
Abstract
Unveiling the determinants for transferase and hydrolase activity in glycoside hydrolases would allow using their vast diversity for creating novel transglycosylases, thereby unlocking an extensive toolbox for carbohydrate chemists. Three different amino acid substitutions at position 220 of a GH1 β-glucosidase from Thermotoga neapolitana caused an increase of the ratio of transglycosylation to hydrolysis (r s/r h) from 0.33 to 1.45–2.71. Further increase in r s/r h was achieved by modulation of pH of the reaction medium. The wild-type enzyme had a pH optimum for both hydrolysis and transglycosylation around 6 and reduced activity at higher pH. Interestingly, the mutants had constant transglycosylation activity over a broad pH range (5–10), while the hydrolytic activity was largely eliminated at pH 10. The results demonstrate that a combination of protein engineering and medium engineering can be used to eliminate the hydrolytic activity without affecting the transglycosylation activity of a glycoside hydrolase. The underlying factors for this success are pursued, and perturbations of the catalytic acid/base in combination with flexibility are shown to be important factors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glycosylation is an important source of structural diversity of natural products. It can alter the properties of compounds in a multitude of ways, e.g., changing the flavor or smell (Ribeiro 2011; Roitner et al. 1984), improving water solubility (Chen et al. 2011) or stability (Yamamoto et al. 1990), or reducing skin irritation (Kurosu et al. 2002). Glycosylation can additionally be used to produce attractive biosurfactants, alkyl glycosides (van Rantwijk et al. 1999). In nature, glycosylation is mainly performed by Leloir glycosyltransferases. However, they are not well suited for glycosylation in vitro as they require expensive nucleotide-activated sugars as glycosyl donors and are often difficult to express. Glycoside hydrolases (GH) would be ideal enzymes for glycosylation with regard to their natural abundance, robustness, and wide acceptor specificity. For synthetic purposes, retaining GH are the most studied, since they all possess the machinery making transglycosidic activity possible. Most of them function via a double displacement mechanism, first proposed by Koshland (1953). In the first step, the catalytic nucleophile attacks the anomeric carbon of the glycosyl donor and forms a glycosyl enzyme intermediate, with the help of the catalytic acid that protonates the glycosidic oxygen (Fig. 1). Upon formation of the glycosyl enzyme, the pKa of the catalytic acid is reduced, allowing it to act as a general base in the second reaction step (McIntosh et al. 1996). The intermediate is subsequently deglycosylated by either water (hydrolysis) or another hydroxyl-containing acceptor (transglycosylation). Unfortunately, the use of GH for glycosylation is impaired by their naturally dominant hydrolytic activity.

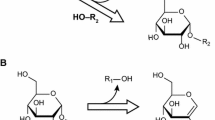
Reaction mechanism. The double displacement mechanism of retaining glycoside hydrolases. In this study, the leaving group (LG) is p-nitrophenol and the acceptor (A) is hexanol. The catalytic nucleophile is E164 and the catalytic acid/base is E349
A significant step toward utilizing GH for glycosylation was taken in 1998, with the development of the first glycosynthases (Mackenzie et al. 1998; Malet and Planas 1998). For retaining glycosynthases, the general strategy is to mutate the catalytic nucleophile, rendering the enzymes inactive. Using an activated donor, commonly glycosyl fluorides of the anomeric configuration opposite to the normal substrates, reactivates the enzyme. The glycosynthase no longer acts via a double-displacement mechanism but utilizes general base catalysis in combination with a good leaving group. With no glycosyl enzyme, hydrolysis can be avoided and the product cannot be hydrolyzed since it lacks good leaving group. However, glycosynthases do not always work efficiently and rely on activated glycosyl donors.
An alternative strategy to constructing glycosynthases is to limit the hydrolytic reaction of GH, transforming them into transglycosylases (TG). TG are rare in nature (Bissaro et al. 2015b; Larsbrink et al. 2012; Luang et al. 2013) but have been successfully produced from retaining GH through directed evolution (Bissaro et al. 2015a; Feng et al. 2005; Kone et al. 2009; Teze et al. 2014). Nevertheless, to be able to utilize the vast array of GH for creation of TG, we need to understand what governs their propensity for transglycosylation or hydrolysis.
