correlation between EBV latency promoter methylation and promoter activity (Guo and Gewurz 2022).
DNA methylation of Wp
Wp is the first EBV latency promoter used for transcription of the genes encoding EBNA2 and EBNA leader protein upon infection, and its regulatory elements are sensitive to CpG methylation. During early EBV infection, the EBV genome is packaged into virions and enters human cells; the entire genome is nonmethylated, Cp and Qp are in the off state, and Wp is in the on state (Belleau et al. 2018; Ling et al. 1993; Ponnusamy et al. 2019). Several days after EBV infection, the transcriptional activity of Wp closes, and Cp begins to activate and becomes the major latency promoter (Chelouah et al. 2018). The Cp of the LCL cell lines also shows a certain degree of methylation over time, but methylation cannot silence promoter transcription (Tao et al. 2002). In one study, sulfite genome sequencing was used to study Wp, and the transcriptional repression of viral genes was found to be closely associated with the methylation of Wp. Two kinds of cellular transcription factors, BSAP/Pax5 and CREB, are important in the transcriptional activity of Wp and are sensitive to methylation of DNA-binding sites in the promoter region. Wp methylation was detected seven days after infection, and almost all sites in the Wp region were methylated 18 days after infection (infectious virus particles showed extensive hypomethylation of the virus genome) (Chelouah et al. 2018). In the latency I program, Qp is the active promoter, while Wp and Cp are silent promoters (Fig. 1). When BL cells in latency I are transformed into latency III in vitro, Cp is activated. However, the degree of EBV methylation in LCLs and latency III-infected BL cells is generally lower than that in type I latent-infected BL cells. Wp also maintained stable silencing and methylation in type III latent infection BL cells (Paulson and Speck 1999; Robert et al. 2020). It has been found that DNMT3b plays a critical role in Wp methylation, and the methyltransferase inhibitor 5-azacytidine (5-AzaC) can induce Wp demethylation in EBV-positive cells and reactivate the transcriptional activity of Wp. DNA methylation is the main means of regulatory “switching from Wp to Cp” in B cells after primary infection (Table 1).

Epigenetic markers of EBV gene promoters. The EBV circular episome genome is shown with viral promoters (left). Promoter usage and expression during latent and lytic infection depend on DNA methylation (right). Symbols: arrow, directions of the encoding; solid squares, active promoters; dotted squares, silent promoters
DNA methylation of Cp
The main transcriptional regulatory elements of Cp include the viral origin of replication OriP and EBNA2 effector elements (Li et al. 2020a; Robertson 2000). Sulfite genome sequencing and methylation-sensitive polymerase chain reaction confirmed that Cp shows extensive unmethylation in latency III infection and that all EBNAs are expressed by Cp. Cp is hypermethylated and silent in latency I and II infection (Li et al. 2021; Salamon et al. 2001). It has been shown that Wp and Cp are hypermethylated in B lymphocytes of healthy carriers (Robertson and Ambinder 1997), and no transcripts initiated by Wp and Cp are detected. De novo DNA methyltransferase 3b (DNMT3b) is also an important regulator of Cp methylation. David et al. found that DNMT3b was upregulated in BL cell lines that maintained latency I, while reduction of DNMT3b levels failed to induce Cp reactivation (Hughes et al. 2012). However, other research has found that treatment of BL cells with 5-azaC can lead to the activation of Cp and the expression of EBNAs (Chau and Lieberman 2004; Tempera and Lieberman 2014). Lu et al. found that the DNA demethylase TET2 cooperates with the viral oncogene EBNA2 at RBP-jκ sites to regulate the demethylation of the EBV epigenome in latency III (Lu et al. 2017). Cp methylation is important for viruses to evade immune recognition because all viral immunodominant proteins are expressed from Cp. It has been reported that cells with “Cp on” latency only survive when cytotoxic T cells targeting EBNAs are lost or defective (Niller et al. 2016). This means that activating Cp and inducing EBNAs expression through demethylating drugs may have therapeutic potential for EBV-associated diseases.
DNA methylation of Qp
Similar to the host housekeeping gene, Qp is a promoter that is rich in CpG and lacks a TATA box and is also regulated by cellular Sp1 transcription factors (Bristol et al. 2018). Unlike other latent EBV promoters, it always remains unmethylated, regardless of its activity. During latency I, Qp transcribes EBNA1, and Qp is active and does not require methylation; in latency III (such as LCLs), the activity of Qp is silent but still not methylated (Paulson et al. 2002). It is suggested that DNA methylation is not involved in the silencing of Qp in latency III-infected cells, and the silencing may be regulated by other mechanisms. Tempera et al. found that the chromatin insulator protein CTCF prevents the encroachment of the CpG methylation at the Qp initiation site (Takacs et al. 2010; Tempera et al. 2010).
Unmethylation of the EBER promoter
The EBER promoter is located upstream of OriP, and it is non-CpG methylated. EBERs are expressed in all latency states, and this property may help EBV drive B cell immortalization (Volk et al. 2020). The patterns of EBV genome methylation are specific and selected. Certain viral promoters, such as the Qp and EBER promoters and flanking sequences, appear to never be methylated (Takacs et al. 2010).
DNA methylation of LMP promoters
LMP1, LMP2A, and LMP2B are three expression products of the LMP gene in latencies II and III. LMP1 serves as the main oncogene product in virus-infected cells and functions in tumor invasion, migration, and angiogenesis (Ai et al. 2012; Nkosi et al. 2020). LMP2A can suppress the expression of EBV lytic proteins BZLF1 and g350/220, helping EBV maintain a long-term latent infection in the host. LMP2B can regulate the distribution and function of LMP2A and jointly maintain latent EBV infection (Hino et al. 2009). LMP promoters are methylated in latency I infection. However, LMP promoters are not methylated and express LMP1 and LMP2 in latency II and III infection. LMP1 activates DNMT1 through the JNK pathway and downregulates the expression of TETs, leading to DNA methylation of host tumor suppressor genes (TSGs) in NPC. It can also increase the expression of DNMT1 by upregulating miRNA-155 (Stanland and Luftig 2020; Zhang et al. 2021). These results indicate that EBV regulates the mechanism of DNA methylation in host cells mainly through LMPs activation of DNMTs.
Epigenetic silence of lytic promoters in latency
During latent EBV infection, lytic gene regulatory elements are usually silent. EBV lytic infection is triggered by the expression of two immediate early genes, BZLF1 (Zta) and BRLF1 (Rta), which are initiated by Zp and Rp, respectively (Jin et al. 2010). Zta can also be transcribed by Rp. Zta is a transcriptional activator that activates viral lytic gene expression and initiates the switch of EBV from latency to the lytic life cycle; thus, the activation of Rp triggers the lytic cascade (Germini et al. 2020). Rp and Zp are highly methylated in most latency I and III cell lines. The physiological triggering mechanism of EBV reactivation in vivo is not clear. In in vitro cell culture, EBV lytic reactivation can be triggered by a variety of drugs, such as demethylation drugs, histone deacetylase inhibitors and phorbol ester, and directly or indirectly activate BZLF1 and BRLF1 gene promoters, triggering lytic replication (Honeywell et al. 2018).
BZLF1 preferentially recognizes methylated binding sites in lytic activation
Anne et al. reported that BZLF1 preferentially binds to meZRE CpG-methylated motifs in key viral promoters and overcomes heavily repressed chromatin without the need for active DNA demethylation (Woellmer et al. 2012; Woellmer and Hammerschmidt 2013). Marisa et al. reported that BZLF1 interacts with the chromatin remodeller INO80. Upon BZLF1 binding, nucleosomes are removed, Polycomb repression is lost, and RNA polymerase II is recruited to activate early promoters, promoting efficient lytic viral gene expression (Buschle and Hammerschmidt 2020; Schaeffner et al. 2019). These findings document that DNA methylation is a prerequisite for EBV lytic reactivation.
