
Abstract
Many cases of avian influenza A(H7N9) virus infection in humans have been reported since its first emergence in 2013. The disease is of concern because most patients have become severely ill with roughly 30% mortality rate. Because the threat in public health caused by H7N9 virus remains high, advance preparedness is essentially needed. In this study, the recombinant H7N9 hemagglutinin (HA) was expressed in insect cells and purified for generation of two monoclonal antibodies, named F3-2 and 1C6B. F3-2 can only recognize the H7N9 HA without having cross-reactivity with HA proteins of H1N1, H3N2, H5N1, and H7N7. 1C6B has the similar specificity with F3-2, but 1C6B can also bind to H7N7 HA. The binding epitope of F3-2 is mainly located in the region of H7N9 HA(299–307). The binding epitope of 1C6B is located in the region of H7N9 HA(489–506). F3-2 and 1C6B could not effectively inhibit the hemagglutination activity of H7N9 HA. However, F3-2 can prevent H7N9 HA from trypsin cleavage and can bind to H7N9 HA which has undergone pH-induced conformational change. F3-2 also has the ability of binding to H7N9 viral particles and inhibiting H7N9 virus infection to MDCK cells with the IC50 value of 22.18 μg/mL. In addition, F3-2 and 1C6B were utilized for comprising a lateral flow immunochromatographic test strip for specific detection of H7N9 HA.
Key points
• Two mouse monoclonal antibodies, F3-2 and 1C6B, were generated for recognizing the novel binding epitopes in H7N9 HA.
• F3-2 can prevent H7N9 HA from trypsin cleavage and inhibit H7N9 virus infection to MDCK cells.
• F3-2 and 1C6B were developed as a lateral flow immunochromatographic test for specific detection of H7N9 HA.
Similar content being viewed by others

Introduction
In February 2013, China first reported human infection with a novel avian influenza A(H7N9) virus (Chen et al. 2013), which is more suitable for infecting humans (Dortmans et al. 2013). Since then, H7N9 infections continue to occur, and most human infections are mainly through contact with infected poultry or exposure to contaminated environments. Among critically ill patients infected with the H7N9 virus, 20% died with acute respiratory distress syndrome or multiple organ failure (Li et al. 2014). If an H7N9 infection can be diagnosed early, effective treatment can be applied by using neuraminidase inhibitors, such as Oseltamivir and Zanamivir (Chang et al. 2013).
Hemagglutinin (HA) is a surface glycoprotein of influenza virus. Its main function is to mediate infection with target cells. HA can recognize the sialic acid at the end of the receptor (Matrosovich et al. 2004), and when the bound HA is at a low pH value, a conformational change will occur to help the virus outer membrane and the endosomal membrane fuse (Stegmann 2000). In addition, HA can make red blood cells form agglutination, called hemagglutination. In the host cell, the precursor of the HA protein, called HA0, needs to be cleaved by protease to form two subunits, HA1 and HA2. It is noted that HA1 and HA2 are still assembled together by disulfide bonds (Mair et al. 2014). This cleavage event observed on HA may vary according to the number of basic amino acids contained in the cleavage sites. The efficiency of HA0 cleavage also limits the ability of the virus to spread to different tissues of the host (Steinhauer 1999). In the highly pathogenic H5 and H7 subtypes of avian influenza viruses (AIVs), the cleavage site usually contains multiple basic amino acid residues, such as multiple arginine or lysine residues. Proteases such as subtilisin-like proteases, furin, and PC6 usually cleave and activate HA0 (Bottcher et al. 2006; Horimoto and Kawaoka 2005).
The receptor-binding domain of HA is located at the top of the globular head domain of HA1 (Skehel and Wiley 2000). The transmembrane domain of HA is located at the bottom of HA2, and this structure is very important for stabilizing the HA trimer immobilized on the outer membrane of the virus (Mair et al. 2014). Therefore, the N-terminus of HA2 is speculated to play a very important function during membrane fusion (Wiley and Skehel 1987). After the influenza virus binds to the host cell receptor, the viral genome will be delivered into the cell through membrane fusion. However, there are kinetic energy barriers during membrane fusion, but influenza viruses can overcome this barrier through the irreversible conformational change of HA (Huang et al. 2002; Huang et al. 2003; Xu and Wilson 2011).
The three-dimensional structure of avian influenza A(H7N9) virus HA has been resolved, and its structure is similar to the HA of the highly pathogenic AIV H7N7 (A/Netherlands/219/2003) (Xu et al. 2013; Yang et al. 2012). Although the Q226 site in the H7 subtype is very conserved (Kageyama et al. 2013), the Q226L mutation on the novel H7N9 virus further greatly increases the ability of the virus to bind to the human sialic acid receptor (Barman et al. 2017). Therefore, the novel H7N9 virus has a stronger ability to infect humans than other H7 viruses (Dortmans et al. 2013). Notably, the HA Q226L mutation of H7N9 virus is similar with that of the H2N2(1957) and H3N2(1968) pandemic influenza viruses (Barman et al. 2017). Variation at this site is also considered to be highly correlated with human-to-human transmission (Imai and Kawaoka 2012).
Current evidence indicates that the novel H7N9 virus does not have the ability to continuously spread from human to human, but the H7N9 virus may possibly mutate and cause a pandemic (Chen et al. 2013; Dortmans et al. 2013). It is worried that H7N9 mutations will be more adapted to human-to-human transmission, leading to cause a sudden pandemic. Therefore, the development of vaccines and diagnostic reagents for prevention of H7N9 pandemic is of great importance. So far, there are limited studies on neutralizing monoclonal antibodies (mAbs) targeting to the antigenic epitopes of H7N9 HA which are away from the receptor-binding sites or stalk domain (Henry Dunand et al. 2016; Krammer et al. 2014; Tharakaraman et al. 2015; Yang et al. 2020). In the present study, two mouse mAbs, F3-2 and 1C6B, targeting to the recombinant H7N9 HA protein (H7N9 rHA) were isolated by the conventional hybridoma technology. Interestingly, F3-2 and 1C6B can specifically bind to H7N9 rHA through recognition of two novel antigenic epitopes which are located in the connection region between HA1 and HA2 and the upstream region of the transmembrane domain. The functionality of F3-2 and 1C6B in trypsinization prevention, hemagglutination inhibition, and blocking neutralization of virus infection were also characterized. In addition, F3-2 and 1C6B were utilized to develop a lateral flow immunochromatographic test (ICT) strip for specific detection of H7N9 rHA.
