
Abstract
Pentacyclic triterpenes are diverse plant secondary metabolites derived from the mevalonate (MVA) pathway. Many of these molecules are potentially valuable, particularly as pharmaceuticals, and research has focused on their production in simpler and more amenable heterologous systems such as the yeast Saccharomyces cerevisiae. We have developed a new heterologous platform for the production of pentacyclic triterpenes in S. cerevisiae based on a combinatorial engineering strategy involving the overexpression of MVA pathway genes, the knockout of negative regulators, and the suppression of a competing pathway. Accordingly, we overexpressed S. cerevisiae ERG13, encoding 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) synthase, and a truncated and deregulated variant of the rate-limiting enzyme HMG-CoA reductase 1 (tHMGR). In the same engineering step, we deleted the ROX1 gene, encoding a negative regulator of the MVA pathway and sterol biosynthesis, resulting in a push-and-pull strategy to enhance metabolic flux through the system. In a second step, we redirected this enhanced metabolic flux from late sterol biosynthesis to the production of 2,3-oxidosqualene, the direct precursor of pentacyclic triterpenes. In yeast cells transformed with a newly isolated sequence encoding lupeol synthase from the Russian dandelion (Taraxacum koksaghyz), we increased the yield of pentacyclic triterpenes by 127-fold and detected not only high levels of lupeol but also a second valuable pentacyclic triterpene product, β-amyrin.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Isoprenoids are a diverse group of natural compounds found in all living organisms, with at least 50,000 different structures already reported (Hemmerlin et al. 2012; Liao et al. 2016). In plants, these products are derived from the plastididial 2C-methyl-d-erythritol 4-phosphate (MEP) and the cytosolic mevalonate (MVA) pathway. In the latter, acetyl-CoA is converted to the isoprenoid precursor isopentenyl diphosphate (IPP) via six enzymatic steps. Two important MVA pathway enzymes are the sequentially acting 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) synthase (HMGS) and HMG-CoA reductase (HMGR), the latter representing the rate-limiting step (Demierre et al. 2005). IPP is isomerized to form dimethylallyl pyrophosphate (DMAPP), and together, IPP and DMAPP can act as substrates for various isoprenoid-derived pathways. For example, two molecules of IPP and one of DMAPP can be converted into farnesyl pyrophosphate (FPP) which in turn can be converted into squalene by squalene synthase (SQS). The oxidized form of squalene (2,3-oxidosqualene) is a precursor for the synthesis of sterols (leading to the production of lanosterol in fungi and animals, or cycloartenol in plants) and also pentacyclic triterpenes (Fig. 1a), the latter involving various oxidosqualene cyclases (OSCs) such as lupeol synthase in the dandelion Taraxacum officinale and β-amyrin synthase in the wormwood plant Artemisia annua (Shibuya et al. 1999; Kirby et al. 2008). The products of these enzymes can be further metabolized by acylation or oxidation. The efficient triterpene oxidation of, e.g., lupeol to betulin and betulinic acid by P450 enzymes could be demonstrated in yeast (Zhou et al. 2016). FPP is also a precursor for the synthesis of sesquiterpenes, e.g., farnesene or amorpha-4,11-diene, a precursor of the anti-malarial drug artemisinin (Martin et al. 2003).

Triterpene accumulation in the yeast S. cerevisiae expressing TkLUP. a Schematic representation of the MVA pathway leading to the synthesis of sterols and pentacyclic triterpenes via oxidosqualene cyclases (OSCs). Dashed arrows represent multiple enzymatic reactions. AACT = acetyl-CoA C-acetyltransferase; DMAPP = dimethylallyl pyrophosphate; IPP = isopentenyl diphosphate; HMGS = 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) synthase; HMGR = HMG-CoA reductase; SQS = squalene synthase; SQE = squalene epoxidase. b Schematic representation of the TkLUP coding sequence under the control of the GAL1 promoter (PGAL1) and CYC1 terminator (TCYC1). c Yeast cells carrying the TkLUP coding sequence showed two additional peaks in the GC-MS spectrum (m/z = 218, arrows), probably representing β-amyrin (retention time = 17.95 min) and lupeol (retention time = 18.25 min) because they match the corresponding standards. d Yeasts carrying the TkLUP coding sequence accumulated 0.16 mg/g CDW of the putative lupeol but the quantification of the β-amyrin peak was not possible. Wild-type (WT) and pAG424_PGAL1-ccdb vector control CEN.PK2-1C cells served as controls. The standard deviation was calculated from n = 3 individual transformants; CDW = cell dry weight
The value of isoprenoids, particularly as pharmaceuticals, has prompted the development of heterologous production systems including the yeast Saccharomyces cerevisiae (reviewed by Liao et al. 2016 and Vickers et al. 2017). The potential of the yeast MVA pathway for the production of isoprenoids was first demonstrated by overexpressing the catalytic domain of HMGR (tHMGR), which increased the yield of squalene (Donald et al. 1997). The consequences of overexpressing other MVA pathway genes were determined by combinatorial library screening for the overexpression of ERG10 (acetoacetyl CoA thiolase; AACT), ERG13 (HMGS), and ERG12 (mevalonate kinase) which enhanced the production of amorpha-4,11-diene (Yuan and Ching 2014). The MVA pathway has also been targeted using the CRISPR/Cas9 system, revealing loci that trigger the accumulation of mevalonate and triterpenes when knocked out (Jakočiūnas et al. 2015; Arendt et al. 2017). The targets included ROX1, encoding a transcriptional regulator that inhibits genes involved in the MVA pathway and sterol biosynthesis (Henry et al. 2002; Montañés et al. 2011; Özaydin et al. 2013; Jakočiūnas et al. 2015). The insertion of regulated promoters into the yeast genome can also suppress genes involved in endogenous but competitive isoprenoid pathways to redirect the metabolic flux in a more precise and desirable manner. Accordingly, the methionine sensitive MET3 promoter was used to downregulate the expression of the lanosterol synthase gene (ERG7), which represents the first committed step in the late sterol biosynthesis pathway, enhancing the production of β-amyrin (Kirby et al. 2008). A similar strategy was used to suppress the squalene synthase gene (ERG9), allowing the enhanced production of artemisinin (Ro et al. 2006; Westfall et al. 2012) until the authors changed their strategy and used the copper transporter 3 (CTR3) promoter, which allows transcriptional repression to be induced by adding CuSO4 to the medium, thus reducing the costs of industrial processes (Paddon et al. 2013).