From the numerous mutational studies aimed at improving the ratio of transglycosylation, three main mechanisms can be identified. The first mechanism is reducing the binding in glycone (−) subsites. Teze et al. improved disaccharide yield from 36 to 82 % through mutations in the −1 subsite (Teze et al. 2014). They hypothesized that the reduced interactions destabilize the transition states of the reaction, which affects hydrolysis more than transglycosylation and thereby improves the ratio of transglycosylation. The same effect has been seen in several other mutational studies (Arab-Jaziri et al. 2015; Aronson et al. 2006; Bissaro et al. 2015a; Feng et al. 2005).
The second common mechanism for improving transglycosylation is increasing the affinity in aglycone (+) subsites. Armand et al. (2001) found that several aromatic residues in the aglycone subsite of a GH10 xylanase were crucial for transglycosylation. The importance of aromatic residues in the aglycone subsite for transglycosylation has been seen also in GH16 and GH18 enzymes (Johansson et al. 2004; Taira et al. 2010). Moreover, Feng et al. (2005) reasoned that since deglycosylation is rate limiting, increased affinity of the acceptor would improve transglycosylation, which it is also found for a double mutant of a GH1 β-glucosidase. Higher ratios of transglycosylation have also been seen in other mutational studies with improved affinity in aglycone subsites (Champion et al. 2012; Lu et al. 2009; Osanjo et al. 2007).
The third popular strategy for promoting transglycosylation in GH is disrupting the binding of the catalytic water, which can be done in a multitude of ways. Honda et al. (2008) removed a known hydrogen-bonding interaction with the catalytic water and thereby reduced the hydrolytic reactivity of an inverting xylanase. Other studies have focused on improving the hydrophobicity of the entrance to the active site (Frutuoso and Marana 2013; Kuriki et al. 1996) or acceptor subsite (Lundemo et al. 2013). Natural TG could instead function through locking water molecules in hydrogen-bonding networks offset from the preferred position for a catalytic water, as demonstrated for a GH31 α-transglucosylase (Larsbrink et al. 2012).
Common in all studies mentioned above is that the reaction rate of the transglycosylation is reduced, although not as much as the hydrolysis. Even natural TG are commonly much less efficient catalysts than their hydrolytic counterparts (Bissaro et al. 2015b).
Apart from mutational studies, the propensity for transglycosylation has been affected through a variety of other methods. These methods include increasing reactant concentrations (Giacomini et al. 2002; Mala et al. 1999), changing reaction temperature (Ribeirao et al. 1997) or pH (Bonnin et al. 1997; Oikawa et al. 2001; Saitoh et al. 1995; Seidle and Huber 2005), adding cosolvent (Perez-Sanchez et al. 2011), and reducing water activity (Lundemo et al. 2014).
One of these methods showed that the hydrolytic activity of a β-glucosidase from Aspergillus niger could be effectively eliminated without affecting the rate of transglycosylation, through utilizing the enzymes’ seemingly constant transglycosylation rate across a wide pH range (Seidle and Huber 2005). Unfortunately, this property has not been found in any other glycoside hydrolase, to the best of our knowledge.
In this paper, we show that although transglycosylation is highly dependent on pH for the wild-type β-glucosidase from Thermotoga neapolitana, it can be changed by single mutations. The transglycosylation rate of three single mutant enzyme variants is unaffected at high pH, while the hydrolytic reaction is reduced. Subsequently, complete elimination of the hydrolytic side reaction and quantitative yields for our model reaction were made possible, without reducing the rate of transglycosylation. Furthermore, the molecular determinants for this success are pursued, in order to enable a novel strategy for improving the synthetic usefulness of glycosidases.
Materials and methods
Material
Hexyl-β-d-glucoside (HG), p-nitrophenol (pNP), and p-nitrophenol-β-d-glucoside (pNPG) were obtained from Sigma-Aldrich (St Louis, Missouri, USA) and all other chemicals from VWR International (Stockholm, Sweden).