Materials and methods
The gene sequence of H7N9 HA
The cDNA sequence encoding for the HA protein of A/Taiwan/1/2013(H7N9) is identical to the cDNA sequence of NCBI accession number AGL43637. H7N9 rHA1 is the fragment of the amino acid residues 1–320 of H7N9 HA. H7N9 rHA2 is the fragment of the amino acid residues 321–506 of H7N9 HA. H7N9 rHA is the fragment of the amino acid residues 18–524 of H7N9 HA without containing the N-terminal signal peptide, the C-terminal transmembrane domain, and the cytoplasmic tail.
Expression and purification of the H7N9 rHA
The baculovirus expression vector pFastBac HTA-HA(H7N9) or pFastBac HTA-HA1(H7N9) was transformed into DH10bac competent cells for preparation of the bacmid. The purified bacmid was subsequently transfected into Sf21 insect cells by using the Cellfectin II reagent according to the instruction of the Bac-to-Bac baculovirus expression system (Thermo Fisher Scientific Inc.). Sf21 insect cells were infected by the baculovirus with a multiplicity of infection (MOI) of 1 pfu/cell. The culture media were collected by centrifugation (20,000×g) for 10 min at 4°C. H7N9 rHA was purified by using a HisTrap HP column (GE Healthcare) pre-equilibrated with a binding buffer (20 mM sodium phosphate, 20 mM imidazole, 0.5 M NaCl, pH 7.4), and bound proteins were eluted with a linear gradient of 20–500 mM imidazole in 20 mM sodium phosphate and 0.5 M NaCl (pH 7.4). Protein purity was examined by SDS-PAGE, and concentration was determined by the Bradford dye-binding method (Bradford 1976).
Preparation of mouse monoclonal antibody
For generation of the mouse mAb against H7N9 rHA, two BALB/c male mice were immunized through intraperitoneal injection one time with a mixture of 50 μg purified H7N9 rHA (for generation of 1C6B) or rHA1 (for generation of F3-2) and complete Freund’s adjuvant (Sigma-Aldrich, Product Number F5881) and three booster injections in a 2-week interval with a mixture of 25 μg purified H7N9 rHA or rHA1 and incomplete Freund’s adjuvant (Sigma-Aldrich, Product Number F5506) followed by a final booster injection of 25 μg purified H7N9 rHA or rHA1 in phosphate buffered saline (PBS). The procedures for screening of hybridoma were performed as described previously (Chen et al. 2002). Briefly, Sp2/0-Ag14 cells (ATCC CRL-1581) were mixed with mouse splenocytes derived from the mouse immunized with the purified H7N9 rHA or rHA1 in a 1:5 ratio of cell numbers. The 0.7 mL of polyethylene glycol 1500 (Sigma-Aldrich, USA) was added to the cell mixture and incubated in a 37°C water bath for 2 min with gentle shaking. The additional 10 mL of Dulbecco’s Modified Eagle Medium (DMEM) was then added to the cell mixture within 4 min. After centrifugation, cells were collected and resuspended in 30 mL of DMEM containing 15% fetal bovine serum, 1% penicillin-streptomycin, 1 mM sodium pyruvate (Thermo Scientific, USA), and HAT Media Supplement (Sigma-Aldrich, USA). Fusion cells (0.1 mL) were aliquoted into each wells of 96-well cell culture plates and grown at 37°C in the 5% CO2 incubator. The 50 μL of HT Media Supplement (Sigma-Aldrich, USA) was added to each wells of 96-well cell culture plates on day 7 after cell fusion. The culture media were collected on day 14 for analysis of the target antibodies by ELISA using 100 ng of purified H7N9 rHA as the antigen. The hybridoma cells which can produce the antibodies against H7N9 rHA were further subcloned into a monoclonal antibody by limiting dilution method.
Purification of mouse monoclonal antibody
The hybridoma cell culture media were collected by centrifugation at 6000×g for 10 min. The supernatant was filtered with 0.45-μm membrane disc and then loaded on HiTrap Protein G HP column (GE Healthcare) pre-equilibrated with PBS. Bound mAbs were eluted with 0.1 M glycine-HCl (pH 3.0) and mixed with the neutralization buffer (1 M Tris-HCl, pH 9.0). The purified antibody samples were loaded on the PD-10 desalting column (GE Healthcare) pre-equilibrated with PBS for exchanging buffer. Antibody purity was examined by SDS-PAGE, and concentration was determined by the Bradford dye-binding method using mouse IgG as the standard.
Recombinant HA proteins of various influenza viruses
The recombinant HA proteins of A/Puerto Rico/8/1934(H1N1), A/California/07/2009(H1N1), A/Victoria/361/2011(H3N2), A/Hong Kong/483/97(H5N1), A/chicken/Netherlands/1/03(H7N7), and A/Shanghai/1/2013(H7N9) were purchased from Sino Biological Inc. (Catalog Number: 11684-V08B, 11085-V08B, 40145-V08B, 11689-V08B, 11212-V08B, and 40104-V08H, respectively).
Site-directed mutagenesis
All of the pGEX-4T-3 plasmids for expression of the H7N9 rHA mutants in Escherichia coli (E. coli) were generated by the PCR-based QuikChange II Site-Directed Mutagenesis Kit (Stratagene, Agilent Technologies) according to the manufacturer’s instruction. All mutations were confirmed by DNA sequencing. All of the plasmids utilized in the study will be provided upon request.
Expression of the GST-tagged truncated H7N9 HA fragments
The cDNAs encoding for the truncated H7N9 HA fragments or mutants were cloned into the pGEX-4T-3 vector and transformed into E. coli BL21(DE3) competent cells. The protein expression procedure was conducted as described previously (Shin et al. 2011) with slight modifications. Briefly, E. coli BL21(DE3) cells were cultured in LB medium with ampicillin (50 μg/mL) and incubated at 37°C on an orbital shaker at 150 rpm. Expression of the recombinant GST-tagged truncated H7N9 HA fragments was induced at an A600 of 0.6–0.7 growing condition by adding IPTG to a final concentration of 1 mM for 4 h. Cells were collected by centrifugation at 6000×g for 10 min. The cell pellet was washed three times with PBS and then subjected to SDS-PAGE and Western blotting.