Aberrant DNA methylation of the host genome in patients with EBV-associated diseases
Global DNA hypermethylation and TSG silencing in EBV-associated neoplasms
After EBV infection, the EBV genome is first regulated by DNA methyltransferase methylation, which leads to extensive methylation of the virus genome and restricts the expression of lytic genes to establish latent infection (Scott 2017). Subsequently, EBV expresses a limited number of viral genes that affect the expression of host genes, including tumour suppressor genes, by regulating DNA methylation enzymes (Fiches et al. 2020). This regulation plays a central role in the maintenance of EBV latent programs in different neoplasms. EBV-regulated methylation plays an important role in EBV carcinogenesis. It forms a self-defense mechanism through its own gene methylation and induces aberrant methylation of host cell genes, leading to the cell cycle disorder and cell transformation and promoting the occurrence and development of EBV-associated neoplasms through epigenetic mechanisms.
EBVaGC
According to the Cancer Genome Atlas (TCGA) classification of gastric cancer, EBVaGC has unique molecular characteristics compared with EBV-negative gastric carcinoma (EBVnGC), such as PIK3CA mutations, DNA hypermethylation, ARID1A and BCOR mutations, and JAK and PD-L1/2 amplifications (Zheng et al. 2020). Liang et al. identified 216 genes downregulated by EBV-induced host DNA hypermethylation in AGS-EBV cells through epigenome and transcriptome sequencing (Liang et al. 2014). Among these genes, CDKN2A is a tumor suppressor that blocks the progression of the cell cycle (Saito et al. 2013), and its silencing is related to a poor prognosis and survival rate (Shi et al. 2012). Another tumor suppressor, FHIT, which is implicated in tumour growth suppression, as well as in the induction of apoptosis, also has obvious methylation in EBVaGC (He et al. 2015). The promoter of RCOR2 is also methylated in EBVaGC tissue; RCOR2 is mainly highly expressed in embryonic stem cells, and its encoded protein interacts with the histone demethylase LSD1 (Yang et al. 2011). A recent study showed that the tumor suppressor CYLD, which encodes a multifunctional deubiquitinase and negatively regulates multiple signal transduction pathways, is hypermethylated and has decreased expression in EBV-associated gastric adenocarcinoma (Ghadami et al. 2019). In addition, some genes, such as E-cadherin, CTNNBIP1, and PTEN, which are involved in cancer-related cell adhesion, cell migration, the WNT pathway, and the MAPK signaling pathway, have also been found to be hypermethylated in EBVaGC (Kosari-Monfared et al. 2019; Yang et al. 2020). EBVaGC is intermediate between latency I and II and mainly expresses EBERs, EBNA1, and BARTs with almost no or low LMP1 and LMP2B. LMP2A is expressed in approximately 40% of patients (Strong et al. 2013). It has been shown that LMP2A upregulates the expression of the methyltransferase DNMT1 and downregulates the expression of the key demethylase TET2 (Fiches et al. 2020). LMP2A enhances the expression of DNMT1 by inducing phosphorylation of STAT3, resulting in PTEN silencing through hypermethylation of the promoter in an IL-6-independent manner (Wu et al. 2017). It can also induce ERK phosphorylation to upregulate DNMT3a, leading to methylation of the water-glycerol channel protein AQP3 promoter and resulting in downregulation of AQP3 (Wang et al. 2019). LMP1 is expressed at low levels in EBVaGC, and only 10% of patients express LMP1 (Yang et al. 2020). Li et al. concluded that all LMP promoters were methylated to different degrees and that the expression of LMP1 could be recovered using a demethylation agent (Li et al. 2016). Gao et al. found that LMP1 leads to hypermethylation of the tumour suppressor gene RASSF10 by upregulating the expression of DNMT1. RASSF10 encodes a protein that inhibits cell proliferation, invasion, and migration and induces apoptosis. Overexpression of LMP1 in human gastric adenocarcinoma AGS cells promotes cell migration, invasion, and colony formation; when RASSF10 is coexpressed, this effect is counteracted (Gao et al. 2021). Currently, the mechanism by which EBV regulates DNA methylation in GC is still unknown. However, the knowledge summarized at present shows that EBV regulates the mechanism of DNA methylation in host cells mainly through LMP1/2A activation of DNMT1/3 and inhibition of TET2 (Table 2) (Okabe et al. 2017).
EBV-associated NPC
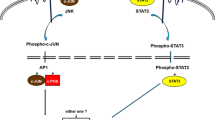
EBV is closely associated with the development of NPC. EBV establishes latency II infection in EBV-associated NPC. The main viral products expressed in NPC are EBNA1, LMP1, and LMP2A, and the CpG sites of LMP are hypermethylated (Ernberg et al. 1989). This methylation is different from silencing of TSG expression and helps EBV evade the surveillance of the immune system; thus, EBV genome methylation plays an indispensable role in maintaining the latent infection state of EBV in NPC (Allday et al. 1990; Lam et al. 2019). It has been found that the YYD domain of COOH terminal activation region 2 (CTAR2) of LMP1 in NPC cells is the key to activating the JNK signaling pathway, which leads to c-Jun phosphorylation. c-Jun (homodimer) or c-Fox (heterodimer) forms AP-1 (Fos/Jun) complexes, which bind to the DNMT1 promoter region and enhance the expression of DNMT1 (Tsai et al. 2006). Tsai et al. confirmed that LMP1 induces DNMT1 to increase the methylation level of E-cadherin and inhibit its expression by activating the JNK/AP-1 signalling pathway (Tsai et al. 2002). As a result, the invasion and metastasis of NPC are regulated. LMP1 can also activate DNMT3b through the NFκB signaling pathway (Ammous-Boukhris et al. 2019; Peng et al. 2016) and activate DNMT1 through miRNA-155, thereby inhibiting TSGs in a DNA methylation-dependent manner (Lu et al. 2008). Recent studies have found that positive expression of LMP1 is significantly correlated with a poor overall survival rate (Shi et al. 2019). In addition, DNA hypomethylation of S100A4 was found in LMP2A-positive NPC tissues. S100A4 can enhance the invasion and metastasis of NPC cells in vitro and in vivo, and researchers found that inhibition of DNA methyltransferase by 5-aza-DC stimulated the expression of S100A4 in cells without ectopically expressing LMP2A. Methylation-specific PCR (MSP) confirmed that ectopic expression of LMP2A led to demethylation of the S100A4 promoter. These results suggest that LMP2A-induced hypomethylation is involved in the regulation of S100A4 expression in NPC (Table 2) (Lin et al. 2016). Consequently, hypomethylation and activation of related genes are also important for the progression of cancer metastasis. Analysis of the epigenetic modification of EBV-associated tumours can help elucidate the pathogenesis of NPC and promote the development of novel treatments (Niller et al. 2009).
EBV-associated HL
Fifty percent of HL is associated with EBV infection (Bu et al. 2019). EBV establishes latency II infection in EBV-associated HL. The main expressed products are EBNA1 and LMP1 (Zhou et al. 2015). EBV Cp can evoke the expression of virus proteins targeting CD8+ cytotoxic T cells, but these proteins are generally not expressed in EBV-related HL. It has been found that EBV can mask the presence of the virus in tumor tissue by inhibiting the expression of virus protein antigen and methylation of transcriptional control sequence, escaping the immune surveillance of CD8+ cytotoxic T cells and promoting tumorigenesis (Dhiab et al. 2015; Kis et al. 2011). Myriam et al. found that tumor suppressor gene (p16, RASSF1A, CDH1, DAPK, GSTP1, SHP1, and MGMT) promoter methylation occurs in both EBV-positive and EBV-negative HL. Surprisingly, most of these gene promoters are more frequently hypermethylated in EBV-negative HL cases than in EBV-positive HL cases. In particular, the DAPK gene promoter had higher methylation levels in LMP1-negative cases than in LMP1-positive cases (Dhiab et al. 2015). This suggests that DAPK gene promoter hypermethylation may represent an alternative mechanism for the survival of neoplastic B cells in the absence of EBV infection. These observations likely reflect the multitude of factors involved in HL development and the complexity of their interactions with epigenetic factors.