Here, we developed a new yeast-based platform for the synthesis of triterpenes, combining the overexpression of the MVA pathway genes ERG13 (HMGS) and a truncated version of HMG1 (tHMGR), the disruption of the ROX1 gene, and the copper-regulated repression of ERG7 using the CTR3 promoter. By implementing this platform, we were able to enhance the productivity of the MVA pathway and redirect metabolic flux from late sterol biosynthesis, resulting in a 127-fold increase in the yield of lupeol using lupeol synthase from the Russian dandelion Taraxacum koksaghyz (TkLUP) as a model enzyme.
Materials and methods
Cloning of constructs
The construct pAG424_PGAL1-TkLUP was prepared by amplifying the TkLUP coding sequence (GenBank MG646375) from T. koksaghyz cDNA using forward primer 5′-AAA GTC GAC TAA AAA AAT GTG GAA GCT GAA AAT AGC-3′ and reverse primer 5′-AAA CTC GAG ATA TAT TTT GAA CAA TAC GA-3′ (restriction sites are underlined). The PCR product was purified, digested, and inserted into pENTR3c (Invitrogen, Carlsbad, USA). The TkLUP coding sequence was then introduced into pAG424_PGAL1-ccdB (Alberti et al. 2007; Addgene, Cambridge, USA) by LR recombination.
The construct pESC-rox1-KlURA3_tHMGR/ERG13 was generated by amplifying the ROX1 coding sequence from S. cerevisiae genomic DNA using forward primer 5′-AAA GCG GCC GCA TGA ATC CTA AAT CCT CTAC-3′ and reverse primer 5′-AAA GCG GCC GCT CAT TTC GGA GAA ACT AGG-3′ (restriction sites are underlined). Furthermore, pESC-URA (Agilent Technologies, Santa Clara, USA) was used as a template to amplify a pESC-URA vector backbone containing NotI restriction sites using forward primer 5′-AAA GCG GCC GCC CAG CTG CAT TAA TGA ATC G-3′ and reverse primer 5′-AAA GCG GCC GCG AAG TTC CTA TTC TCT AGA AA-3′ (restriction sites are underlined). After digestion with NotI, the restriction fragments were ligated to obtain pESC-rox1. This vector was digested with BglII and a synthetic DNA fragment (Invitrogen), consisting of a KlURA3 marker cassette (Gueldener et al. 2002), and an AsiSI/Nb.BsmI uracil-specific excision reaction (USER) cassette (Hansen et al. 2011) was inserted to obtain pESC-rox1-KlURA3. In a parallel approach, the coding sequences of the truncated HMG1 gene (tHMGR) and ERG13 were amplified from S. cerevisiae genomic DNA using forward primer 5′-AAA GGA TCC AAA AAA ATG GTT TTA ACC AAT AAA AC-3′ and reverse primer 5′-AAA GTC GAC TTA GGA TTT AAT GCA GGT GAC-3′ for tHMGR and forward primer 5′-AAA GAA TTC AAA AAA ATG AAA CTC TCA ACT AAA CTT TG-3′ and reverse primer 5′-AAA GCG GCC GCT TAT TTT TTA ACA TCG TAA GAT C-3′ for ERG13 (restriction sites are underlined). The PCR products were digested with BamHI/SalI and EcoRI/NotI, and ligated into pESC-URA to generate pESC-URA-tHMGR/ERG13. The NotI restriction site was removed using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer’s protocol. In the final step, the expression cassette, containing the tHMGR and ERG13 coding sequences, as well as the bidirectional GAL1/GAL10 promoter and the ADH1/CYC1 terminators, was amplified using forward primer 5′-CGT GCG AUT CAG AGC GAC CTC ATG CTA TAC-3′ and reverse primer 5′-CAC GCG AUC TTC GAG CGT CCC AAA ACC-3′ (uracil base for USER cloning shown in bold) and was introduced into pESC-rox1-KlURA3 (digested with AsiSI) according to the USER protocol.