Measurements of antibody affinity by ELISA
The approximate affinity of mAb against H7N9 HA was determined using an indirect ELISA method. Generally, 100 ng of purified H7N9 rHA was coated on the bottom of the 96-well plate for 1 h at room temperature. The plate was blocked with 1% BSA in PBS for 1 h at room temperature. Subsequently, a series of two-fold dilutions (2000–12,800×) of mAbs were added to each well of the plate to incubate with H7N9 rHA for 1 h at room temperature. The 96-well plate was washed three times with PBS containing 0.05% Tween 20 (PBST). Next, the horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG was added to each well of the plate, followed by incubation at room temperature for 1 h. Finally, 100 μL of 3,3’,5,5’-tetramethylbenzidine (TMB) substrate (BD Bioscience, USA) was added to each well of the plate for signal detection. Absorbance at 450 nm was measured and recorded by ELISA reader.
pH-induced conformational change ELISA
The procedure was conducted as described previously (Tan et al. 2012) with slight modifications. Briefly, 96-well EIA plates were coated with 0.1 mL of purified H7N9 rHA (10 μg/mL) for 2 h at room temperature and then blocked with 200 μL of gelatin-PBST buffer. After washing with PBST buffer three times, 100 μL of TPCK-treated trypsin (2.5 ng/mL) was added to activate H7N9 rHA for 30 min at 37°C. Subsequently, 0.2 mL of citrate buffer (adjusted pH to 7.4, 6, or 5, respectively) with 150 mM NaCl was added and then incubated for 1 h at room temperature. To test whether mAb can bind with the H7N9 rHA which had undergone conformational change, 0.1 mL of mAb solution was added to conduct a standard ELISA as described above.
Inhibition of trypsin cleavage of HA by mAb
The procedure was adapted from the protocol described previously (Kallewaard et al. 2016) with slight modifications. The purified H7N9 rHA0 (10 μg) was incubated with 5 μg or 10 μg of F3-2 for 1 h at room temperature and then incubated with TPCK-treated trypsin (100 ng) for 10 min at 37°C. The reaction was terminated by adding SDS-PAGE sample buffer into sample mixtures and heated at 95°C for 20 min. Samples were analyzed on SDS-PAGE and stained with coomassie brilliant blue R-250.
Hemagglutination test and hemagglutination inhibition (HI) test
Hemagglutination tests were performed with standard protocols provided by WHO. Briefly, the hemagglutination activity of the purified H7N9 rHA was tested against 1% (v/v) chicken red blood cells (RBCs), and hemagglutination titers were determined as the highest dilution exhibiting complete hemagglutination (Hu et al. 2017). For conducting HI test, 25 μL of 4 chicken erythrocyte hemagglutination units of purified H7N9 rHA was pre-incubated with 25 μL of different dilutions of F3-2 (1 mg/mL) or 1C6B (1 mg/mL) in a well of a V-bottomed plate for 1 h at room temperature. Twenty-five microliters of 1% (v/v) of chicken RBC suspension was then added to the rHA and mAb mixture, gently shaken, and incubated at room temperature for an additional 40 min. PBS was used as a negative control to replace the mAb for mixing with H7N9 rHA and chicken RBC suspension.
Microneutralization assay
The avian influenza A(H7N9) virus vaccine strain was obtained from the Taiwan Centers for Disease Control (Approval Number: BI1060066). Viruses were propagated in SPF chicken embryonated eggs, and virus titers were determined by mean 50% tissue culture infective dose (TCID50) per milliliter. The microneutralization assay was performed as described previously (Kallewaard et al. 2016). Briefly, MDCK cells were seeded in 96-well plates at the concentration of 2 × 104 cells/well. Two-fold serially diluted antibody solutions (starting from 50 μg/mL to 1.5625 μg/mL) were mixed with an equal volume of virus inoculums (100 TCID50) in the complete DMEM medium containing with 0.75 μg/mL trypsin, followed by 1 h of incubation at 37°C. After incubation, the mixture was added to MDCK monolayers. Cells were cultured for 72 h before the examination of cytopathic effect (CPE). The absence of CPE in individual wells was defined as protection. The assay was performed in six repeats. PBS was used as the negative control. The 50% inhibitory concentration (IC50) of the neutralizing antibody was calculated using the Quest Graph™ IC50 Calculator (AAT Bioquest, Inc.).
Conjugation of mAb with colloidal gold
The mAb-colloidal gold conjugate was prepared as described previously (Shim et al. 2007) with slight modifications. Briefly, 1 mL of colloidal gold (diameter 40 nm) was adjusted to pH 8.0 and mixed with 15 μg of the purified mAb F3-2, followed by an additional 30 min of stirring with 150 μL of 10% (w/v) bovine serum albumin (BSA) for 1 h at room temperature to block the excess reactivity of colloidal gold. After centrifugation at 10,000 rpm for 15 min, the F3-2-colloidal gold pellet was washed with 2 mM borate buffer containing 0.2% PEG-8000 (pH 9.0) twice and then resuspended in 1 mL of 2 mM borate buffer containing 0.2% PEG-8000 and 10% sucrose (pH 8.0).
Preparation and operation of ICT for H7N9 HA detection
The ICT is composed of four main materials, including sample pad, gold pad, nitrocellulose membrane, and absorbent pad (Excelsior Bio-System, Inc., Taipei, Taiwan). The sample pad and gold pad were first treated with 20 mM phosphate buffer containing 1% BSA, 0.5% Tween 20, 0.05% sodium azide, and 5% sucrose at pH 7.4 and subsequently air dried at 37°C. Then, 12 μL of the F3-2 gold conjugate was loaded on the gold pad. Subsequently, 4.5 μg of 1C6B solution or 7 μg of goat anti-mouse IgG (H+L) solution (KPL, USA) was loaded on the NC membrane, serving as the test line or the control line, respectively. Upon use, the test sample was added on the sample pad to start the ICT. The results were interpreted within 10 min.