EBV-associated BL
BL tumours are most frequently associated with EBV infection, as 15–85% of BL shows EBV infection (Hammerl et al. 2019). EBV replicates in latency I in EBV-associated BL and mainly expresses EBNA1. It often shows epigenetic changes, and the viral genomes Cp and Wp are hypermethylated and inactivated. Qp plays a role in maintaining the unmethylated state and directs the transcription of EBNA1. Tao et al. used bisulfite genome sequencing to detect the methylation of frozen endemic BL tissue sections and found no methylation of Qp but complete methylation of Cp in EBNA1(Tao et al. 1998; Zheng et al. 2020). Paschos et al. suggested that the epigenetic mechanism of BL is related to the downregulation of Bim, a member of the apoptotic Bcl-2 family. EBV triggers a series of events to suppress the epigenetics of the tumor suppressor gene Bim in infected B cells and inhibit their passage (Paschos et al. 2009).
Some studies have documented that hypomethylation of certain promoter regions occurs in the early stages of EBV-mediated transformation from resting B cells (RBLs) to proliferating LCLs (Hernando et al. 2013). Further analysis of these hypomethylated regions showed enrichment of specific transcription factors, such as the NFκB/p65 pathway, and high expression of these genes. These results indicate that hypomethylation related to EBV-mediated transformation of RBLs to LCLs is related to proliferation. Hagman et al. also found significantly increased expression of the transcription factors EBF1, IRF4, and MEF2C, and EBF1 plays an essential role in B lymphocyte production and B cell function (Hagman et al. 2012). IRF4 is a key transcription factor for the production of normal plasma B cells (Ikeda and Tagawa 2021). MEF2C is necessary for the proliferation and survival of B cells after antigen receptor stimulation (Wang et al. 2020). Additionally, drug-induced demethylation improved the transformation efficiency and proliferation ability of resting B cells to lymphoblasts. EBV regulates methylation of the NFκB pathway, which plays an important role in EBV infection as well as in the occurrence and development of malignant tumours (Tempera and Lieberman 2014). A recent study found that overexpression of EBNA3C in the EBV-negative BL cell line BJAB can induce DNMT1 protein expression, and EBNA3C can interact with DNMT1 (Pandey and Robertson 2018). Another study found that overexpression of EBNA3C in LCLs could induce the expression of DNMT1 and DNMT3a, which further shows that the promoter of TSG RASSF1 was hypermethylated in BJAB and LCLs overexpressing EBNA3C, suggesting that EBNA3C could downregulate the expression of RASSF1 by enhancing the methylation activity of DNMTs (Fig. 2) (Zhang et al. 2019). Taken together, it seems that EBV can regulate the expression of host genes through methylation mechanisms in order to promote transformation and proliferation of BL.

Schematic presentation of EBV regulation of host genome methylation in EBV-associated neoplasms
EBV-associated BC
Some studies have shown that some BC is associated with EBV infection, and its epigenetic characteristics are characterized by the low frequency of oncogene mutations and the high frequency of epigenetic silencing of BC-related TSGs (Yahia et al. 2014). Through genomic DNA methylation sequencing analysis of EBV-associated BC tissues, Mohammad et al. found that key TSGs of the host genome were hypermethylated, such as BRCA1/2, p14, p16, and hMLH (Table 2) (Yahia et al. 2014). In addition, the main developmental pathways, including the Hippo signaling pathway, are also highly methylated (Abdallah et al. 2018). However, the mechanism of the role of EBV proteins in regulating the methylation of BC requires further investigation.
EBV-associated nonneoplastic diseases
Currently, epigenetic modification research on EBV infection is mostly focused on EBV-associated tumours, and there are very few studies on EBV infection-associated nontumor diseases. In particular, the mechanism underlying the regulatory effect of EBV protein on host genome DNA methylation in these diseases has not been reported.
IM
IM is a self-limited lymphoproliferative disease caused by EBV infection. Latency III is characteristic of IM, and EBV expresses all latent genes. Tierney et al. found that in most IM patients, Wp was hypermethylated and Cp was nonmethylated (Tierney et al. 2000). Cp activity is the main difference between latency III and other latent forms. However, the role of DNA methylation in the development of IM, especially at specific genomic regions, such as host gene promoter CpG islands, has not yet been deeply investigated and warrants further research.
CAEBV
CAEBV is a nonneoplastic lymphoproliferative disease associated with EBV infection. Patients with CAEBV have a poor prognosis, and its pathogenesis and the role of EBV are unclear. CAEBV is a type II latency disease in which only EBNA1 is expressed in EBNAs. Yoshioka et al. detected DNA methylation in spleen tissues of five patients with CAEBV and found that the Cp and Wp promoters were hypermethylated and that Qp was not methylated (Yoshioka et al. 2003). However, they did not find the Qp transcript. Surprisingly, EBNA1 was transcribed by Cp in CAEBV samples. These data suggest that the hypermethylation of the Cp region may not be sufficient to prevent the transcription of Cp, and the transcripts initiated by Cp may have a unique splicing mode and prioritize the induction of transcriptional EBNA1 in most CAEBV cases, but its mechanism needs to be explored further. The seemingly contradictory and unique relationship between the hypermethylation of the CAEBV Cp region and the transcriptional activation of Cp provides an idea for further study of the pathogenic mechanism of CAEBV.
Application of methylation modification in the diagnosis and treatment of EBV-associated diseases
Early diagnosis and prognosis assessment
Hypermethylation of DNA in the peripheral blood provides new insight into the early diagnosis and screening of tumors. Dying cells release DNA fragments into the bloodstream, and this plasma-derived circulating cell-free DNA (cfDNA), also known as “liquid biopsy”, is a promising tool for minimally invasive molecular diagnostics and disease monitoring (Dor and Cedar 2018). cfDNA can be used to track cancer progression or response to treatment even if the tumour is inaccessible or its location is unknown. Some studies have reported that methylation of the promoter of the tumour suppressor genes RUNX3, RASSF1A, and Reprimo may be a powerful and potential noninvasive biomarker for the detection and diagnosis of gastric cancer (Wen et al. 2017). Promoter methylation of p16 can often be detected in tumor samples but not in normal tissues. Therefore, the detection of serum p16 methylation may be a useful marker of early gastric cancer (Liu et al. 2019). Plasma EBV DNA methylation detection has important potential in the diagnosis of NPC (Lam et al. 2019). Therefore, DNA methylation sequencing may be used as a new method for early and noninvasive tumour diagnosis. Moreover, methylated cancer-specific changes can also provide a sensitive indicator of severity and an auxiliary marker of prognosis (Kim et al. 2014). However, the DNA of circulating tumor cells may not be completely released into the blood, and the proportion of cfDNA in the total blood DNA varies widely from very rare (0.01%) to highly prevalent (> 90%) and is histology- and tumor burden-dependent (Liu et al. 2021). Therefore, it has become an obstacle to reliably identifying the methylation level of cfDNA in the blood. Nevertheless, whether “fluid biopsy” could provide sufficient sensitivity and specificity for clinical diagnosis and whether it could correctly distinguish patients with cancer remain unconfirmed.
Treatment
DNA demethylation is promising as a new target for tumor therapy. The genetic damage of tumors with DNA methylation is easier to correct than that of tumors with DNA sequence mutation because of the reversibility of epigenetic modification. This concept has led to the development of drugs that are useful in the treatment of specific tumors (Jones et al. 2016). Although DNA methylation may not play a predominant role in all cancers, the modification patterns will affect cell tendency and tumor phenotype to varying degrees (Dor and Cedar 2018). In cancer treatment, drugs such as azacytidine administered alone or in combination with other compounds could cause global demethylation among target cells (Pulecio et al. 2017). 5-AzaC, a demethylating drug, mainly binds to DNA methyltransferase through covalent bonds to form an irreversible complex, which reduces the level of genomic methylation and reactivates hypermethylated tumour suppressor genes. At present, many studies have confirmed that most of the CpG islands in the promoter region of many tumour suppressor genes are in a state of hypermethylation, and the expression of protein can be restored after intervention with the demethylated drug 5-AzaC (Sun et al. 2020). Additionally, it has been reported that zebularine can activate the tumor suppressor genes p15, p16, and p57 by demethylation in gastric cancer cells (Pan et al. 2016). Moreover, zebularine treatment sensitized the cGAS-STING pathway to recruit CD8+ T cells and NK cells into the tumor microenvironment by demethylating the STING promoter (Lai et al. 2021). However, demethylating drugs not only restore the expression of tumor suppressor genes but also affect the expression of oncogenes. Therefore, the pros and cons of demethylating drugs warrant further investigation.