The construct pESC_PERG7_KlLEU2_PCTR3_erg7 was generated in a three-step process. In the first step, an ERG7 fragment was amplified from S. cerevisiae genomic DNA using forward primer 5′-CAC ATT TAA GGG CTA TAC AAA GAT GAC AGA ATT TTA TTC TGA CA-3′ and reverse primer 5′-AAA GCG GCC GCC CCA ATA AAC GTA AGA TTA CA-3′, and a CTR3 promoter fragment was amplified using forward primer 5′-AAA GCG GCC GCC AGC TGA AGG ATC CGG TAT TCC AAT GAG AAT CGC-3′ and reverse primer 5′-TGT CAG AAT AAA ATT CTG TCA TCT TTG TAT AGC CCT TAA ATG T-3′ (italic letters indicate the overlapping region; restriction sites are underlined). The products were fused by overlapping PCR using the CTR3 promoter forward primer and the ERG7 reverse primer. The spliced product was digested with NotI and transferred to the pESC-URA vector linearized with the same enzyme to obtain pESC_PCTR3_erg7. In the second step, the upstream ERG7 promoter fragment was amplified from S. cerevisiae genomic DNA using forward primer 5′-AAA CAG CTG AAT CTG CTG CTA TTC GTG-3′ and reverse primer 5′-AAA GGA TCC CCT GCA GGT CCG CAG ATA TCA AAT CTA G-3′ and was transferred to the PvuII/BamHI sites of pESC_PCTR3_erg7 to obtain pESC_PERG7_PCTR3_erg7. In the final step, a synthetic DNA fragment (Invitrogen) containing a KlLEU2 auxotrophy cassette (Gueldener et al. 2002) was ligated at the SbfI/BamHI restriction sites to obtain pESC_PERG7_KlLEU2_PCTR3_erg7.
The integrity of all constructs was verified by sequencing (Sanger et al. 1977) on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, USA). Yeast strain CEN.PK2-1C was obtained from EUROSCARF (Oberursel, Germany). Restriction enzymes were obtained from New England Biolabs GmbH (Frankfurt a.M., Germany).
Strain construction and culture conditions
The S. cerevisiae strain CEN-PK2-1C was transformed using the lithium acetate method (Gietz and Schiestl 2007) with URA3 (pESC-rox1-KlURA3_tHMGR/ERG13), LEU2 (pESC_PERG7_KlLEU2_PCTR3_erg7), and TRP1 (pAG424_PGAL1-TkLUP) as selectable markers. For stable integration into the yeast genome, pESC-rox1-KlURA3_tHMGR/ERG13 and pESC_PERG7_KlLEU2_PCTR3_erg7 were digested with NotI to remove the plasmid backbone. The yeast cells were plated on minimal synthetic defined (SD) medium (Clontech, Mountain View, USA) and incubated at 30 °C. Clones were checked for integrity by colony PCR using primers spanning both ends of the integrated construct if needed.
For the expression of galactose-inducible genes, a single colony was picked, inoculated into 5 ml SD medium, and cultivated overnight at 30 °C on a rolling platform. From this culture, 50 ml of fresh SD medium (containing 150 μM CuSO4 when repressing the expression of ERG7) was inoculated to a final cell density of 105 cells/ml and incubated at 30 °C shaking at 140 rpm in a 250-ml Erlenmeyer flask. When the culture reached a cell density of 0.4 × 106 cells/ml, the medium was changed to SD medium containing galactose instead of glucose to induce gene expression. The cells were grown to a density of 4 × 106 cells/ml and harvested by centrifugation (10 min, 1000×g).
Squalene and triterpene extraction and quantitation
Yeast metabolites were extracted as described by Rodriguez et al. (2014). Briefly, freeze-dried yeast cells were incubated at 80 °C in a water bath for 5 min after adding 1 ml 6% [w/v] KOH in methanol (Carl Roth, Karlsruhe, Germany) and 100 μg cholesterol as an internal standard (Sigma, St. Louis, USA) to each sample. To extract the metabolites from the methanol mixture, 1 ml of n-hexane (Carl Roth) was added. After vortexing, the upper phase was transferred to a new vial and the extraction was repeated two times using 500 μl n-hexane. The n-hexane of the pooled extracts was removed by evaporation. The samples were re-solubilized in 1 ml acetone (Carl Roth) and analyzed by gas chromatography mass spectrometry (GC-MS) using a GC-MS-QP 2010 Ultra (Shimadzu, Duisburg, Germany) equipped with a 30-m Rtx-5MS column. After a 1-min hold at 120 °C, the temperature was increased to 330 °C at 21 °C per min (pressure = 58.8 kPa) followed by a hold of 330 °C for 10 min. Different compounds were identified according to their ion mass/charge ratios (43, 55, 69, 95, 109, 189, 204, 207, 218, 271, 285, and 411 m/z) by peak integration using LabSolution software (Shimadzu) and matching to the National Institute of Standards and Technology library. The total ion current of the detected substances was normalized against the cholesterol internal standard and the dry weight of the sample. The statistical significance of the results was confirmed using a two-sample t test at p < 0.05.
Results
Identification of TkLUP as a lupeol synthase from T. koksaghyz
We chose a lupeol synthase from the rubber-producing dandelion T. koksaghyz as a model enzyme for the new yeast platform. Using primers designed for the amplification of TRX, a lupeol synthase gene from T. officinale (Shibuya et al. 1999), we were able to amplify a 2277-bp open reading frame from T. koksaghyz cDNA encoding a polypeptide of 759 amino acids, showing 99.3% sequence identity to the T. officinale lupeol synthase. For expression in the wild-type (WT) S. cerevisiae strain CEN.PK2-1C, we inserted the sequence into pAG424_PGAL1-ccdB, which allows expression under the control of the GAL1 promoter (Fig. 1 b). The empty vector pAG424PGAL1_ccdB served as the vector control in the expression experiments. After cultivation and triterpene extraction, GC-MS analysis revealed two additional peaks (m/z = 218) in the extracts from three independent transformants expressing the lupeol synthase sequence, but these peaks were not observed in the WT or vector control samples (Fig. 1c). The retention times matched those of the β-amyrin and lupeol standards, indicating that the peaks reflected the accumulation of trace amounts of β-amyrin and ~ 0.16 mg/g cell dry weight (CDW) of lupeol (Fig. 1d). These data strongly supported the annotation of the new T. koksaghyz sequence as lupeol synthase (TkLUP).