Results
Specificity and epitope mapping of mAb F3-2
To examine whether F3-2 can only recognize H7N9 HA, the HA proteins of H1N1, H3N2, H5N1, and H7N9 were analyzed on SDS-PAGE and transferred to PVDF membranes for Western blotting. The results showed that F3-2 can only recognize H7N9 HA and H7N9 HA1 proteins (Fig. 1, lanes 1, 2, and 7) without having any cross-reactivity with HAs derived from other subtypes. To further determine the epitope sequence recognized by F3-2, several truncated H7N9 HA fragments with an N-terminal GST tag (as illustrated in Fig. 2a) were generated for Western blotting with F3-2. The results showed that F3-2 can bind to HA(1–320), HA(246–320), and HA(148–320) (Fig. 2d), indicating that the binding epitope of F3-2 is located in the region of residues 246–320. The amino acid sequences of HA(256–320) was aligned with the corresponding sequences of H7N7 HA, revealing that residues 261, 289, and 303 are not conserved in these two HAs (Fig. 3a). The residues 261, 289, and 303 of H7N9 HA were mutated to the corresponding residues of H7N7 HA and further subjected to Western blotting with F3-2. The result showed that the mutation of R303E can inhibit F3-2 binding, indicating that R303 of H7N9 HA was involved in interacting with F3-2 (Fig. 3b). The alanine substitution experiment by the single mutation of residues 299–307 of HA1 to alanine was also applied for further identifying the important residues of H7N9 HA1 recognized by F3-2. To our surprise, the R303A did not inhibit F3-2 binding, whereas Q302A, L305A, and L306A markedly inhibited F3-2 binding (Fig. 3e). Moreover, the residues 299–307 deletion mutant of H7N9 HA1 cannot be recognized by F3-2 (Fig. 3e), indicating that the binding epitope of F3-2 is mainly located in the region of residues 299–307 of H7N9 HA1. In addition, the R303K did not inhibit F3-2 binding, whereas R303E greatly inhibited F3-2 binding (Fig. 3e), suggesting that a positively charged side chain of residue 303 of H7N9 HA1 is also important for F3-2 binding.

mAb F3-2 specifically recognizes H7N9 HA. The recombinant HA proteins of H1N1, H3N2, H5N1, H7N7, and H7N9 were analyzed on SDS-PAGE and subjected to Western blotting with mAb F3-2 (a) and amido black staining (b). Molecular weight standards were marked with small ticks. The number labeled on the left of the tick is the indicated molecular weight in kDa unit. Lane 1, the recombinant HA protein of A/Taiwan/1/2013 (H7N9). Lane 2, the recombinant HA1 protein of A/Taiwan/1/2013 (H7N9). Lane 3, the recombinant HA protein of A/Puerto Rico/8/1934 (H1N1). Lane 4, the recombinant HA protein of A/California/07/2009 (H1N1). Lane 5, the recombinant HA protein of A/Victoria/361/2011 (H3N2). Lane 6, the recombinant HA protein of A/Hong Kong/483/97 (H5N1). Lane 7, the recombinant HA protein of A/Shanghai/1/2013 (H7N9). Lane 8, the recombinant HA protein of A/chicken/Netherlands/1/03 (H7N7). WB, Western blotting

The epitope mapping analysis of mAb F3-2. a The schematic diagram illustrates the truncated H7N9 HA fragments utilized in the experiment. The N-terminal GST tag is not shown in the diagram. The HA1 regions are shown in dark gray. The HA2 regions are shown in light gray. The GST-tagged truncated H7N9 HA fragments as indicated were analyzed on SDS-PAGE and then subjected to coomassie staining (b), or Western blotting with anti-GST antibody (c) or mAb F3-2 (d). WB, Western blotting

The Q302, R303, L305, and L306 of H7N9 HA are critical residues for specific recognition by mAb F3-2. a The sequences of residues 256–311 of H7N9 and H7N9 HAs are aligned. The majority of the sequences in the aligned regions is identical among H7N9 and H7N7 HAs despite that residues 261, 289, and 303 are different. b The residues 261, 289, and 303 of H7N9 HA are substituted by the corresponding residues of H7N7 to generate the H7N9 HA mutants for conducting Western blotting with mAb F3-2. The alanine substitution method was performed to determine the critical residues which were involved in interacting with mAb F3-2. A series of GST-tagged HA1 mutants as indicated were analyzed on SDS-PAGE and then subjected to coomassie staining (c) and Western blotting with anti-GST antibody (d) or mAb F3-2 (e). WB, Western blotting
Specificity and epitope mapping of mAb 1C6B
To characterize the binding specificity of 1C6B, the HA proteins of H1N1, H3N2, H5N1, and H7N9 were analyzed on SDS-PAGE and transferred to PVDF membranes for Western blotting with 1C6B. The result showed that 1C6B can only recognize H7N9 HA and H7N7 HA without having any cross-reactivity with HAs derived from other subtypes (Fig. 4a, lanes 1, 7, and 8). To further determine the binding epitope recognized by 1C6B, several truncated H7N9 HA fragments with an N-terminal GST tag (as illustrated in Fig. 5a) were generated for Western blotting with 1C6B. The results showed that 1C6B can bind to HA(321–506), HA(388–506), HA(454–506), and HA(489–506) (Fig. 5c), indicating that residues 489–506 of H7N9 HA are the major region recognized by 1C6B. By comparing the amino acid sequences of residues 489–506 of H7N9 HA with other H7-subtuype HAs in the NCBI database, it was found that the indicated sequences are highly conserved among H7-subtuype HAs. Therefore, it was concluded that 1C6B can be applied for specific detection of the H7-subtype influenza viruses.

mAb 1C6B specifically recognizes H7N9 HA and H7N7 HA. The recombinant HA proteins of H1N1, H3N2, H5N1, H7N7, and H7N9 were analyzed on SDS-PAGE and then subjected to amido black staining (bottom panel), or Western blotting with mAb 1C6B (top panel). Lane 1, the recombinant HA protein of A/Taiwan/1/2013 (H7N9). Lane 2, the recombinant HA1 protein of A/Taiwan/1/2013 (H7N9). Lane 3, the recombinant HA protein of A/Puerto Rico/8/1934 (H1N1). Lane 4, the recombinant HA protein of A/California/07/2009 (H1N1). Lane 5, the recombinant HA protein of A/Victoria/361/2011 (H3N2). Lane 6, the recombinant HA protein of A/Hong Kong/483/97 (H5N1). Lane 7, the recombinant HA protein of A/Shanghai/1/2013 (H7N9). Lane 8, the recombinant HA protein of A/chicken/Netherlands/1/03 (H7N7). WB, Western blotting

The major binding epitope of mAb 1C6B is located in the region of residues 489–506. a The schematic diagram illustrated the construction of the truncated HA fragments with an N-terminal GST tag (not shown in the diagram). The HA1 regions are shown in dark gray. The HA2 regions are shown in light gray. The GST-tagged HA fragments as indicated were analyzed on SDS-PAGE and then subjected to coomassie staining (b), or Western blotting with anti-GST antibody (c) or mAb 1C6B (d). WB, Western blotting
F3-2 and 1C6B can bind to the pH-induced conformationally changed H7N9 rHA
During the influenza virus infection to host cells, HA will undergo the configurational change to mediate the membrane fusion. To test whether F3-2 and 1C6B can bind to the pH-induced conformationally changed H7N9 rHA, the H7N9 rHA samples pre-treated with trypsin and low pH buffer were coated on the bottom of an EIA plate for performing the ELISA experiment. The ELISA results showed that F3-2 even more preferred to bind to the pH 5-induced conformationally changed H7N9 rHA than the samples in the pH 6 and pH 7.4 assay conditions (Fig. 6a). In contrast, 1C6B can bind to the H7N9 rHAs in the pH 5, pH 6, and pH 7.4 assay conditions without having significant difference (Fig. 6b). Furthermore, it was found that the ELISA experiments using F3-2 as the primary antibody can obtain larger absorbances at 450 nm than the data using 1C6B as the primary antibody, suggesting that F3-2 has higher affinity against H7N9 rHA than does 1C6B (Fig. 6).