Conclusion and prospects
Selective activation of EBV promoters is regulated by DNA methylation. All promoters are nonmethylated upon infection, and the EBNA genes are transcribed via Wp. Wp becomes inactive due to DNA methylation shortly after infection. Cp shows extensive unmethylation in latency III infection, and all EBNAs are transcribed by Cp. However, Cp is hypermethylated and silent in latency I and II infection, Qp is active, and only EBNA1 is expressed during latency I. The silencing of the EBV genome facilitates immune evasion in latently infected cells through DNA methylation, while Qp remains unmethylated to support the latent state, which is probably also supported by the binding of chromatin insulator protein CTCF that prevents the encroachment of CpG methylation at the Qp initiation site. Moreover, DNA methylation is a prerequisite for escape from viral latency, and the immediate early protein BZLF1 preferentially binds to CpG-methylated motifs meZRE in key viral promoters to promote efficient lytic viral gene expression without the need for active DNA demethylation. Thus, EBV regulates its latent infection and reactivation with the help of cell-mediated DNA methylation.
Epigenetic markers and differentially methylated genes may have diagnostic and prognostic value in EBV-related diseases. EBV infection results in hypermethylation of EBV promoters and the host genome in NPC and EBVaGC, and is associated with downregulation of TSGs. Due to the reversibility of epigenetics, drugs that affect the epigenetic characteristics of EBV-associated tumors may be developed for treatment. Hypomethylating agents have demonstrated the potential to drive hypomethylation of the viral genome and initiate re-expression of immunogenic viral genes in EBV-associated lymphoma cells. Therefore, targeting DNA methylation is a potential novel therapeutic strategy for EBV-associated diseases.
Change history
07 October 2022
A Correction to this paper has been published: https://doi.org/10.1007/s00253-022-12216-2
References
Abdallah MOE, Algizouli UK, Suliman MA, Abdulrahman RA, Koko M, Fessahaye G, Shakir JH, Fahal AH, Elhassan AM, Ibrahim ME, Mohamed HS (2018) EBV associated breast cancer whole methylome analysis reveals viral and developmental enriched pathways. Front Oncol 8:316. https://doi.org/10.3389/fonc.2018.00316
Ai J, Xie Z (2018) Epstein-Barr Virus-positive T/NK-cell lymphoproliferative diseases in Chinese Mainland. Front Pediatr 6:289. https://doi.org/10.3389/fped.2018.00289
Ai J, Xie Z, Liu C, Huang Z, Xu J (2012) Analysis of EBNA-1 and LMP-1 variants in diseases associated with EBV infection in Chinese children. Virol J 9:13. https://doi.org/10.1186/1743-422X-9-13
Allday MJ, Kundu D, Finerty S, Griffin BE (1990) CpG methylation of viral DNA in EBV-associated tumours. Int J Cancer 45(6):1125–1130. https://doi.org/10.1002/ijc.2910450623
Ammous-Boukhris N, Mosbah A, Ayadi W, Sahli E, Chevance S, Bondon A, Gargouri A, Baudy-Floc'h M, Mokdad-Gargouri R (2019) B1.12: a novel peptide interacting with the extracellular loop of the EBV oncoprotein LMP1. Scientific reports 9(1):4389 https://doi.org/10.1038/s41598-019-39732-y
Belleau P, Deschênes A, Scott-Boyer MP, Lambrot R, Dalvai M, Kimmins S, Bailey J, Droit A (2018) Inferring and modeling inheritance of differentially methylated changes across multiple generations. Nucleic Acids Res 46(14):e85. https://doi.org/10.1093/nar/gky362
Bristol JA, Djavadian R, Albright ER, Coleman CB, Ohashi M, Hayes M, Romero-Masters JC, Barlow EA, Farrell PJ, Rochford R, Kalejta RF, Johannsen EC, Kenney SC (2018) A cancer-associated Epstein-Barr virus BZLF1 promoter variant enhances lytic infection. PLoS Pathog 14(7):e1007179. https://doi.org/10.1371/journal.ppat.1007179
Bu W, Joyce MG, Nguyen H, Banh DV, Aguilar F, Tariq Z, Yap ML, Tsujimura Y, Gillespie RA, Tsybovsky Y, Andrews SF, Narpala SR, McDermott AB, Rossmann MG, Yasutomi Y, Nabel GJ, Kanekiyo M, Cohen JI (2019) Immunization with components of the viral fusion apparatus elicits antibodies that neutralize Epstein-Barr virus in B cells and epithelial cells. Immunity 50(5):1305-1316.e6. https://doi.org/10.1016/j.immuni.2019.03.010
Buschle A, Hammerschmidt W (2020) Epigenetic lifestyle of Epstein-Barr virus. Semin Immunopathol 42(2):131–142. https://doi.org/10.1007/s00281-020-00792-2
Chau CM, Lieberman PM (2004) Dynamic chromatin boundaries delineate a latency control region of Epstein-Barr virus. J Virol 78(22):12308–12319. https://doi.org/10.1128/JVI.78.22.12308-12319.2004
Chelouah S, Cochet E, Couve S, Balkaran S, Robert A, May E, Ogryzko V, Wiels J (2018) New interactors of the truncated EBNA-LP protein identified by mass spectrometry in P3HR1 Burkitt's lymphoma cells. Cancers (Basel) 10(1) https://doi.org/10.3390/cancers10010012
Chiu YF, Sugden B (2016) Epstein-Barr Virus: the path from latent to productive infection. Annu Rev Virol 3(1):359–372. https://doi.org/10.1146/annurev-virology-110615-042358
Cohen JI, Jaffe ES, Dale JK, Pittaluga S, Heslop HE, Rooney CM, Gottschalk S, Bollard CM, Rao VK, Marques A, Burbelo PD, Turk SP, Fulton R, Wayne AS, Little RF, Cairo MS, El-Mallawany NK, Fowler D, Sportes C, Bishop MR, Wilson W, Straus SE (2011) Characterization and treatment of chronic active Epstein-Barr virus disease: a 28-year experience in the United States. Blood 117(22):5835–5849. https://doi.org/10.1182/blood-2010-11-316745
Dhiab MB, Ziadi S, Mestiri S, Gacem RB, Ksiaa F, Trimeche M (2015) DNA methylation patterns in EBV-positive and EBV-negative Hodgkin lymphomas. Cell Oncol (dordr) 38(6):453–462. https://doi.org/10.1007/s13402-015-0242-8
Dor Y, Cedar H (2018) Principles of DNA methylation and their implications for biology and medicine. Lancet 392(10149):777–786. https://doi.org/10.1016/s0140-6736(18)31268-6
Epstein MA, Achong BG, Barr YM (1964) Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1(7335):702–703. https://doi.org/10.1016/s0140-6736(64)91524-7
Ernberg I, Falk K, Minarovits J, Busson P, Tursz T, Masucci MG, Klein G (1989) The role of methylation in the phenotype-dependent modulation of Epstein-Barr nuclear antigen 2 and latent membrane protein genes in cells latently infected with Epstein-Barr virus. J Gen Virol 70(Pt 11):2989–3002. https://doi.org/10.1099/0022-1317-70-11-2989
Esteller M (2007) Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet 8(4):286–298. https://doi.org/10.1038/nrg2005