The deletion of ROX1 and the overexpression of tHMGR and ERG13 enhance the accumulation of lupeol by 16.5-fold
To enhance the production of lupeol, we overexpressed two yeast MVA pathway genes that have previously been shown to improve the yield of isoprenoids in heterologous yeast systems (Kirby et al. 2008; Asadollahi et al. 2010; Paddon et al. 2013). HMGR (HMG1) catalyzes the rate-limiting step of the pathway and is subject to strict feedback control, so we overexpressed a truncated form of the enzyme (tHMGR) which no longer responds to feedback inhibition by enzyme degradation due to a missing ubiquitination signal (DeBose-Boyd 2008). We also overexpressed HMGS (ERG13) which promotes isoprenoid biosynthesis by supplying the substrate for HMGR (Yuan and Ching 2014). Finally, we knocked out the ROX1 gene, which encodes a negative regulator of the MVA pathway and late sterol biosynthesis (Henry et al. 2002; Montañés et al. 2011; Özaydin et al. 2013; Jakočiūnas et al. 2015). We expressed the truncated HMG1 gene and EGR13 under the control of the bidirectional GAL1/GAL10 promoter and integrated the entire overexpression cassette into the ROX1 locus to concurrently knock out this gene (Fig. 2a). As expected, the overexpression of tHMGR and HMGS and the knockout of ROX1 in yeast cells already expressing TkLUP resulted in a significant 8.2-fold increase in the squalene content as well as a 16.5-fold higher yield of lupeol compared to cells carrying the TkLUP sequence alone (Fig. 2b). There was also a 3.6-fold increase in the abundance of lanosterol (Table 1).

Accumulation of squalene and lupeol after the deletion of ROX1 and the overexpression of MVA pathway genes. a Schematic representation of the construct for the deletion of ROX1 and the overexpression of tHMGR and ERG13. The tHMGR and ERG13 coding sequences were placed under the control of a bidirectional GAL1/GAL10 promoter (PGAL1; PGAL10). KlURA3 was used to complement the uracil auxotrophy during the integration of the construct into the yeast genome. The locus for integration was defined by sequences flanking the construct (target-up; target-down) that were homologous to the ROX1 target site. Transformation of the NotI-linearized construct led to the knockout of ROX1 by homologous recombination. b Yeast strains carrying the integrated construct in addition to the TkLUP coding sequence (rox1::PGAL1-tHMGR PGAL10-ERG13 TkLUP) accumulated higher levels of the lupeol precursor squalene in contrast to cells carrying only the TkLUP sequence (p = 0.0137). Cells containing the empty vector pAG424_PGALl_ccdB served as controls (rox1::PGAL1-tHMGR PGAL10-ERG13 vector control). The deletion of ROX1 and the overexpression of tHMGR and ERG13 resulted in a 16.5-fold increase in the lupeol content (p = 0.00893). c Mass spectrum of the designated lupeol peak. d Mass spectrum of the measured external lupeol standard. Standard deviation was calculated from n = 3 individual transformants. CDW = cell dry weight; one asterisk = p ≤ 0.05; two asterisks = p ≤ 0.01
The transformed yeast cells grew at a slower rate, as the time to reach the cell density for harvesting extended from 17 to 18.5 h, perhaps reflecting the toxicity of the higher squalene content (Donald et al. 1997; Asadollahi et al. 2010). However, this engineering step allowed us to compare the putative lupeol peak (Fig. 2c) with that of the lupeol standard (Fig. 2d). The comparable masses of the peaks supported our annotation of the TkLUP sequence. The putative β-amyrin peak was still too weak for detailed mass analysis.
ERG7 repression using the CTR3 promoter causes 2,3-oxidosqualene to accumulate and achieves a further 7.6-fold increase in the lupeol content
The engineering steps outlined above resulted in the accumulation of squalene, but not of 2,3-oxidosqualene (the substrate of the TkLUP). This indicates that squalene synthase (ERG9) may have a higher metabolic capacity than squalene epoxidase (ERG1) as previously suggested by Asadollahi et al. (2010), perhaps exacerbated by the rapid conversion of the limited 2,3-oxidosqualene pool by TkLUP itself. Alternatively, competition from the endogenous late sterol biosynthesis pathway could draw flux away from the pentacyclic triterpenes. Yeast cells overexpressing ERG1 accumulate 2,3-oxidosqualene but not downstream pentacyclic triterpenes (Veen et al. 2003), suggesting that competition from the endogenous late sterol biosynthesis pathway is primarily responsible for the low pentacyclic triterpene content in our cells. We therefore set out to suppress the expression of ERG7 (lanosterol synthase) representing the first committed step in the late sterol biosynthesis pathway.
We inserted a 735-bp promoter fragment from the copper transporter gene CTR3 upstream of the endogenous ERG7 coding sequence and, in the same step, deleted a 196-bp fragment of the endogenous ERG7 promoter (Fig. 3a). The inserted CTR3 promoter fragment contains two cis-acting copper responsive elements (TTTGCTC) which inhibit gene expression in the presence of CuSO4 (Labbé et al. 1997). A CTR3 promoter fragment was used successfully to repress ERG9 expression and thus enhance artemisinin production, thereby lowering the costs of this industrial process, indicating that the CTR3 promoter is a suitable replacement for the MET3 promoter that was used in previous studies by the same authors (Paddon et al. 2013).