mAb F3-2 and mAb 1C6B can bind to the pH-induced conformationally changed H7N9 rHA. The 96-well EIA plates were coated with 0.1 mL of purified H7N9 rHA (10 μg/mL). After blocking with BSA and washing with PBST, 100 μL of TPCK-treated trypsin (2.5 ng/mL) was added to activate H7N9 rHA. Subsequently, 0.2 mL of citrate buffer (adjusted pH to 7.4, 6, or 5, respectively) was added to the wells to trigger H7N9 HA undergoing conformational change. The ELISA using serial dilutions of mAb F3-2 or mAb 1C6B solutions as the primary antibodies was conducted to determine the binding efficiency by measuring the absorbance at 450 nm
F3-2 can protect H7N9 rHA0 from trypsin cleavage
The full-length intact HA, termed HA0, needs to be cleaved by trypsin-like protease to turn into a functional HA by formation of two interconnected subunits of HA1 and HA2. To examine whether F3-2 can protect H7N9 rHA0 from trypsin cleavage, the H7N9 rHA0 was firstly pre-incubated with F3-2 and then subjected to trypsinization for mimicking the cleavage process. The experimental results showed that 10 μg of H7N9 rHA0 was completely cleaved by trypsin in the absence of F3-2 (Fig. 7, lane 2). However, when 5 μg of F3-2 was pre-incubated with H7N9 HA0 and then subjected to trypsinization, HA1 and HA2 subunits were observed (Fig. 7, lane 3). Moreover, when 10 μg of F3-2 was utilized in the experiment, more HA0 can be clearly observed (Fig. 7, lane 4), indicating that F3-2 has the ability to protect H7N9 rHA0 from trypsin cleavage. However, 1C6B was not able to protect H7N9 rHA0 from trypsin cleavage (Fig. 7, lanes 5 and 6).

mAb F3-2 can protect H7N9 HA from trypsinization. The purified H7N9 rHA0 (10 μg) was incubated with 5 μg or 10 μg of mAb F3-2 or 1C6B at room temperature for 1 h and then incubated with TPCK-treated trypsin (100 ng) for 10 min at 37°C. The reaction was terminated by adding SDS-PAGE sample buffer into sample mixtures and heated at 95°C for 20 min. Samples were analyzed on SDS-PAGE and subjected to coomassie staining. HC, antibody heavy chain. LC, antibody light chain
F3-2 has slight hemagglutination inhibition activity
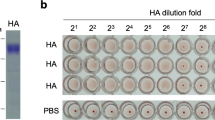
Although the binding epitope of F3-2 or 1C6B is not located in the head domain of H7N9 rHA, the HI test was still conducted to analyze whether F3-2 or 1C6B can have hemagglutination inhibition activity. It is not surprised that 1C6B does not have any hemagglutination inhibition activity (Fig. 8, rows 1–3). In contrast, F3-2 has slight hemagglutination inhibition activity, but its ability of inhibiting the hemagglutination function of H7N9 rHA was gone while the antibody dilution fold was larger than 23. It was concluded that the hemagglutination inhibition activity was not significant (Fig. 8, rows 4–6).

Analysis of the hemagglutination inhibition activity of mAb F3-2 and mAb 1C6B by HI test. Twenty-five microliters of mAb F3-2 or mAb 1C6B (1 mg/mL) was subjected to two-fold serial dilution (starting from 21 to 212) and then mixed with 25 μL of H7N9 rHA (2 μg/mL), followed by mixing with 25 μL of 1% (v/v) chicken RBCs. After incubation at room temperature for 40 min, the hemagglutination phenomena were observed. In the control experiments, mAb solutions were replaced by PBS
F3-2 can bind to H7N9 viral particles and inhibit H7N9 virus infection to MDCK cells
Since F3-2 can protect H7N9 rHA0 from trypsin cleavage and has slight hemagglutination inhibition activity, it is expected that F3-2 may have the ability to inhibit H7N9 virus infection to host cells even though its binding epitope is not located in the receptor-binding domain of H7N9 HA. Therefore, H7N9 viral particles were utilized in the ELISA experiments to examine whether F3-2 can bind to H7N9 viral particles. The results clearly showed that the absorbance at 450 nm was increased along with the higher concentration of F3-2 and was utilized in the experiments, indicating that F3-2 can efficiently bind to H7N9 viral particles (Fig. 9a). In contrast, 1C6B does not have the ability for binding to H7N9 viral particles (Fig. 9a). Furthermore, the results of microneutralization assay showed that PBS and 1C6B cannot inhibit H7N9 virus infection to MDCK cells, therefore the H7N9 viruses were produced normally, leading to strong hemagglutination phenomena in the experiments (Fig. 9b, right panel). However, when 50 μg/mL or 25 μg/mL of F3-2 was pre-incubated with H7N9 viruses in the microneutralization assay, the hemagglutination inhibition phenomena were clearly observed (Fig. 9b, left panel, rows 1 and 2), indicating that the H7N9 viruses produced by MDCK cells was markedly reduced. Additionally, the more diluted concentration of F3-2 (12.5, 6.25, 3.125, and 1.5625 μg/mL) did not show neutralizing activity (Fig. 9b, left panel, rows 3–6). The IC50 of the neutralizing antibody was calculated using the Quest Graph™ IC50 Calculator (AAT Bioquest, Inc.). The IC50 value of F3-2 for neutralizing H7N9 virus infection to MDCK cells is 22.18 μg/mL.