Mutagenesis
The gene encoding TnBgl1A was previously cloned into PET22b(+) (Novagen, Madison, WI, USA) (Turner et al. 2006). A reduced site saturation mutagenesis was generated using the QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA), using the sequence with GenBank accession number KC776911 as the template and the primer 5′-GGCAAAATTGGCATTGTGTTCNDTAATGGCTACTTCGAACCAGC-3′. The resulting plasmid library was retransformed into Escherichia coli Nova Blue cells for storage and into E. coli BL21 (Novagen, Madison, WI, USA) for expression. Functional mutants were selected from a 96-well assay, and the complete gene was sequenced by GATC Biotech AG (Konstanz, Germany) to confirm the mutations. Selected single mutants were produced in larger scale for characterization.
Expression and purification
The enzymes were synthesized in 0.5-l cultivations of E. coli BL21 (Novagen, Madison, WI, USA) in Erlenmeyer flasks at 37 °C, pH 7, in Luria-Bertani (LB) media containing 100 μg/ml ampicillin and inoculated with 1 % overnight precultures. After reaching 0.6 in optical density, measured at 620 nm, TnBgl1A expression was induced by addition of 0.5 ml 100 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) and production was continued for 20 h. Cells were harvested by centrifugation for 10 min (4 °C, 5500×g), re-suspended in binding buffer (20 mM imidazole, 20 mM Tris-HCl, 0.75 M NaCl, pH 7.5), and lysed by sonication 6 × 3 min at 60 % amplitude and a cycle of 0.5 using a 14-mm titanium probe (UP400 S, Dr. Hielscher, Teltow, Germany). Heat treatment (70 °C, 30 min) and centrifugation (30 min, 4 °C, 15,000×g) were used to remove most of the native E. coli proteins before purification by immobilized metal affinity chromatography using an ÄKTA prime system (Amersham Biosciences, Uppsala, Sweden). The protein slurry was applied to a HisTrap FF crude column (GE Healthcare, Uppsala, Sweden) pretreated with 0.1 M copper(II) sulfate. Bound proteins were eluted using elution buffer (250 mM imidazole, 20 mM Tris-HCl, 0.75 M NaCl, pH 7.5). Fractions containing protein were pooled and dialyzed against 50 mM citrate phosphate buffer, pH 5.6, overnight using a 3500-Da molecular weight cutoff dialysis membrane (Spectrum Laboratories, Rancho Dominguez, CA, USA) and stored at −20 °C until use. Purity of the expressed proteins was estimated using SDS-PAGE according to Laemmli (1970).
Water activity control
Substrate solutions (34 mM pNPG in hexanol) were incubated over saturated salt solutions to define water activities. The salts used for equilibration were KCH3CO2 (a w = 0.23), MgCl2 (a w = 0.33), Mg(NO3)2 (a w = 0.53), NaCl (a w = 0.75), KCl (a w = 0.84), and K2SO4 (a w = 0.97) (Greenspan 1977) (Goderis et al. 1987). Triplicate samples from each equilibrated hexanol sample were injected on an 899 Karl Fischer coulometer (Metrohm, Herisau, Switzerland). The obtained relation between water activity and water amount was used to estimate water activity in the transferase reactions.
Transglycosylation in monophasic hexanol
Reactions of 2 ml 34 mM pNPG were started and concurrently set to the desired water activity by addition of 2–20 ng enzyme in a determined volume of 0.1 M citrate phosphate buffer, pH 5.6, based on the abovementioned Karl-Fisher calibration curve. The reactions were kept in a ThermoMixer (HLC Biotech, Bovenden, Germany) set to 70 °C and 700 rpm.
Buffers used
The buffers used in this study are 0.1 M of the following, unless otherwise stated: citrate phosphate (pH 3.0–6.0), sodium phosphate (pH 6.0–8.5), Tris-HCl (pH 8.0–9.0), glycine-NaOH (pH 9.0–10.5), and phosphate-NaOH (pH 11.0–11.5).
Biphasic transglycosylation
In a ThermoMixer (HLC Biotech, Bovenden, Germany) set to 70 °C and 800 rpm, 2550 μl 34 mM pNPG in water-saturated n-hexanol was preheated. Reactions were started by addition of 450 μl enzyme (≈10 ng/ml) in 0.1 M citrate phosphate buffer, pH 5.5.