Fiches GN, Zhou D, Kong W, Biswas A, Ahmed EH, Baiocchi RA, Zhu J, Santoso N (2020) Profiling of immune related genes silenced in EBV-positive gastric carcinoma identified novel restriction factors of human gammaherpesviruses. PLoS Pathog 16(8):e1008778. https://doi.org/10.1371/journal.ppat.1008778
Gao Y, Fu Y, Wang J, Zheng X, Zhou J, Ma J (2021) EBV as a high infection risk factor promotes RASSF10 methylation and induces cell proliferation in EBV-associated gastric cancer. Biochem Biophys Res Commun 547:1–8. https://doi.org/10.1016/j.bbrc.2021.02.014
Germi R, Guigue N, Lupo J, Semenova T, Grossi L, Vermeulen O, Epaulard O, de Fraipont F, Morand P (2016) Methylation of Epstein-Barr virus Rta promoter in EBV primary infection, reactivation and lymphoproliferation. J Med Virol 88(10):1814–1820. https://doi.org/10.1002/jmv.24524
Germini D, Sall FB, Shmakova A, Wiels J, Dokudovskaya S, Drouet E, Vassetzky Y (2020) Oncogenic Properties of the EBV ZEBRA Protein. Cancers (Basel) 12(6) https://doi.org/10.3390/cancers12061479
Ghadami E, Nikbakhsh N, Fattahi S, Kosari-Monfared M, Ranaee M, Taheri H, Amjadi-Moheb F, Godazandeh G, Shafaei S, Nosrati A, Pilehchian Langroudi M, Samadani AA, Amirbozorgi G, Mirnia V, Akhavan-Niaki H (2019) Epigenetic alterations of CYLD promoter modulate its expression in gastric adenocarcinoma: a footprint of infections. J Cell Physiol 234(4):4115–4124. https://doi.org/10.1002/jcp.27220
Guo R, Gewurz BE (2022) Epigenetic control of the Epstein-Barr lifecycle. Curr Opin Virol 52:78–88. https://doi.org/10.1016/j.coviro.2021.11.013
Hagman J, Ramírez J, Lukin K (2012) B lymphocyte lineage specification, commitment and epigenetic control of transcription by early B cell factor 1. Curr Top Microbiol Immunol 356:17–38. https://doi.org/10.1007/82_2011_139
Hammerl L, Colombet M, Rochford R, Ogwang DM, Parkin DM (2019) The burden of Burkitt lymphoma in Africa. Infectious Agents and Cancer 14:17. https://doi.org/10.1186/s13027-019-0236-7
He D, Zhang YW, Zhang NN, Zhou L, Chen JN, Jiang Y, Shao CK (2015) Aberrant gene promoter methylation of p16, FHIT, CRBP1, WWOX, and DLC-1 in Epstein-Barr virus-associated gastric carcinomas. Medical Oncology (northwood, London, England) 32(4):92. https://doi.org/10.1007/s12032-015-0525-y
Hernando H, Shannon-Lowe C, Islam AB, Al-Shahrour F, Rodríguez-Ubreva J, Rodríguez-Cortez VC, Javierre BM, Mangas C, Fernández AF, Parra M, Delecluse HJ, Esteller M, López-Granados E, Fraga MF, López-Bigas N, Ballestar E (2013) The B cell transcription program mediates hypomethylation and overexpression of key genes in Epstein-Barr virus-associated proliferative conversion. Genome Biol 14(1):R3. https://doi.org/10.1186/gb-2013-14-1-r3
Hino R, Uozaki H, Murakami N, Ushiku T, Shinozaki A, Ishikawa S, Morikawa T, Nakaya T, Sakatani T, Takada K, Fukayama M (2009) Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res 69(7):2766–2774. https://doi.org/10.1158/0008-5472.Can-08-3070
Honeywell RJ, Sarkisjan D, Kristensen MH, de Klerk DJ, Peters GJ (2018) DNA methyltransferases expression in normal tissues and various human cancer cell lines, xenografts and tumors. Nucleosides Nucleotides Nucleic Acids 37(12):696–708. https://doi.org/10.1080/15257770.2018.1498516
Hughes DJ, Marendy EM, Dickerson CA, Yetming KD, Sample CE, Sample JT (2012) Contributions of CTCF and DNA methyltransferases DNMT1 and DNMT3B to Epstein-Barr virus restricted latency. J Virol 86(2):1034–1045. https://doi.org/10.1128/JVI.05923-11
Hutt-Fletcher LM (2007) Epstein-Barr virus entry. J Virol 81(15):7825–7832. https://doi.org/10.1128/JVI.00445-07
Ikeda S, Tagawa H (2021) Impact of hypoxia on the pathogenesis and therapy resistance in multiple myeloma. Cancer Sci 112(10):3995–4004. https://doi.org/10.1111/cas.15087
Jair KW, Bachman KE, Suzuki H, Ting AH, Rhee I, Yen RW, Baylin SB, Schuebel KE (2006) De novo CpG island methylation in human cancer cells. Cancer Res 66(2):682–692. https://doi.org/10.1158/0008-5472.CAN-05-1980
Jin Y, Xie Z, Lu G, Yang S, Shen K (2010) Characterization of variants in the promoter of BZLF1 gene of EBV in nonmalignant EBV-associated diseases in Chinese children. Virol J 7:92. https://doi.org/10.1186/1743-422X-7-92
Jones PA, Baylin SB (2007) The epigenomics of cancer. Cell 128(4):683–692. https://doi.org/10.1016/j.cell.2007.01.029
Jones PA, Issa JP, Baylin S (2016) Targeting the cancer epigenome for therapy. Nat Rev Genet 17(10):630–641. https://doi.org/10.1038/nrg.2016.93
Kalla M, Göbel C, Hammerschmidt W (2012) The lytic phase of epstein-barr virus requires a viral genome with 5-methylcytosine residues in CpG sites. J Virol 86(1):447–458. https://doi.org/10.1128/jvi.06314-11
Kartika AV, Iizasa H, Ding D, Kanehiro Y, Tajima Y, Kaji S, Yanai H, Yoshiyama H (2020) Application of biopsy samples used for helicobacter pylori urease test to predict Epstein-Barr Virus-associated cancer. Microorganisms 8(6) https://doi.org/10.3390/microorganisms8060923
Kim K, Shin DG, Park MK, Baik SH, Kim TH, Kim S, Lee S (2014) Circulating cell-free DNA as a promising biomarker in patients with gastric cancer: diagnostic validity and significant reduction of cfDNA after surgical resection. Ann Surg Treat Res 86(3):136–142. https://doi.org/10.4174/astr.2014.86.3.136
Kis LL, Gerasimcik N, Salamon D, Persson EK, Nagy N, Klein G, Severinson E, Klein E (2011) STAT6 signaling pathway activated by the cytokines IL-4 and IL-13 induces expression of the Epstein-Barr virus-encoded protein LMP-1 in absence of EBNA-2: implications for the type II EBV latent gene expression in Hodgkin lymphoma. Blood 117(1):165–174. https://doi.org/10.1182/blood-2010-01-265272
Kosari-Monfared M, Nikbakhsh N, Fattahi S, Ghadami E, Ranaei M, Taheri H, Amjadi-Moheb F, Godazandeh GA, Shafaei S, Pilehchian-Langroudi M, Samadani AA, Akhavan-Niaki H (2019) CTNNBIP1 downregulation is associated with tumor grade and viral infections in gastric adenocarcinoma. J Cell Physiol 234(3):2895–2904. https://doi.org/10.1002/jcp.27106
Kwok H, Chiang AK (2016) From Conventional to Next Generation Sequencing of Epstein-Barr Virus Genomes. Viruses 8(3):60. https://doi.org/10.3390/v8030060
Lai J, Fu Y, Tian S, Huang S, Luo X, Lin L, Zhang X, Wang H, Lin Z, Zhao H, Lin S, Zhao J, Xu S, Li D, Cai S, Dong L, Qian J, Liang J, Li Q, Zhang Y, Fan J, Balderas R, Chen Q (2021) Zebularine elevates STING expression and enhances cGAMP cancer immunotherapy in mice. Mol Ther 29(5):1758–1771. https://doi.org/10.1016/j.ymthe.2021.02.005
Lam WKJ, Jiang P, Chan KCA, Peng W, Shang H, Heung MMS, Cheng SH, Zhang H, Tse OYO, Raghupathy R, Ma BBY, Hui EP, Chan ATC, Woo JKS, Chiu RWK, Lo YMD (2019) Methylation analysis of plasma DNA informs etiologies of Epstein-Barr virus-associated diseases. Nat Commun 10(1):3256. https://doi.org/10.1038/s41467-019-11226-5