Analysis of the capability of mAb F3-2 and mAb 1C6B for binding to H7N9 viral particles and inhibiting H7N9 virus infection to MDCK cells. a mAb F3-2 can bind to the H7N9 viral particles. The H7N9 viral particles (100 ng/well) were coated on the 96-well plate for performing the ELISA experiments with mAb F3-2 or mAb 1C6B (0.01, 0.1, 1, or 10 μg/mL). Black bars represent the ELISA results (absorbance 450 nm) of mAb F3-2. Gray bars represent the ELISA results of mAb 1C6B. b H7N9 virus (100 TCID50) was mixed with two-fold serially diluted antibody solutions of mAb F3-2 (left panel) or mAb 1C6B (right panel) solutions (starting from 50 to 1.5625 μg/mL) in six wells of a 96-well plate containing complete DMEM medium and 0.75 μg/mL trypsin. After incubation at 37°C for 1 h, the mixture was added to confluent MDCK monolayers. Cells were cultured for 72 h before the examination of the absence of CPE in individual wells. The absence of CPE in individual wells was defined as protection. The assay was performed in six repeats. PBS was used as the negative control
Development of ICT with F3-2 and 1C6B for detection of H7N9 rHA
The design of the ICT strip was illustrated in a schematic diagram (Fig. 10a). The ICT experiments started from adding PBS, H7N9 rHA, H1N1 rHA, or H7N7 rHA on the sample pad, respectively. The results showed that only a strong signal was observed in the test line when H7N9 rHA was loaded on the sample pad (Fig. 10b). In contrast, there is not any detection signal observed when H1N1 rHA or H7N7 rHA was loaded on the sample pad (Fig. 10b), indicating that the ICT strip composed of F3-2 and 1C6B has very high specificity. Thus, the combination of F3-2 and 1C6B can be applied for specific detection of H7N9 HA in the sample. Furthermore, the detection signal was not observed when H7N9 viral particles were tested in the experiment (Fig. 10b). Recall that the previous data also showed that 1C6B cannot bind to the H7N9 viral particles in the ELISA experiments (Fig. 9a).

The lateral flow ICT for detection of H7N9 rHA. a The schematic diagram illustrates the design of the ICT strip. mAb F3-2 was conjugated with colloidal gold particles and loaded on the gold pad. mAb 1C6B was loaded on the test line. Goat anti-mouse IgG antibody was loaded on the control line. b The assay was conducted by adding PBS, H7N9 rHA, H1N1 rHA, H7N7 rHA, or H7N9 viral particles on the sample pad to start the ICT. The results were interpreted within 10 min. The colloidal gold particles were seen in all of the control lines, indicating that all of the assays were valid. However, the colloidal gold particles can only be seen on the test line of the ICT strip while using H7N9 rHA as the test sample
Discussion
Since 2013, many cases of AIV H7N9 infection in humans have been reported, making the previously unconcerned low pathogenic AIV H7 subtype once again aroused public attention. The present study showed that both of mAb 1C6B and mAb F3-2 are highly specific to H7 HA, especially F3-2 only recognizes H7N9 HA (Fig. 1a). The epitope mapping experiments showed that F3-2 recognizes residues Q302, L305, and L306 of H7N9 HA (Fig. 3e). Compared the amino acid sequences of residues 296–314 of H7N9 HA with other H7-subtuype HAs in the NCBI database, Q302 and L305 are very conservative residues; most of the residue 306 are leucine and a few are methionine. However, the residue 303 of H7N2 and H7N7 HA is E, and only H7N9 is mainly R. Therefore, if F3-2 is utilized in a diagnostic kit, it can be applied for specific detection of the novel H7N9 influenza virus. As a result, our ICT strip using F3-2 and 1C6B as the capture antibodies can specifically distinguish H7N9 rHA and H7N7 rHA (Fig. 10b). Unfortunately, since 1C6B cannot bind to H7N9 viral particles (Fig. 9a), the ICT strip composed of F3-2 and 1C6B cannot be applied for detecting natural H7N9 viral particles (Fig. 10b). It is also demonstrated that the ICT strip can provide very high specificity for discriminating H7N9 virus from other viral subtypes, but the sensitivity of the ICT strip may vary depending on what kinds of samples are loaded on the sample pad. If the sample is viral particles, the ICT strip will get an invalid result, leading to a false negative conclusion possibly.
Previous studies mentioned that the antibody targeting to the HA stalk region cannot inhibit hemagglutination and lacks the ability to directly neutralize the virus (DiLillo et al. 2016; Henry Dunand et al. 2016). Since the binding epitopes of F3-2 and 1C6B are not located in the receptor-binding site of H7N9 HA, it is expected that these two mAbs may not be able to form a steric obstacle to inhibit the hemagglutination activity of HA. The HI test demonstrated that only F3-2 has slight hemagglutination inhibition activity and 1C6B has no function in hemagglutination inhibition (Fig. 8). To our surprise, F3-2 performed neutralizing activity for inhibiting H7N9 virus infection to MDCK cells in the microneutralization assay (Fig. 9). Thus, we proposed that when the connection region between HA1 and HA2 was bound with F3-2, the C-terminus of HA1 and the N-terminus of HA2 may be trapped together, and the protease cleavage site before the fusion peptide will be hindered for not being able to trigger the conformational change and the subsequent membrane fusion process. Moreover, the capability of F3-2 or 1C6B for binding to conformationally changed H7N9 rHA was not affected under the assay condition of pH 5 (Fig. 6a). In fact, F3-2 even more preferred to bind to H7N9 rHA which has undergone the conformational change (Fig. 6a), implying that the binding epitope of F3-2 on H7N9 rHA is probably more accessible at low pH.