Repression of ERG7 leads to the accumulation of 2,3-oxidosqualene. a Schematic representation of the construct used for the integration of the copper-sensitive CTR3 promoter (PCTR3). To introduce the promoter into the yeast genome, leucine auxotrophy was complemented by the KlLEU2 gene. The construct was flanked by sequences homologous to the ERG7 promoter (target-up; PERG7) and coding sequence (target-down; ERG7) to place the CTR3 promoter upstream of the endogenous ERG7 coding sequence. b The yeast strain carrying the CTR3 promoter construct showed a reduction in squalene levels and the accumulation of 2,3-oxidosqualene even without exposure to CuSO4 (rox1::PGAL1-tHMGR PGAL10-ERG13 PERG7Δ::PCTR3 0 μM CuSO4). This effect was significantly enhanced in the presence of 150 μM CuSO4 (rox1::PGAL1-tHMGR PGAL10-ERG13 PERG7Δ::PCTR3 150 μM CuSO4; p = 0.00613 for the reduction of squalene; p = 0.00507 for the accumulation of 2,3-oxidosqualene). Higher concentrations of copper (375 μM CuSO4) reduced squalene levels and increased the abundance of 2,3-oxidosqualene (rox1::PGAL1-tHMGR PGAL10-ERG13 PERG7Δ::PCTR3 375 μM CuSO4). Standard deviation was calculated from n = 3 individual transformants. CDW = cell dry weight; one asterisk = p ≤ 0.05; two asterisks = p ≤ 0.01; three asterisks = p ≤ 0.001
To test the activity of the construct, we introduced the corresponding fragment of pESC_PERG7_KlLEU2_PCTR3_erg7, containing the CTR3 promoter fragment, into the yeast strain carrying the fragment of pESC-rox1-KlURA3_tHMGR/ERG13. Positive transformants were cultured in the presence of 0, 150, and 375 μM CuSO4, and the expression of tHMGR and ERG13 was induced as described in the “Materials and Methods,” resulting in a shift from squalene to 2,3-oxidosqualene accumulation (Fig. 3b). Even in the absence of copper, we detected the accumulation of small amounts of 2,3-oxidosqualene, indicating that the CTR3 promoter fragment was weaker than the endogenous ERG7 promoter. This effect was even stronger in the yeast growing in the presence of 150 μM CuSO4. The amount of squalene declined significantly (p = 0.00613) as the accumulation of 2,3-oxidosqualene increased by 4.7-fold (p = 0.00507), which may reflect the absence of ERG9 and ERG1 repression by ergosterol, as well as the enhanced induction of ERG1 due to the lower levels of lanosterol (Table 1) (M’Baya et al. 1989). A further significant decrease in squalene levels (p = 0.00061) was observed when the yeast were cultivated in the presence of 375 μM CuSO4. However, there was no significant change in 2,3-oxidosqualene levels but the time to reach the cell density for harvesting extended from 20 to 35 h. This indicates that high levels of CuSO4 are toxic. We concluded that sufficient repression of ERG7 was achieved in the presence of 150 μM CuSO4, allowing us to overcome the bottleneck in 2,3-oxidosqualene synthesis. We therefore chose 150 μM CuSO4 for the enhanced production of lupeol in a strain combining rox1::PGAL1-tHMGR PGAL10-ERG13 PERG7Δ::PCTR3 and the coding sequence of TkLUP, or the pAG424_PGAL1_ccdB empty plasmid serving as a control (Fig. 4).

The repression of ERG7 enhances the accumulation of lupeol while reducing the abundance of sterols. a The accumulation of 2,3-oxidosqualene and the lower levels of squalene were observed in the yeast strain carrying the TkLUP sequence in addition to the rox1::PGAL1-tHMGR PGAL10-ERG13 and the PERG7Δ::PCTR3 cassettes, following exposure to 150 μM CuSO4 during growth (rox1::PGAL1-tHMGR PGAL10-ERG13 PERG7Δ::PCTR3 TkLUP) compared to the parental strain (rox1::PGAL1-tHMGR PGAL10-ERG13 TkLUP; p = 0.04422 for squalene). b The sterol content was reduced as anticipated, as shown by the amounts of lanosterol and ergosterol as representatives for sterol biosynthesis. The repression of ERG7 resulted in a 6.5-fold decrease in lanosterol levels (p = 0.02384) and a 3.9-fold decrease in ergosterol levels (p = 0.00941). c The repression of sterol biosynthesis enhances lupeol production by 7.6-fold (p = 0.00637), suggesting the redirection of the metabolic flux from sterol biosynthesis to lupeol production. d Comparison of the total ion count of different yeast strains. (A) = rox1::PGAL1-tHMGR PGAL10-ERG13 PERG7Δ::PCTR3 TkLUP; (B) = rox1::PGAL1-tHMGR PGAL10-ERG13 PERG7Δ::PCTR3 vector control; (C) = TkLUP; (1) = squalene peak; (2) = 2,3-oxidosqualene peak; (3) = cholesterol peak; (4) = ergosterol peak; (5) = lanosterol peak; (6) = β-amyrin peak; asterisk = unidentified yeast metabolites. e Detailed comparison of the β-amyrin and lupeol peak [m/z = 218] from different yeast strains. (A) = rox1::PGAL1-tHMGR PGAL10-ERG13 PERG7Δ::PCTR3 TkLUP; (B) = rox1::PGAL1-tHMGR PGAL10-ERG13 PERG7Δ::PCTR3 vector control; (C) = TkLUP. f Identification of β-amyrin in yeast following the repression of ERG7. Mass spectrum of the designated β-amyrin peak from the engineered yeast strain (rox1::PGAL1-tHMGR PGAL10-ERG13 PERG7Δ::PCTR3 TkLUP) (upper part). Mass spectrum of the measured external β-amyrin standard (lower part). Standard deviation was calculated from n = 3 individual transformants. CDW = cell dry weight; one asterisk = p ≤ 0.05; two asterisks = p ≤ 0.01
As expected, the yeast strain carrying all three constructs, when cultivated in the presence of CuSO4, showed a shift in squalene and 2,3-oxidosqualene levels compared to its parental strain lacking the CTR3 promoter fragment. The squalene content of these cells was 2.6-fold lower (p = 0.04422), but 2,3-oxidosqualene accumulated instead. Furthermore, the sterol content declined, as shown by the 6.5-fold lower levels of lanosterol (p = 0.02384) and 3.9-fold lower levels of ergosterol (p = 0.00941), confirming that copper-based repression was sufficient for the regulation of ERG9 and ERG1 in the strain expressing TkLUP. Moreover, introducing the copper-repressible promoter enhanced the lupeol content by a further 7.6-fold (p = 0.00637). No additional peaks representing products of TkLUP, but the already described β-amyrin peak, could be observed in the comparison of the total ion count (Fig. 4d) and the m/z 218 scan (Fig. 4e). Furthermore, the identification of β-amyrin became possible as shown in the corresponding mass spectra for the sample (Fig. 4f, upper part) and the external β-amyrin standard (Fig. 4f, lower part). The accumulation of 2,3-oxidosqualene and lupeol, as well as the lower levels of sterols, confirmed the redirection of metabolic flux from sterol biosynthesis to lupeol production in this engineered yeast strain.