Leong MML, Lung ML (2021) The impact of Epstein-Barr Virus infection on epigenetic regulation of host cell gene expression in epithelial and lymphocytic malignancies. Front Oncol 11:629780. https://doi.org/10.3389/fonc.2021.629780
Li SS, Yang S, Yin DH, Li P (2016) Inhibition of PI3K-Akt pathway reverses LMP1 induced TRAIL resistance in nasopharyngeal carcinoma cell. Lin Chung Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 30(9):744–747. https://doi.org/10.13201/j.issn.1001-1781.2016.09.019
Li C, Romero-Masters JC, Huebner S, Ohashi M, Hayes M, Bristol JA, Nelson SE, Eichelberg MR, Van Sciver N, Ranheim EA, Scott RS, Johannsen EC, Kenney SC (2020a) EBNA2-deleted Epstein-Barr virus (EBV) isolate, P3HR1, causes Hodgkin-like lymphomas and diffuse large B cell lymphomas with type II and Wp-restricted latency types in humanized mice. PLoS Pathog 16(6):e1008590. https://doi.org/10.1371/journal.ppat.1008590
Li W, He C, Wu J, Yang D, Yi W (2020b) Epstein barr virus encodes miRNAs to assist host immune escape. J Cancer 11(8):2091–2100. https://doi.org/10.7150/jca.42498
Li W, Okabe A, Usui G, Fukuyo M, Matsusaka K, Rahmutulla B, Mano Y, Hoshii T, Funata S, Hiura N, Fukayama M, Tan P, Ushiku T, Kaneda A (2021) Activation of EHF via STAT3 phosphorylation by LMP2A in Epstein-Barr virus-positive gastric cancer. Cancer Sci 112(8):3349–3362. https://doi.org/10.1111/cas.14978
Liang Q, Yao X, Tang S, Zhang J, Yau TO, Li X, Tang CM, Kang W, Lung RW, Li JW, Chan TF, Xing R, Lu Y, Lo KW, Wong N, To KF, Yu C, Chan FK, Sung JJ, Yu J (2014) Integrative identification of Epstein-Barr virus-associated mutations and epigenetic alterations in gastric cancer. Gastroenterology 147(6):1350-62e4. https://doi.org/10.1053/j.gastro.2014.08.036
Lin Z, Deng L, Ji J, Cheng C, Wan X, Jiang R, Tang J, Zhuo H, Sun B, Chen Y (2016) S100A4 hypomethylation affects epithelial-mesenchymal transition partially induced by LMP2A in nasopharyngeal carcinoma. Mol Carcinog 55(10):1467–1476. https://doi.org/10.1002/mc.22389
Ling PD, Rawlins DR, Hayward SD (1993) The Epstein-Barr virus immortalizing protein EBNA-2 is targeted to DNA by a cellular enhancer-binding protein. Proc Natl Acad Sci U S A 90(20):9237–9241. https://doi.org/10.1073/pnas.90.20.9237
Liu Z, Lin H, Gan Y, Cui C, Zhang B, Gu L, Zhou J, Zhu G, Deng D (2019) P16 methylation leads to paclitaxel resistance of advanced non-small cell lung cancer. J Cancer 10(7):1726–1733. https://doi.org/10.7150/jca.26482
Liu T, Yao Q, Jin H (2021) Plasma circulating tumor DNA sequencing predicts minimal residual disease in resectable esophageal squamous cell carcinoma. Front Oncol 11:616209. https://doi.org/10.3389/fonc.2021.616209
Lu F, Weidmer A, Liu CG, Volinia S, Croce CM, Lieberman PM (2008) Epstein-Barr virus-induced miR-155 attenuates NF-kappaB signaling and stabilizes latent virus persistence. J Virol 82(21):10436–10443. https://doi.org/10.1128/jvi.00752-08
Lu F, Wiedmer A, Martin KA, Wickramasinghe P, Kossenkov AV, Lieberman PM (2017) Coordinate regulation of TET2 and EBNA2 controls the DNA methylation state of latent Epstein-Barr Virus. J Virol 91(20) https://doi.org/10.1128/jvi.00804-17
Ma SD, Hegde S, Young KH, Sullivan R, Rajesh D, Zhou Y, Jankowska-Gan E, Burlingham WJ, Sun X, Gulley ML, Tang W, Gumperz JE, Kenney SC (2011) A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J Virol 85(1):165–177. https://doi.org/10.1128/jvi.01512-10
McKenzie J, El-Guindy A (2015) Epstein-Barr Virus lytic cycle reactivation. Curr Top Microbiol Immunol 391:237–261. https://doi.org/10.1007/978-3-319-22834-1_8
Moore LD, Le T, Fan G (2013) DNA methylation and its basic function. Neuropsychopharmacol official publication Am Coll Neuropsychopharmacol 38(1):23–38. https://doi.org/10.1038/npp.2012.112
Navari M, Etebari M, De Falco G, Ambrosio MR, Gibellini D, Leoncini L, Piccaluga PP (2015) The presence of Epstein-Barr virus significantly impacts the transcriptional profile in immunodeficiency-associated Burkitt lymphoma. Front Microbiol 6:556. https://doi.org/10.3389/fmicb.2015.00556
Niller HH, Wolf H, Minarovits J (2009) Epigenetic dysregulation of the host cell genome in Epstein-Barr virus-associated neoplasia. Semin Cancer Biol 19(3):158–164. https://doi.org/10.1016/j.semcancer.2009.02.012
Niller HH, Banati F, Salamon D, Minarovits J (2016) Epigenetic alterations in Epstein-Barr Virus-associated diseases. Adv Exp Med Biol 879:39–69. https://doi.org/10.1007/978-3-319-24738-0_3
Nkosi D, Sun L, Duke LC, Patel N, Surapaneni SK, Singh M, Meckes DG, Jr. (2020) Epstein-Barr Virus LMP1 promotes syntenin-1- and Hrs-induced extracellular vesicle formation for its own secretion to increase cell proliferation and migration 11(3) doi:https://doi.org/10.1128/mBio.00589-20
Odumade OA, Hogquist KA, Balfour HH Jr (2011) Progress and problems in understanding and managing primary Epstein-Barr virus infections. Clin Microbiol Rev 24(1):193–209. https://doi.org/10.1128/CMR.00044-10
Ok CY, Li L, Young KH (2015) EBV-driven B-cell lymphoproliferative disorders: from biology, classification and differential diagnosis to clinical management. Exp Mol Med 47(1):e132. https://doi.org/10.1038/emm.2014.82
Okabe A, Funata S, Matsusaka K, Namba H, Fukuyo M, Rahmutulla B, Oshima M, Iwama A, Fukayama M, Kaneda A (2017) Regulation of tumour related genes by dynamic epigenetic alteration at enhancer regions in gastric epithelial cells infected by Epstein-Barr virus. Sci Rep 7(1):7924. https://doi.org/10.1038/s41598-017-08370-7
Pan FP, Zhou HK, Bu HQ, Chen ZQ, Zhang H, Xu LP, Tang J, Yu QJ, Chu YQ, Pan J, Fei Y, Lin SZ, Liu DL, Chen L (2016) Emodin enhances the demethylation by 5-Aza-CdR of pancreatic cancer cell tumor-suppressor genes P16, RASSF1A and ppENK. Oncol Rep 35(4):1941–1949. https://doi.org/10.3892/or.2016.4554
Pandey S, Robertson ES (2018) Oncogenic Epstein-Barr virus recruits Nm23-H1 to regulate chromatin modifiers. Lab Invest J Techn methods apathology 98(2):258–268. https://doi.org/10.1038/labinvest.2017.112
Paschos K, Smith P, Anderton E, Middeldorp JM, White RE, Allday MJ (2009) Epstein-barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene Bim. PLoS Pathog 5(6):e1000492. https://doi.org/10.1371/journal.ppat.1000492
Paulson EJ, Speck SH (1999) Differential methylation of Epstein-Barr virus latency promoters facilitates viral persistence in healthy seropositive individuals. J Virol 73(12):9959–9968. https://doi.org/10.1128/JVI.73.12.9959-9968.1999