Several studies on characterization of mAbs which can efficiently neutralize H7N9 virus have been reported. The major targeting epitopes of these mAbs are located at the receptor-binding site or a conserved helical region at the stalk domain. Two human mAbs, HNIgGD5 and HNIgGH8, possessed high neutralizing activity against H7N9, depending on two residues in the receptor-binding site at positions V186 and L226 of HA (Wang et al. 2015). Two murine mAbs, 1H10 and 2D1, have been identified to have therapeutic and prophylactic efficacy against the highly pathogenic H7N9 strain in mouse passive transfer-viral challenge experiments. The binding epitopes of 1H10 and 2D1 are located at the residues 128–153 of HA1 head domain (Yang et al. 2020). VIS410, a broadly neutralizing human mAb that binds the HA stem of influenza A viruses, is effective against H7N9 (Tharakaraman et al. 2015). A broadly neutralizing antibody, MEDI8852 (Kallewaard et al. 2016), targeting the stalk domain of HA, can effectively inhibit the cleavage of HA by protease (Kallewaard et al. 2016) and was also effective for prophylaxis and treatment of H7N9 and H5N1 infection in mice (Paules et al. 2017). In the study, F3-2 can protect H7N9 rHA from trypsinization and further prevent HA from being trimmed into HA1 and HA2 (Fig. 7). The novel F3-2 binding epitope is located in the connection region between HA1 head domain and HA2 stalk domain, instead of locating in the receptor-binding domain. Interestingly, F3-2 has slight hemagglutination inhibition activity and can inhibit H7N9 virus infection to MDCK cells with the IC50 value of 22.18 μg/mL. Notably, the murine neutralizing antibody 1H10 can neutralize A/Zhejiang/DTID-ZJU01/2013(H7N9) and A/Guangdong/HP001/2017(H7N9) with the IC50 values of 0.15 μg/mL and 0.075 μg/mL, respectively (Yang et al. 2020). Therefore, the neutralizing antibody, such as F3-2, targeting to the epitope away from the receptor-binding domain of H7N9 HA seems to have higher IC50 value.
The functionality and the mechanism for F3-2 to inhibit hemagglutination and block H7N9 virus infection might be different from the neutralizing antibody which binds to the receptor-binding domain of HA. Although F3-2 does not bind to the receptor-binding domain of H7N9 HA, its binding epitope is still located in the C-terminal region of the HA head domain. Thus, we hypothesize that the special orientation of the complex of F3-2 antibody molecule and H7N9 HA may cause certain steric hindrance to inhibit the formation of hemagglutination. Furthermore, since the H7N9 viruses utilized in the neutralization experiments have been previously activated by trypsin to process HA0 into HA1 and HA2 in the culture medium, and F3-2 also performed better ability for binding to H7N9 HA which has undergone a conformational change, we speculate that the mechanism for F3-2 to inhibit virus infection might function through blocking the membrane fusion between the virus and the host cell. These unique functions of F3-2 might reinforce it to inhibit the conformational change of HA and the subsequent membrane fusion. This study paved a new road for designing potent vaccines and developing neutralizing antibodies against H7N9 or other influenza virus subtypes.
Data availability
All data associated with this study are included in the paper. All plasmids generated in the study are available from the corresponding author on reasonable request.
References
Barman S, Krylov PS, Turner JC, Franks J, Webster RG, Husain M, Webby RJ (2017) Manipulation of neuraminidase packaging signals and hemagglutinin residues improves the growth of A/Anhui/1/2013 (H7N9) influenza vaccine virus yield in eggs. Vaccine 35(10):1424–1430
Bottcher E, Matrosovich T, Beyerle M, Klenk HD, Garten W, Matrosovich M (2006) Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol 80(19):9896–9898
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chang SY, Lin PH, Tsai JC, Hung CC, Chang SC (2013) The first case of H7N9 influenza in Taiwan. Lancet 381(9878):1621
Chen HM, Chang SC, Wu CC, Cuo TS, Wu JS, Juang RH (2002) Regulation of the catalytic behaviour of L-form starch phosphorylase from sweet potato roots by proteolysis. Physiol Plant 114(4):506–515
Chen Y, Liang W, Yang S, Wu N, Gao H, Sheng J, Yao H, Wo J, Fang Q, Cui D, Li Y, Yao X, Zhang Y, Wu H, Zheng S, Diao H, Xia S, Zhang Y, Chan KH, Tsoi HW, Teng JL, Song W, Wang P, Lau SY, Zheng M, Chan JF, To KK, Chen H, Li L, Yuen KY (2013) Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: clinical analysis and characterisation of viral genome. Lancet 381(9881):1916–1925
DiLillo DJ, Palese P, Wilson PC, Ravetch JV (2016) Broadly neutralizing anti-influenza antibodies require Fc receptor engagement for in vivo protection. J Clin Invest 126(2):605–610
Dortmans JC, Dekkers J, Wickramasinghe IN, Verheije MH, Rottier PJ, van Kuppeveld FJ, de Vries E, de Haan CA (2013) Adaptation of novel H7N9 influenza A virus to human receptors. Sci Rep 3:3058
Henry Dunand CJ, Leon PE, Huang M, Choi A, Chromikova V, Ho IY, Tan GS, Cruz J, Hirsh A, Zheng NY, Mullarkey CE, Ennis FA, Terajima M, Treanor JJ, Topham DJ, Subbarao K, Palese P, Krammer F, Wilson PC (2016) Both neutralizing and non-neutralizing human H7N9 influenza vaccine-induced monoclonal antibodies confer protection. Cell Host Microbe 19(6):800–813
Horimoto T, Kawaoka Y (2005) Influenza: lessons from past pandemics, warnings from current incidents. Nat Rev Microbiol 3(8):591–600
Hu CJ, Chien CY, Liu MT, Fang ZS, Chang SY, Juang RH, Chang SC, Chen HW (2017) Multi-antigen avian influenza A (H7N9) virus-like particles: particulate characterizations and immunogenicity evaluation in murine and avian models. BMC Biotechnol 17(1):2
Huang Q, Opitz R, Knapp EW, Herrmann A (2002) Protonation and stability of the globular domain of influenza virus hemagglutinin. Biophys J 82(2):1050–1058
Huang Q, Sivaramakrishna RP, Ludwig K, Korte T, Bottcher C, Herrmann A (2003) Early steps of the conformational change of influenza virus hemagglutinin to a fusion active state: stability and energetics of the hemagglutinin. Biochim Biophys Acta 1614(1):3–13