Steady-state kinetics for hydrolysis
In 96-well PCR plates, each enzyme variant was assayed at nine different pHs using seven different concentrations of pNPG with triplicate samples for each concentration. On ice, 195-μl pNPG solution was added to each well and 5-μl enzyme solution (0.67 ng/ml). Triplicate pNP standards were loaded on each plate as well as blanks for each combination of pH and pNPG concentration, to account for spontaneous hydrolysis. The reactions were started by moving the plate to a preheated thermomixer set to 70 °C and 300 rpm mixing for a set time, cooling on ice for 5 min before transferring 40 μl to a flat-bottomed 96-well plate, and reading the absorbance at 400 nm using a Multiskan GO plate reader (Thermo Scientific, USA). Two replicates were run for each plate to ensure keeping in the linear range. The incubation times were 10 and 30 min for low pH, 10 and 20 min for medium pH, and 5 and 10 min for high pH plates due to the higher absorbance of pNP in its deprotonated state.
HPLC analysis
Samples were withdrawn from the hexanol phase through a septum lid to follow the reaction by analysis on HPLC. Transferase and reverse hydrolysis reactions were monitored using reverse-phase HPLC (L-7100 pump, L-7000 interface, L-7250 autosampler with a 20-ml injection loop, and L-7400 UV detector; LaChrom; Hitachi Ltd., Tokyo, Japan) with the HPLC equipped with an evaporative light-scattering detector (500 ELSD; Alltech Associates Inc., Deerfield, IL) with an evaporator temperature of 94 °C and a nebulizer gas flow of 2.5 standard liters per minute and a Kromasil 100-5C18 column (4.6 μm by 250 mm; Kromasil; Eka Chemicals AB, Separation Products, Bohus, Sweden). A gradient was applied from 50 to 70 % methanol in Milli-Q water over 5 min and kept at 70 % methanol for 1 min before returning to initial conditions for reequilibration. A constant flow rate of 1.0 ml/min was used. pNPG elutes after 3.5 min and was followed with the ELSD detector. HG and pNP both have a retention time of 7.5 min, but HG does not absorb at 405 nm and pNP is too volatile to be detected by the ELSD detector. Sample chromatograms can be found in Supplementary Fig. S1. Concentrations were determined by use of eight-point external standard curves. Sampling was done only from the hexanol phase, and partitioning to the aqueous phase was accounted for by using experimentally determined apparent partitioning coefficients (Lundemo et al. 2013). After compensating for withdrawn sample volume, the total specific initial reaction rate and the ratio of transglycosylation over hydrolysis (r s/r h) were calculated. The synthetic reaction rate equals the formation of HG, and the hydrolytic reaction rate is calculated by subtracting the HG formation rate from the total reaction rate (formation rate of pNP). No self-condensation of pNPG was observed at the pNPG concentrations used.
Molecular dynamic simulation
As a starting point for the simulations, the coordinates from the crystal structure of TnBgl1A were used (PDB code 5IDI). Point mutations were introduced using Scwrl4 (Krivov et al. 2009). The molecular dynamics (MD) simulation was performed using GROMACS 5.0 (Pronk et al. 2013) and the OPLS-AA force field (Kaminski et al. 2001) at 343 K and with ionization states corresponding to pH 6 and pH 10, predicted by PROPKA 3.1 (Olsson et al. 2011). The enzyme variants were placed in the center of a cubic box filled with 21,289–21,750 water molecules from a simple point charge water model (Hermans et al. 1984). Before MD, the system was energy minimized and equilibrated with constant volume and temperature followed by constant pressure and temperature. Simulations of 500 ps were performed with a time step of 2 fs, and the coordinates were saved every 5000 steps (10 ps).
Results
In a previous study, a single mutation, N220F (Fig. 2), with substantial effect on the ratio of transglycosylation over hydrolysis was identified (Lundemo et al. 2013). N220 is positioned in the second layer of subsite +1, and the homologous position in ZmBgl1 has a water-mediated interaction to the bound oligosaccharides (Chuenchor et al. 2011). This position was investigated in detail in this study with the aim of understanding the reason for its importance and evaluating whether further improvements to the ratio of transglycosylation are possible.