Paulson EJ, Fingeroth JD, Yates JL, Speck SH (2002) Methylation of the EBV genome and establishment of restricted latency in low-passage EBV-infected 293 epithelial cells. Virology 299(1):109–121. https://doi.org/10.1006/viro.2002.1457
Peng H, Chen Y, Gong P, Cai L, Lyu X, Jiang Q, Wang J, Lu J, Yao K, Liu K, Li J, Li X (2016) Higher methylation intensity induced by EBV LMP1 via NF-κB/DNMT3b signaling contributes to silencing of PTEN gene. Oncotarget 7(26):40025–40037. https://doi.org/10.18632/oncotarget.9474
Pierard V, Guiguen A, Colin L, Wijmeersch G, Vanhulle C, Van Driessche B, Dekoninck A, Blazkova J, Cardona C, Merimi M, Vierendeel V, Calomme C, Nguyen TL, Nuttinck M, Twizere JC, Kettmann R, Portetelle D, Burny A, Hirsch I, Rohr O, Van Lint C (2010) DNA cytosine methylation in the bovine leukemia virus promoter is associated with latency in a lymphoma-derived B-cell line: potential involvement of direct inhibition of cAMP-responsive element (CRE)-binding protein/CRE modulator/activation transcription factor binding. J Biol Chem 285(25):19434–19449. https://doi.org/10.1074/jbc.M110.107607
Ponnusamy R, Khatri R, Correia PB, Wood CD, Mancini EJ, Farrell PJ, West MJ (2019) Increased association between Epstein-Barr virus EBNA2 from type 2 strains and the transcriptional repressor BS69 restricts EBNA2 activity. PLoS Pathog 15(7):e1007458. https://doi.org/10.1371/journal.ppat.1007458
Pulecio J, Verma N, Mejía-Ramírez E, Huangfu D, Raya A (2017) CRISPR/Cas9-based engineering of the epigenome. Cell Stem Cell 21(4):431–447. https://doi.org/10.1016/j.stem.2017.09.006
Robert A, Pujals A, Favre L, Debernardi J, Wiels J (2020) The BCL-2 family protein inhibitor ABT-737 as an additional tool for the treatment of EBV-associated post-transplant lymphoproliferative disorders. Mol Oncol 14(10):2520–2532. https://doi.org/10.1002/1878-0261.12759
Robertson KD (2000) The role of DNA methylation in modulating Epstein-Barr virus gene expression. Curr Top Microbiol Immunol 249:21–34. https://doi.org/10.1007/978-3-642-59696-4_2
Robertson KD, Ambinder RF (1997) Methylation of the Epstein-Barr virus genome in normal lymphocytes. Blood 90(11):4480–4484
Rowles DL, Tsai YC, Greco TM, Lin AE, Li M, Yeh J, Cristea IM (2015) DNA methyltransferase DNMT3A associates with viral proteins and impacts HSV-1 infection. Proteomics 15(12):1968–1982. https://doi.org/10.1002/pmic.201500035
Saito M, Nishikawa J, Okada T, Morishige A, Sakai K, Nakamura M, Kiyotoki S, Hamabe K, Okamoto T, Oga A, Sasaki K, Suehiro Y, Hinoda Y, Sakaida I (2013) Role of DNA methylation in the development of Epstein-Barr virus-associated gastric carcinoma. J Med Virol 85(1):121–127. https://doi.org/10.1002/jmv.23405
Sakamoto K, Sekizuka T, Uehara T, Hishima T, Mine S, Fukumoto H, Sato Y, Hasegawa H, Kuroda M, Katano H (2017) Next-generation sequencing of miRNAs in clinical samples of Epstein-Barr virus-associated B-cell lymphomas. Cancer Med 6(3):605–618. https://doi.org/10.1002/cam4.1006
Salamon D, Takacs M, Ujvari D, Uhlig J, Wolf H, Minarovits J, Niller HH (2001) Protein-DNA binding and CpG methylation at nucleotide resolution of latency-associated promoters Qp, Cp, and LMP1p of Epstein-Barr virus. J Virol 75(6):2584–2596. https://doi.org/10.1128/JVI.75.6.2584-2596.2001
Schaeffner M, Mrozek-Gorska P, Buschle A, Woellmer A, Tagawa T, Cernilogar FM, Schotta G, Krietenstein N, Lieleg C, Korber P, Hammerschmidt W (2019) BZLF1 interacts with chromatin remodelers promoting escape from latent infections with EBV. Life Sci Alliance 2(2) https://doi.org/10.26508/lsa.201800108
Scott RS (2017) Epstein-Barr virus: a master epigenetic manipulator. Curr Opin Virol 26:74–80. https://doi.org/10.1016/j.coviro.2017.07.017
Sharma S, Kelly TK, Jones PA (2010) Epigenetics in cancer. Carcinogenesis 31(1):27–36. https://doi.org/10.1093/carcin/bgp220
Shi J, Zhang G, Yao D, Liu W, Wang N, Ji M, He N, Shi B, Hou P (2012) Prognostic significance of aberrant gene methylation in gastric cancer. Am J Cancer Res 2(1):116–129
Shi F, Zhou M, Shang L, Du Q, Li Y, Xie L, Liu X, Tang M, Luo X, Fan J, Zhou J, Gao Q, Qiu S, Wu W, Zhang X, Bode AM, Cao Y (2019) EBV(LMP1)-induced metabolic reprogramming inhibits necroptosis through the hypermethylation of the RIP3 promoter. Theranostics 9(9):2424–2438. https://doi.org/10.7150/thno.30941
Stanland LJ, Luftig MA (2020) The role of EBV-induced hypermethylation in gastric cancer tumorigenesis. Viruses 12(11) https://doi.org/10.3390/v12111222
Strong MJ, Xu G, Coco J, Baribault C, Vinay DS, Lacey MR, Strong AL, Lehman TA, Seddon MB, Lin Z, Concha M, Baddoo M, Ferris M, Swan KF, Sullivan DE, Burow ME, Taylor CM, Flemington EK (2013) Differences in gastric carcinoma microenvironment stratify according to EBV infection intensity: implications for possible immune adjuvant therapy. PLoS Pathog 9(5):e1003341. https://doi.org/10.1371/journal.ppat.1003341
Sun X, Li H, Zhu Y, Xu P, Zuo Q, Li B, Gu X (2020) 5-Azacytidine-induced cardiomyocyte differentiation of very small embryonic-like stem cells. Stem Cells Int 2020:5162350. https://doi.org/10.1155/2020/5162350
Takacs M, Banati F, Koroknai A, Segesdi J, Salamon D, Wolf H, Niller HH, Minarovits J (2010) Epigenetic regulation of latent Epstein-Barr virus promoters. Biochim Biophys Acta 1799(3–4):228–235. https://doi.org/10.1016/j.bbagrm.2009.10.005
Tao Q, Robertson KD, Manns A, Hildesheim A, Ambinder RF (1998) Epstein-Barr virus (EBV) in endemic Burkitt’s lymphoma: molecular analysis of primary tumor tissue. Blood 91(4):1373–1381
Tao Q, Huang H, Geiman TM, Lim CY, Fu L, Qiu GH, Robertson KD (2002) Defective de novo methylation of viral and cellular DNA sequences in ICF syndrome cells. Hum Mol Genet 11(18):2091–2102. https://doi.org/10.1093/hmg/11.18.2091
Tempera I, Lieberman PM (2014) Epigenetic regulation of EBV persistence and oncogenesis. Semin Cancer Biol 26:22–29. https://doi.org/10.1016/j.semcancer.2014.01.003
Tempera I, Wiedmer A, Dheekollu J, Lieberman PM (2010) CTCF prevents the epigenetic drift of EBV latency promoter Qp. PLoS Pathog 6(8):e1001048. https://doi.org/10.1371/journal.ppat.1001048
Tierney RJ, Kirby HE, Nagra JK, Desmond J, Bell AI, Rickinson AB (2000) Methylation of transcription factor binding sites in the Epstein-Barr virus latent cycle promoter Wp coincides with promoter down-regulation during virus-induced B-cell transformation. J Virol 74(22):10468–10479. https://doi.org/10.1128/jvi.74.22.10468-10479.2000
Tonoyan L, Chevalier M, Vincent-Bugnas S, Marsault R, Doglio A (2020) Detection of Epstein-Barr Virus in periodontitis: a review of methodological approaches. Microorganisms 9(1) https://doi.org/10.3390/microorganisms9010072