Imai M, Kawaoka Y (2012) The role of receptor binding specificity in interspecies transmission of influenza viruses. Curr Opin Virol 2(2):160–167
Kageyama T, Fujisaki S, Takashita E, Xu H, Yamada S, Uchida Y, Neumann G, Saito T, Kawaoka Y, Tashiro M (2013) Genetic analysis of novel avian A(H7N9) influenza viruses isolated from patients in China, February to April 2013. Euro Surveill 18(15):20453
Kallewaard NL, Corti D, Collins PJ, Neu U, McAuliffe JM, Benjamin E, Wachter-Rosati L, Palmer-Hill FJ, Yuan AQ, Walker PA, Vorlaender MK, Bianchi S, Guarino B, De Marco A, Vanzetta F, Agatic G, Foglierini M, Pinna D, Fernandez-Rodriguez B, Fruehwirth A, Silacci C, Ogrodowicz RW, Martin SR, Sallusto F, Suzich JA, Lanzavecchia A, Zhu Q, Gamblin SJ, Skehel JJ (2016) Structure and function analysis of an antibody recognizing all influenza A subtypes. Cell 166(3):596–608
Krammer F, Jul-Larsen A, Margine I, Hirsh A, Sjursen H, Zambon M, Cox RJ (2014) An H7N1 influenza virus vaccine induces broadly reactive antibody responses against H7N9 in humans. Clin Vaccine Immunol 21(8):1153–1163
Li Q, Zhou L, Zhou M, Chen Z, Li F, Wu H, Xiang N, Chen E, Tang F, Wang D, Meng L, Hong Z, Tu W, Cao Y, Li L, Ding F, Liu B, Wang M, Xie R, Gao R, Li X, Bai T, Zou S, He J, Hu J, Xu Y, Chai C, Wang S, Gao Y, Jin L, Zhang Y, Luo H, Yu H, He J, Li Q, Wang X, Gao L, Pang X, Liu G, Yan Y, Yuan H, Shu Y, Yang W, Wang Y, Wu F, Uyeki TM, Feng Z (2014) Epidemiology of human infections with avian influenza A(H7N9) virus in China. N Engl J Med 370(6):520–532
Mair CM, Ludwig K, Herrmann A, Sieben C (2014) Receptor binding and pH stability — how influenza A virus hemagglutinin affects host-specific virus infection. Biochim Biophys Acta 1838(4):1153–1168
Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk HD (2004) Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc Natl Acad Sci U S A 101(13):4620–4624
Paules CI, Lakdawala S, McAuliffe JM, Paskel M, Vogel L, Kallewaard NL, Zhu Q, Subbarao K (2017) The hemagglutinin A stem antibody MEDI8852 prevents and controls disease and limits transmission of pandemic influenza viruses. J Infect Dis 216(3):356–365
Shim WB, Yang ZY, Kim JS, Kim JY, Kang SJ, Woo GJ, Chung YC, Eremin SA, Chung DH (2007) Development of immunochromatography strip-test using nanocolloidal gold-antibody probe for the rapid detection of aflatoxin B1 in grain and feed samples. J Microbiol Biotechnol 17(10):1629–1637
Shin YC, Tang SJ, Chen JH, Liao PH, Chang SC (2011) The molecular determinants of NEDD8 specific recognition by human SENP8. PLoS One 6(11):e27742
Skehel JJ, Wiley DC (2000) Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69:531–569
Stegmann T (2000) Membrane fusion mechanisms: the influenza hemagglutinin paradigm and its implications for intracellular fusion. Traffic 1(8):598–604
Steinhauer DA (1999) Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology 258(1):1–20
Tan GS, Krammer F, Eggink D, Kongchanagul A, Moran TM, Palese P (2012) A pan-H1 anti-hemagglutinin monoclonal antibody with potent broad-spectrum efficacy in vivo. J Virol 86(11):6179–6188
Tharakaraman K, Subramanian V, Viswanathan K, Sloan S, Yen HL, Barnard DL, Leung YH, Szretter KJ, Koch TJ, Delaney JC, Babcock GJ, Wogan GN, Sasisekharan R, Shriver Z (2015) A broadly neutralizing human monoclonal antibody is effective against H7N9. Proc Natl Acad Sci U S A 112(35):10890–10895
Wang J, Chen Z, Bao L, Zhang W, Xue Y, Pang X, Zhang X, Qin C, Jin Q (2015) Characterization of two human monoclonal antibodies neutralizing influenza A H7N9 viruses. J Virol 89(17):9115–9118
Wiley DC, Skehel JJ (1987) The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu Rev Biochem 56:365–394
Xu R, Wilson IA (2011) Structural characterization of an early fusion intermediate of influenza virus hemagglutinin. J Virol 85(10):5172–5182
Xu R, de Vries RP, Zhu X, Nycholat CM, McBride R, Yu W, Paulson JC, Wilson IA (2013) Preferential recognition of avian-like receptors in human influenza A H7N9 viruses. Science 342(6163):1230–1235
Yang H, Carney PJ, Donis RO, Stevens J (2012) Structure and receptor complexes of the hemagglutinin from a highly pathogenic H7N7 influenza virus. J Virol 86(16):8645–8652
Yang F, Xiao Y, Lu R, Chen B, Liu F, Wang L, Yao H, Wu N, Wu H (2020) Generation of neutralizing and non-neutralizing monoclonal antibodies against H7N9 influenza virus. Emerg Microbes Infect 9(1):664–675
Acknowledgements
The authors are thankful to the excellent technical assistance from Technology Commons, College of Life Science, National Taiwan University.
Funding
This work was supported by grants from the Ministry of Science and Technology, Taiwan (MOST103-2321-B-002-061, MOST104-2321-B-002-019, MOST107-2313-B-002-045, and MOST108-2313-B-002-011).
Author information
Authors and Affiliations
Contributions
YWC, CJL, HYS, HWC, and SCC conceived and designed the study. HWC and SCC acquired funds and conducted experiments. YWC, CJL, HYS, KTH, and CWW performed the experiments. YWC, CJL, HYS, HWC, and SCC analyzed the data and wrote the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval
The animal experiment was approved by the Institutional Animal Care and Use Committee (IACUC) of National Taiwan University (IACUC Approval Numbers: NTU102-EL-93, NTU107-EL-00023, and NTU107-EL-00211) and implemented in accordance with the animal care and ethics guidelines. The article does not contain any studies with human participants performed by any of the authors.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yi-Wei Chiang, Chia-Jung Li, and Heng-Yi Su are joint first authors.
Rights and permissions
About this article

Cite this article
Chiang, YW., Li, CJ., Su, HY. et al. Development of mouse monoclonal antibody for detecting hemagglutinin of avian influenza A(H7N9) virus and preventing virus infection. Appl Microbiol Biotechnol 105, 3235–3248 (2021). https://doi.org/10.1007/s00253-021-11253-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-021-11253-7