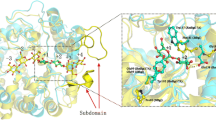
Active site. Position of N220 (green) in TnBgl1A in relation to the catalytic acid/base E168 (top, red) and nucleophile E349 (bottom, red), position Y293 (blue), and a docked pNPG molecule (black) (Color figure online)
Improved transglycosylation of mutants at position N220
A reduced site saturation library for position 220 was generated, and a selected group was further characterized. Specifically, one small nucleophilic (Cys, C), one small hydrophobic (Leu, L), an acidic (Asp, D), and a basic (Arg, R) along with two aromatic (Phe, F, and Tyr, Y) amino acids were selected.
To study transglycosylation, aqueous enzyme solution was added to pNPG in water-saturated hexanol, creating a two-phase system. As seen in Table 1, at optimal pH for total activity (pH 5.5), N220L lowered the reaction rate and specificity for transglycosylation significantly, while all other mutations improved the ratio of synthetic over hydrolytic activity (r s/r h). The positive effects on specificity for a hydrophilic amino acid, arginine, in Table 1 suggest that merely making the acceptor subsite more hydrophobic is not the only way to favor transglycosylation and warrants further investigation.
Influence of pH on transglycosylation and hydrolysis in hexanol/water
For each enzyme variant, the two-phase reaction system was run at 18 different pH values and the total initial reaction rates of the hydrolytic and transglycosidic reactions were measured and are presented in Fig. 3. For the wild-type, N220C and N220D mutants, the hydrolytic activity dominates across the pH spectrum and the activity profiles have the expected bell shape. Interestingly, the most beneficial mutants (N220Y, N220F, and, to some extent, N220R) only display a strong dependence on pH for the hydrolytic reaction, while the transglycosylation is largely unaffected. This leads to improved r s/r h with increased pH, and the effect persists until no hydrolytic side reaction is observed, similar to the results by Seidle and Huber (2005).

pH dependence of transglycosylation and hydrolysis. Specific initial reaction rate (r) for hydrolysis (diamonds) and transglycosylation (squares) for TnBgl1A in 85 % hexanol 15 % 0.1 M buffer systems with p-nitrophenyl-β-d-glucopyranoside as glycosyl donor. Alongside the wild-type enzyme (a), N220C (b), N220L (c), N220D (d), N220R (e), N220F (f), and N220Y (g) variants are presented. Error bars represent 1σ
In the practical application of enzymatic reactions, the fraction of the substrate being converted to the desired product is of crucial importance. When using glycosidases for the synthesis of alkyl glycosides, the competition between transglycosylation and hydrolysis reactions is the key issue. To illustrate the influence of changes in the enzyme and modification of the reaction conditions, the ratio of the transglycosylation rate and the total rate of substrate (pNPG) conversion (η) were plotted as a function of pH for the wild-type enzyme and the N220F mutant (Fig. 4). In absence of secondary hydrolysis, η is a good estimate of the reaction yield. Secondary hydrolysis occurs at a rate of less than 1 % compared to the total reaction rate of all enzyme variants in this study (data not shown). Hence, it is obvious that the combination of a single mutation and a change in reaction pH is enough to provide an excellent catalyst for the transglycosylation reaction with a minimum of substrate being lost in the hydrolytic side reaction. In an attempt to uncover the mechanistic reason for this favorable result and thereby unlock a novel strategy for the construction of non-Leloir glycosyltransferases, a battery of additional tests was performed.