Discussion
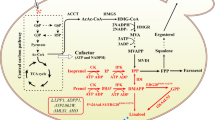
We have established a new platform for the production of pentacyclic triterpenes in yeast, using the lupeol synthase of T. koksaghyz as a model enzyme. This platform was based on a push-and-pull strategy combining the overexpression of MVA pathway genes with the deletion of a negative regulator of the MVA pathway and late sterol biosynthesis. Furthermore, we were able to use a copper-regulated promoter to enhance the accumulation of pentacyclic triterpenes by redirecting metabolic flux from late sterol biosynthesis, starting with the formation of lanosterol, to the direct production of the pentacyclic triterpene precursor 2,3-oxidosqualene (Fig. 5).

Engineered yeast platform for the enhanced production of pentacyclic triterpenes. The MVA pathway was pushed by the overexpression of ERG13 and tHMGR, and pulled by the knockout of ROX1 to enhance the productivity of MVA pathway and late sterol biosynthesis. To redirect metabolic flux from late sterol biosynthesis into the synthesis of pentacyclic triterpenes by the T. koksaghyz lupeol synthase (TkLUP), the CTR3 promoter (PCTR3) was used to inhibit the expression of ERG7 following the addition of copper (Cu2+). An additional effect on 2,3-oxidosqualene accumulation was achieved by the missing repression of ERG9 and ERG1 due to the lower sterol content (lanosterol and ergosterol). Dashed arrows represent multiple enzymatic reactions
To accomplish our push-and-pull strategy, we overexpressed the MVA pathway genes ERG13 (HMGS) and a truncated form of HMG1 (tHMGR) because others have shown that HMGR is the key rate-limiting step in the pathway and its deregulated form tHMGR can enhance squalene accumulation and isoprenoid levels in yeast platforms (Kirby et al. 2008; Asadollahi et al. 2010; Westfall et al. 2012; Scalcinati et al. 2012; Paddon et al. 2013; Lv et al. 2014; Yuan and Ching 2014). Furthermore, the overexpression of ERG13 increases flux through the pathway, given that a 13-fold increase in amorpha-4,11-diene production was observed when ERG13 was overexpressed in combination with tHMGR, ERG10, and ERG12 (Yuan and Ching 2014). We chose the ROX1 locus as an integration site for our overexpression cassette, thus knocking out this negative regulator of the MVA pathway and late sterol biosynthesis (Henry et al. 2002; Montañés et al. 2011; Özaydin et al. 2013; Jakočiūnas et al. 2015). The knockout should upregulate the MVA pathway in general, although the higher levels of squalene resulting from the increased flux were in part consumed by the simultaneously upregulated late sterol biosynthesis pathway. Using this strategy, we were able to increase the accumulation of squalene, lanosterol, ergosterol, and lupeol (Fig. 2b and Table 1). This came at the cost of a slight reduction in growth of our squalene-accumulating yeast strain, consistent with some previous studies (Donald et al. 1997; Asadollahi et al. 2010) but not others (Veen et al. 2003). We did not observe the accumulation of 2,3-oxidosqualene, the direct precursor of pentacyclic triterpene synthesis, so we redirected the flux from late sterol biosynthesis towards the production of pentacyclic triterpenes by replacing the endogenous ERG7 promoter with the copper-repressible CTR3 promoter (Labbé et al. 1997). The absence of 2,3-oxidosqualene is therefore likely to reflect the lower capacity of ERG1 compared to ERG9 (Asadollahi et al. 2010) and the rapid conversion of 2,3-oxidosqualene into lanosterol by ERG7 (Veen et al. 2003) or into pentacyclic triterpenes by TkLUP (this study).