Tsai CN, Tsai CL, Tse KP, Chang HY, Chang YS (2002) The Epstein-Barr virus oncogene product, latent membrane protein 1, induces the downregulation of E-cadherin gene expression via activation of DNA methyltransferases. Proc Natl Acad Sci U S A 99(15):10084–10089. https://doi.org/10.1073/pnas.152059399
Tsai CL, Li HP, Lu YJ, Hsueh C, Liang Y, Chen CL, Tsao SW, Tse KP, Yu JS, Chang YS (2006) Activation of DNA methyltransferase 1 by EBV LMP1 involves c-Jun NH(2)-terminal kinase signaling. Cancer Res 66(24):11668–11676. https://doi.org/10.1158/0008-5472.Can-06-2194
Tsao SW, Tsang CM, Lo KW (2017) Epstein-Barr virus infection and nasopharyngeal carcinoma. Philos Trans R Soc Lond B Biol Sci 372:1732. https://doi.org/10.1098/rstb.2016.0270
Vivekanandan P, Daniel HD, Kannangai R, Martinez-Murillo F, Torbenson M (2010) Hepatitis B virus replication induces methylation of both host and viral DNA. J Virol 84(9):4321–4329. https://doi.org/10.1128/JVI.02280-09
Volk V, Theobald SJ, Danisch S, Khailaie S, Kalbarczyk M, Schneider A, Bialek-Waldmann J, Kronke N, Deng Y, Eiz-Vesper B, Dragon AC, von Kaisenberg C, Lienenklaus S, Bleich A, Keck J, Meyer-Hermann M, Klawonn F, Hammerschmidt W, Delecluse HJ, Munz C, Feuerhake F, Stripecke R (2020) PD-1 blockade aggravates Epstein-Barr Virus(+) post-transplant lymphoproliferative disorder in humanized mice resulting in central nervous system involvement and CD4(+) T cell dysregulations. Front Oncol 10:614876. https://doi.org/10.3389/fonc.2020.614876
Wang J, Liu W, Zhang X, Zhang Y, Xiao H, Luo B (2019) LMP2A induces DNA methylation and expression repression of AQP3 in EBV-associated gastric carcinoma. Virology 534:87–95. https://doi.org/10.1016/j.virol.2019.06.006
Wang Z, Zhang Y, Zhu S, Peng H, Chen Y, Cheng Z, Liu S, Luo Y, Li R, Deng M, Xu Y, Hu G, Chen L, Zhang G (2020) A small molecular compound CC1007 induces cross-lineage differentiation by inhibiting HDAC7 expression and HDAC7/MEF2C interaction in BCR-ABL1(-) pre-B-ALL. Cell Death Dis 11(9):738. https://doi.org/10.1038/s41419-020-02949-1
Weber E, Buzovetsky O, Heston L, Yu KP, Knecht KM, El-Guindy A, Miller G, Xiong Y (2019) A noncanonical basic motif of Epstein-Barr Virus ZEBRA protein facilitates recognition of methylated DNA, high-affinity DNA binding, and lytic activation. J Virol 93(14) https://doi.org/10.1128/jvi.00724-19
Wen J, Zheng T, Hu K, Zhu C, Guo L, Ye G (2017) Promoter methylation of tumor-related genes as a potential biomarker using blood samples for gastric cancer detection. Oncotarget 8(44):77783–77793. https://doi.org/10.18632/oncotarget.20782
Woellmer A, Hammerschmidt W (2013) Epstein-Barr virus and host cell methylation: regulation of latency, replication and virus reactivation. Curr Opin Virol 3(3):260–265. https://doi.org/10.1016/j.coviro.2013.03.005
Woellmer A, Arteaga-Salas JM, Hammerschmidt W (2012) BZLF1 governs CpG-methylated chromatin of Epstein-Barr Virus reversing epigenetic repression. PLoS Pathog 8(9):e1002902. https://doi.org/10.1371/journal.ppat.1002902
Wu J, Tang Q, Yang L, Chen Y, Zheng F, Hann SS (2017) Interplay of DNA methyltransferase 1 and EZH2 through inactivation of Stat3 contributes to beta-elemene-inhibited growth of nasopharyngeal carcinoma cells. Sci Rep 7(1):509. https://doi.org/10.1038/s41598-017-00626-6
Yahia ZA, Adam AA, Elgizouli M, Hussein A, Masri MA, Kamal M, Mohamed HS, Alzaki K, Elhassan AM, Hamad K, Ibrahim ME (2014) Epstein Barr virus: a prime candidate of breast cancer aetiology in Sudanese patients. Infectious Agents and Cancer 9(1):9. https://doi.org/10.1186/1750-9378-9-9
Yang P, Wang Y, Chen J, Li H, Kang L, Zhang Y, Chen S, Zhu B, Gao S (2011) RCOR2 is a subunit of the LSD1 complex that regulates ESC property and substitutes for SOX2 in reprogramming somatic cells to pluripotency. Stem Cells dayton Ohio 29(5):791–801. https://doi.org/10.1002/stem.634
Yang J, Liu Z, Zeng B, Hu G, Gan R (2020) Epstein-Barr virus-associated gastric cancer: a distinct subtype. Cancer Lett 495:191–199. https://doi.org/10.1016/j.canlet.2020.09.019
Yoshioka M, Kikuta H, Ishiguro N, Ma X, Kobayashi K (2003) Unique Epstein-Barr virus (EBV) latent gene expression, EBNA promoter usage and EBNA promoter methylation status in chronic active EBV infection. J Gen Virol 84(Pt 5):1133–1140. https://doi.org/10.1099/vir.0.18777-0
Zhang S, Pei Y, Lang F, Sun K, Singh RK, Lamplugh ZL, Saha A, Robertson ES (2019) EBNA3C facilitates RASSF1A downregulation through ubiquitin-mediated degradation and promoter hypermethylation to drive B-cell proliferation. PLoS Pathog 15(1):e1007514. https://doi.org/10.1371/journal.ppat.1007514
Zhang Q, Luo D, Xie Z, He H, Duan Z (2020) The oncogenic role of miR-BART19-3p in Epstein-Barr Virus-associated diseases. Biomed Res Int 2020:5217039. https://doi.org/10.1155/2020/5217039
Zhang Y, Hu S, Li J, Shi D, Luo B (2021) The promoter aberrant methylation status of TMEM130 is associated with gastric cancer. Digestive and liver disease : official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver doi:https://doi.org/10.1016/j.dld.2021.05.035
Zheng XH, Wang RZ, Li XZ, Zhou T, Zhang JB, Zhang PF, Lu LX, Jia WH (2020) Detection of methylation status of Epstein-Barr virus DNA C promoter in the diagnosis of nasopharyngeal carcinoma. Cancer Sci 111(2):592–600. https://doi.org/10.1111/cas.14281
Zhou C, Xie Z, Gao L, Liu C, Ai J, Zhang L, Shen K (2015) Profiling of EBV-encoded microRNAs in EBV-associated hemophagocytic lymphohistiocytosis. Tohoku J Exp Med 237(2):117–126. https://doi.org/10.1620/tjem.237.117
Funding
ZX was supported by the CAMS Innovation Fund for Medical Sciences (CIFMS) (2019-I2M-5-026); RW was supported by the National Natural Science Foundation of China (82002130), Beijing Natural Science Foundation, China (7222059), and the Open Research Fund Program of the State Key Laboratory of Virology of China (2021IOV002).
Author information
Authors and Affiliations
Contributions
LZ drafted the manuscript. RW and ZX revised and edited the manuscript. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Conflict of interests
The authors declare that they have no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, L., Wang, R. & Xie, Z. The roles of DNA methylation on the promotor of the Epstein–Barr virus (EBV) gene and the genome in patients with EBV-associated diseases. Appl Microbiol Biotechnol 106, 4413–4426 (2022). https://doi.org/10.1007/s00253-022-12029-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-022-12029-3