Comparison of wild type and mutant. The ratio of the transglycosylation rate and the total rate of substrate conversion, i.e., estimated yield (η) of TnBgl1A and mutant N220F as a function of pH in 85 % hexanol 15 % 0.1 M buffer systems with p-nitrophenyl-β-d-glucopyranoside as glycosyl donor
Kinetics of hydrolysis in water
The two most prevalent hypotheses for improved ratio of transglycosylation in GH are reduced affinity in glycone-binding sites and improved affinity in aglycone-binding sites (Bissaro et al. 2015b). To acquire information on the binding of the glycone to the enzyme variants, a simplified system, without hexanol phase, was studied and the kinetic constants for pNPG hydrolysis were determined. Few significant effects of the mutations were seen, only the inactivation of N220L and the increased k cat and K M of mutant N220R (Supplementary Fig. S2). Interestingly, the latter effect is not seen for the other two mutants with large effects on specificity for transglycosylation (N220Y and N220F).
Probing the ionization state of catalytic residues using hydrolysis kinetics in water
Further information on the reaction mechanism can be obtained from studies of the pH dependence of kinetic constants for hydrolysis. In a somewhat simplified model, the variation of k cat/K M with pH can be described by the following equation (Cornish-Bowden 2012):
The pKa values pKE1 and pKE2 are assumed to reflect the ionization of the nucleophile and the catalytic acid/base, an assumption supported by NMR for another GH by Joshi et al. (2000, 2001). All mutations lead to more narrow pH optima (seen as the difference between pKE1 and pKE2), but, in general, there were no significant changes to the optimal pH, with the exception of N220D and N220Y (Table 2, Fig. 6, Supplementary Fig. S3). Both show significant perturbation to the pKa of the nucleophile (+1.1 and +1.0 pH units, respectively), while N220Y additionally affected the pKa of the catalytic acid/base (+0.5 pH units). At pH values above 9, very low activity was observed for all enzyme variants in this aqueous system.
Investigating flexibility through molecular dynamic simulation
To investigate if the point mutations influence the overall flexibility of the enzyme, 500-ns MD simulations were performed under standard state conditions. The root-mean-square fluctuations of wild-type and N220R, N220F, and N220Y are presented in Supplementary Fig. S4. No significant changes in protein flexibility were observed. However, distortions of the catalytic acid/base were observed for the mutants N220Y and N220R after 50- to 500-ns simulation (Supplementary Fig. S5).
The role of water for performance of enzyme variants in monophasic hexanol
To further study the possibilities to influence the competition between transglycosylation and hydrolysis, experiments were carried out using hexanol as reaction medium and with the water activity being controlled by equilibration with saturated salt solutions. Figure 5 shows that all enzyme variants display exponential increase of reaction rates with increasing water activity, consistent with previous studies (Hansson et al. 2001; Lundemo et al. 2014; Mladenoska et al. 2007; Mladenoska et al. 2008). Moreover, in agreement with the two-phase system, N220L was significantly slower than the other mutants. For the wild-type enzyme and the N220L, N220D, and N220C, the ratio of transglycosylation to hydrolysis (r s/r h) was largely unaffected by water content, while this ratio increased with increasing water activity for the mutants N220R, N220F, and N220Y. This counter intuitive effect of water on r s/r h has been observed previously for several GH (Hansson et al. 2001; Lundemo et al. 2014; Mladenoska et al. 2007; Mladenoska et al. 2008). The N220F variant reached a higher r s/r h in this system without a macroscopic aqueous phase than in the two-phase system (Table 1), while the two-phase system gave the highest r s/r h for N220R and N220Y.

Water activity dependence. Total specific initial reaction rate (left) and transglycosylation (r s) over hydrolysis (r h) ratio (right) for TnBgl1A and single mutation variants of the same, in micro-aqueous hexanol at varying water activities (a w). Error bars represent 1σ
Discussion
To be able to utilize the vast array of GH for creation of TG, we need to understand what governs their predisposition for transglycosylation or hydrolysis. We show that a single point mutation in the second layer of the aglycone subsite of a β-glucosidase from T. neapolitana, in combination with high pH, can reduce the hydrolytic reaction to undetectable levels, hence transforming the enzyme into a transglycosylase. Moreover, in contrast to previously published studies, the reaction rates of the transglycosylation for these three mutants are maintained. Furthermore, in an attempt to unveil the mechanism behind this success, several factors influencing the propensity for transglycosylation have been studied.