As expected, we observed lower squalene levels and the accumulation of 2,3-oxidosqualene following the repression of endogenous ERG7 in our squalene and pentacyclic triterpene accumulating yeast strain. This may reflect the lower expression of ERG7 itself, but the regulatory mechanism of ERG1 and ERG9 may also contribute to our observations. Because we detected lower levels of ergosterol and lanosterol, the deregulation of a described negative feedback loop may also contribute to the enhanced expression of ERG9 and ERG1. Therefore, the lower sterol content may prevent the suppression of ERG9 and ERG1 due to limited ergosterol levels, and may enhance the expression of ERG1 due to the lack of suppression by lanosterol (M’Baya et al. 1989). By providing the bulk of the direct substrate 2,3-oxidosqualene by the described metabolic engineering steps for the TkLUP enzyme, we were able to enhance the accumulation of lupeol even further and also annotate a thus far uncharacterized peak in the GC-MS spectrum as β-amyrin. In the literature, the product specificity of lupeol synthases ranges from very specific with minor byproducts depending on cultivation conditions (e.g., OEW lupeol synthase from Olea europaea, TRW lupeol synthase from Taraxacum officinale; Shibuya et al. 1999) to moderate specificity with approximately 20% β-amyrin byproduct (lupeol synthase from Arabidopsis thaliana; Herrera et al. 1998). We calculate the β-amyrin byproduct of TkLUP to be approximately 0.6% of total product, classifying it to the higher specificity enzymes.
In conclusion, we were able to enhance the synthesis of pentacyclic triterpenes 127-fold in the case of lupeol within two engineering steps. In addition, we were able to characterize a second pentacyclic triterpene (β-amyrin) synthesized by our model enzyme TkLUP. The unused bulk of 2,3-oxidosqualene could be used as a substrate by other oxidosqualene cyclases to produce even higher amounts of oxidosqualene derivatives. Optimal cultivation techniques could also be used to increase the yields of these molecules (reviewed by Liao et al. 2016).
References
Alberti S, Gitler AD, Lindquist S (2007) A suite of gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast 24:913–919. https://doi.org/10.1002/yea.1502
Arendt P, Miettinen K, Pollier J, De Rycke R, Callewaert N, Goossens A (2017) An endoplasmic reticulum-engineered yeast platform for overproduction of triterpenoids. Metab Eng 40:165–175. https://doi.org/10.1016/j.ymben.2017.02.007
Asadollahi MA, Maury J, Schalk M, Clark A, Nielsen J (2010) Enhancement of farnesyl diphosphate pool as direct precursor of sesquiterpenes through metabolic engineering of the mevalonate pathway in Saccharomyces cerevisiae. Biotechnol Bioeng 106:89–96. https://doi.org/10.1002/bit.22668
DeBose-Boyd RA (2008) Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Research 18(6):609-621
Demierre MF, Higgings PD, Gruber SB, Hawk E, Lippmann SM (2005) Statins and cancer prevention. Nat Rev Cancer 5:930–942. https://doi.org/10.1038/nrc1751
Donald KA, Hampton RY, Fritz IB (1997) Effects of overproduction of the catalytic domain of 3-hydroxy-3-methylglutaryl coenzyme A reductase on squalene synthesis in Saccharomyces cerevisiae. Appl Environ Microbiol 63:3341–3341
Gietz RD, Schiestl RH (2007) High-efficiency yeast transformation using the LiAC/SS carrier DNA/PEG method. Nat Protoc 2:31–34. https://doi.org/10.1038/nprot.2007.13
Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH (2002) A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res 30(6):e23
Hansen BG, Salomonsen B, Nielsen MT, Nielsen JB, Hansen NB, Nielsen KF, Regueira TB, Nielsen J, Patil KR, Mortensen UH (2011) Versatile enzyme expression and characterization system for Aspergillus nidulans, with the Penicillium brevicompactum polyketide synthase gene from the mycophenolic acid gene cluster as a test case. Appl Environ Microbiol 77:3044–3051. https://doi.org/10.1128/AEM.01768-10
Hemmerlin A, Harwood JL, Bach TJ (2012) A raison d’être for two distinct pathways in the early steps of plant isoprenoid biosynthesis. Prog Lipid Res 51:95–148. https://doi.org/10.1016/j.plipres.2011.12.001
Henry KW, Nickels JT, Edling TD (2002) ROX1 and ERG regulation in Saccharomyces cerevisiae: implications for antifungal susceptibility. Eukaryot Cell 1:1041–1044
Herrera JB, Bartel B, Wilson WK, Matsuda SP (1998) Cloning and characterization of the Arabidopsis thaliana lupeol synthase gene. Phytochemistry 49:1905–1911
Jakočiūnas T, Bonde I, Herrgård M, Harrison SJ, Kristensen M, Pedersen LE, Jensen MK, Keasling JD (2015) Multiplex metabolic pathway engineering using CRISPR/Cas9 in Saccharomyces cerevisiae. Metab Eng 28:213–222. https://doi.org/10.1016/j.ymben.2015.01.008
Kirby J, Romanini DW, Paradise EM, Keasling JD (2008) Engineering triterpene production in Saccharomyces cerevisiae-beta-amyrin synthase from Artemisia annua. FEBS J 275:1852–1859. https://doi.org/10.1111/j.1742-4658.2008.06343.x
Labbé S, Zhu Z, Thiele DJ (1997) Copper-specific transcriptional repression of yeast genes encoding critical components in the copper transport pathway. J Biol Chem 272:1591–1598