Alterations to substrate affinity
Kinetic evaluation of pNPG hydrolysis in aqueous media for TnBgl1A variants revealed no indication of improved or reduced affinity for the glycosyl donor, with the exception of variant N220R. This enzyme variant displayed an equal increase in K M and k cat. An increased K M suggests a decreased affinity for pNPG. Decreased affinity in subsite −1 affecting the glycosyl donor has been identified as a key factor for converting GH into TG (Bissaro et al. 2015b). However, position N220 is more likely to affect the aglycone (+1) subsite due to its position in the second layer of this subsite. Furthermore, the absence of an effect for the other two beneficial mutations (N220F and N220R) suggests that this is not the reason for the improved specificity for transglycosylation in this case.
The role of the catalytic acid/base
MD simulations suggest a distortion of the catalytic acid/base for mutants N220Y and N220R after 50- to 500-ns simulation, while no such distortion is seen for wild-type TnBgl1A (Supplementary Fig. S5). This observation can be relevant for different reasons. Firstly, incorrect positioning of the catalytic base would hamper the deglycosylation step and thereby increase the lifetime of the glycosyl-enzyme intermediate. Extending the lifetime of the intermediate complex allows more time for the slower diffusing preferred acceptor, hexanol, to enter the active site. Alternatively, as described for a GH13 CGTase by Kelly et al. (2008), the catalytic conformation could be regained through acceptor binding, thereby making the enzyme unable to use water as acceptor. An alternative explanation is related to the catalytic water. The only non-water-mediated interaction to the potential catalytic water in the crystal structure of TnBgl1A is the acid/base. Distorting the acid/base could therefore lock the catalytic water in a position unfavorable for hydrolysis, similar to the proposed mechanism of natural TG (Larsbrink et al. 2012).
Influence of protein flexibility
Glycosyltransferases are more flexible than GH, indicating that flexibility could be important for transglycosylation (Rojas-Cervellera et al. 2013). In addition, conformational changes have been proposed in the transglycosylation mechanism of both CGTases (Kelly et al. 2008; Uitdehaag et al. 1999) and glucanotransferases (Barends et al. 2007; Kaper et al. 2007). No significant changes in enzyme flexibility due to amino acid substitutions were observed in MD simulations (Supplementary Fig. S4). However, for the enzyme variants having the most promising transglycosylation activities, r s/r h increased with increasing water activity. This suggests that flexibility indeed plays a role also for GH, since water is well established as molecular lubricant increasing the internal flexibility of enzymes (Affleck et al. 1992; Broos et al. 1995; Soares et al. 2003).
Ionization state of the active site residues
N220Y displays significantly altered pKa values for both nucleophile (pKE1) and acid/base (pKE2), as emphasized in Fig. 6, while N220R and N220F do not. Either the three mutations successfully improve r s/r h through different mechanisms, or alternatively, the pKa shifts are of little importance for the improved specificity for transglycosylation and are merely a coincidence. Nevertheless, ionization of the catalytic residues plays a key role in the hydrolytic activity of the enzyme. For all enzyme variants, the hydrolytic activity is reduced at elevated pH, supposedly due to the catalytic acid losing the protonated state required for catalysis. Why this does not influence the transglycosylation is yet to be determined. Alternatively, the reduction of activity at high pH could be caused by Tyr293 (Fig. 2), losing its protonated state. This tyrosine has been suggested to stabilize the ionized state of the catalytic nucleophile and thereby facilitate the deglycosylation through acid catalysis (Seidle and Huber 2005). A stronger nucleophile acceptor may not require this acid catalysis. Furthermore, several mutational studies have indicated the involvement of this tyrosine in modulating the preference for transglycosylation (Bissaro et al. 2015a; Teze et al. 2014).