Liao P, Hemmerlin A, Bach TJ, Chye ML (2016) The potential of the mevalonate pathway for enhanced isoprenoid production. Biotechnol Adv 34:697–713. https://doi.org/10.1016/j.biotechadv.2016.03.005
Lv X, Xie W, Lu W, Guo F, Gu J, Yu H, Ye L (2014) Enhanced isoprene biosynthesis in Saccharomyces cerevisiae by engineering of the native acetyl-CoA and mevalonic acid pathways with a push-pull-restrain strategy. J Biotechnol 186:128–136. https://doi.org/10.1016/j.jbiotec.2014.06.024
M’Baya B, Fequeur M, Servouse M, Karst F (1989) Regulation of squalene synthase and squalene epoxidase activities in Saccharomyces cerevisiae. Lipids 24:1020–1023
Martin VJ, Pitera DJ, Withers ST, Newman JD, Keasling JD (2003) Engineering a mevalonate pathway in Escherichia coli for the production of terpenoids. Nat Biotechnol 21:796–802. https://doi.org/10.1038/nbt833
Montañés FM, Pascual-Ahuir A, Proft M (2011) Repression of ergosterol biosynthesis is essential for stress resistance and is mediated by the Hog1 MAP kinase and the Mot3 and Rox1 transcription factors. Mol Microbiol 79:1008–1023. https://doi.org/10.1111/j.1365-2958.2010.07502.x
Özaydin B, Burd H, Lee TS, Keasling JD (2013) Carotenoid-based phenotypic screen of the yeast deletion collection reveals new genes with roles in isoprenoid production. Metab Eng 15:1744–1783. https://doi.org/10.1016/j.ymben.2012.07.010
Paddon CJ, Westfall PJ, Pitera DJ, Benjamin K, Fisher K, McPhee D, Leavell MD, Tai A, Main A, Eng D, Polichuk DR, Teoh KH, Reed DW, Treynor T, Lenihan J, Fleck M, Bajad S, Dang G, Dengrove D, Diola D, Dorin G, Ellens KW, Fickes S, Galazzo J, Gaucher SP, Geistlinger T, Henry R, Hepp M, Horning T, Iqbal T, Jiang H, Kizer L, Lieu B, Melis D, Moss N, Regentin R, Secrest S, Tsuruta H, Vazquez R, Westblade LF, Xu L, Yu M, Zhang Y, Zhao L, Lievense J, Covello PS, Keasling JD, Reiling KK, Renninger NS, Newman JD (2013) High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 496:528–532. https://doi.org/10.1038/nature12051
Ro DK, Paradise EM, Oulett M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, Chang MC, Withers ST, Shiba Y, Sarpong R, Keasling JD (2006) Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440:940–943. https://doi.org/10.1038/nature04640
Rodriguez S, Kirby J, Denby CM, Keasling JD (2014) Production and quantification of sesquiterpenes in Saccharomyces cerevisiae, including extraction, detection and quantification of terpene products and key metabolites. Nat Protoc 9:1980–1996. https://doi.org/10.1038/nprot.2014.132
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74:5463–5467
Scalcinati G, Partow S, Siewers V, Schalk M, Daviet L, Nielsen J (2012) Combined metabolic engineering of precursor and co-factor supply to increase α-santalene production by Saccharomyces cerevisiae. Microb Cell Factories 31(11):117. https://doi.org/10.1186/1475-2859-11-117
Shibuya M, Zhang H, Endo A, Shishikura K, Kushiro T, Ebizuka Y (1999) Two branches of the lupeol synthase gene in the molecular evolution of plant oxisqualene cyclases. Eur J Biochem 266:307–307
Veen M, Stahl U, Lang C (2003) Combined overexpression of genes of the ergosterol biosynthetic pathway leads to accumulation of sterols in Saccharomyces cerevisiae. FEMS Yeast Res 4:87–95
Vickers CE, Williams TC, Peng B, Cherry J (2017) Recent advances in synthetic biology for engineered isoprenoid production in yeast. Curr Opin Chem Biol 40:47–56. https://doi.org/10.1016/j.cbpa.2017.05.017
Westfall PJ, Pitera DJ, Lenihan JR, Eng D, Woolard FX, Regentin R, Horning T, Tsuruta H, Melis DJ, Owens A, Fickes S, Diola D, Benjamin KR, Keasling JD, Leavell MD, McPhee DJ, Renninger NS, Newman JD, Paddon CJ (2012) Production of amorphadiene in yeast, and its conversion to dihydroartemisinic acid, precursor to the antimalarial agent artemisinin. Proc Natl Acad Sci U S A 109:E111–E118. https://doi.org/10.1073/pnas.1110740109
Yuan J, Ching CB (2014) Combinatorial engineering of mevalonate pathway for improved amorpha-4,11-diene production in budding yeast. Biotechnol Bioeng 111:608–617. https://doi.org/10.1002/bit.25123
Zhou C, Li J, Li C, Zhang Y (2016) Improvement of betulinic acid biosynthesis in yeast employing multiple strategies. BMC Biotechnol 16:59. https://doi.org/10.1186/s12896-016-0290-9
Funding
Jan Niklas Bröker was funded by a grant (FKZ 031B0014D) of the Federal Ministry of Education and Research, Germany.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Jan Niklas Bröker, Nicole van Deenen, Boje Müller, and Christian Schulze Gronover are named as inventors on a submitted patent application (EP18166374.1) which relates to this study. Dirk Prüfer has no conflict of interest, as he is not an inventor of the mentioned patent application.
Ethical statement
This article does not contain any studies involving human participants or animals.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bröker, J.N., Müller, B., van Deenen, N. et al. Upregulating the mevalonate pathway and repressing sterol synthesis in Saccharomyces cerevisiae enhances the production of triterpenes. Appl Microbiol Biotechnol 102, 6923–6934 (2018). https://doi.org/10.1007/s00253-018-9154-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-9154-7