pH dependence of hydrolysis in water. pH dependence of k cat/K M for TnBgl1A (diamonds) and N220Y mutant (circles) along with their model-fitted curves, solid and dashed lines, respectively
In conclusion, we successfully reduced the unwanted hydrolytic activity of a family 1 glycoside hydrolase from T. neapolitana to an undetectable level, without diminishing the efficiency for transglycosylation. This was achieved through single point mutations (N220F, N220R or N220Y), in combination with catalysis at a pH well above the hydrolytic optima. Moreover, the mechanism behind the efficiency of the mutation as well as the dependence on pH was investigated. MD simulations suggest a distortion of the catalytic acid/base for the mutants, which is not only a catalytic residue but also important for directing the catalytic water. In addition, the hydrolytic activity is diminished at high pH due to deprotonation of the catalytic acid, while the transglycosylation is retained. This suggests that transglycosylation with hexanol as acceptor is not dependent on a catalytic acid/base, the mechanistic reason for which is still unclear. Further studies are required to increase the molecular understanding of this phenomenon, to allow general applicability of this novel strategy for the generation of non-Leloir glycosyltransferases.
References
Affleck R, Haynes CA, Clark DS (1992) Solvent dielectric effects on protein dynamics. Proc Natl Acad Sci 89(11):5167–5170. doi:10.1073/pnas.89.11.5167
Arab-Jaziri F, Bissaro B, Tellier C, Dion M, Faure R, O’Donohue MJ (2015) Enhancing the chemoenzymatic synthesis of arabinosylated xylo-oligosaccharides by GH51 α-L-arabinofuranosidase. Carbohydr Res 401:64–72. doi:10.1016/j.carres.2014.10.029
Armand S, Andrews SR, Charnock SJ, Gilbert HJ (2001) Influence of the aglycone region of the substrate binding cleft of Pseudomonas xylanase 10A on catalysis. Biochem 40(25):7404–7409. doi:10.1021/bi002704r
Aronson NN, Halloran BA, Alexeyev MF, Zhou XE, Wang YJ, Meehan EJ, Chen LQ (2006) Mutation of a conserved tryptophan in the chitin-binding cleft of Serratia marcescens chitinase A enhances transglycosylation. Biosci Biotech Bioch 70(1):243–251
Barends TRM, Bultema JB, Kaper T, van der Maarel MJEC, Dijkhuizen L, Dijkstra BW (2007) Three-way stabilization of the covalent intermediate in amylomaltase, an α-amylase-like transglycosylase. J Biol Chem 282(23):17242–17249. doi:10.1074/jbc.M701444200
Bissaro B, Durand J, Biarnes X, Planas A, Monsan P, O’Donohue MJ, Faure R (2015a) Molecular design of non-Leloir furanose-transferring enzymes from an α-L-arabinofuranosidase: a rationale for the engineering of evolved transglycosylases. ACS Catal 5(8):4598–4611. doi:10.1021/acscatal.5b00949
Bissaro B, Monsan P, Faure R, O’Donohue MJ (2015b) Glycosynthesis in a waterworld: new insight into the molecular basis of transglycosylation in retaining glycoside hydrolases. Biochem J 467:17–35. doi:10.1042/Bj20141412
Bonnin E, Vigouroux J, Thibault JF (1997) Kinetic parameters of hydrolysis and transglycosylation catalyzed by an exo-β-(1,4)-galactanase. Enz Microb Tech 20(7):516–522. doi:10.1016/S0141-0229(96)00188-3
Broos J, Visser AJWG, Engbersen JFJ, Verboom W, vanHoek A, Reinhoudt DN (1995) Flexibility of enzymes suspended in organic solvents probed by time-resolved fluorescence anisotropy. Evidence that enzyme activity and enantioselectivity are directly related to enzyme flexibility. J Am Chem Soc 117(51):12657–12663. doi:10.1021/Ja00156a001
Champion E, Guerin F, Moulis C, Barbe S, Tran TH, Morel S, Descroix K, Monsan P, Mourey L, Mulard LA, Tranier S, Remaud-Simeon M, Andre I (2012) Applying pairwise combinations of amino acid mutations for sorting out highly efficient glucosylation tools for chemo-enzymatic synthesis of bacterial oligosaccharides. J Am Chem Soc 134(45):18677–18688. doi:10.1021/ja306845b
Chen S, Xing XH, Huang JJ, Xu MS (2011) Enzyme-assisted extraction of flavonoids from Ginkgo biloba leaves: improvement effect of flavonol transglycosylation catalyzed by Penicillium decumbens cellulase. Enz Microb Tech 48(1):100–105. doi:10.1016/j.enzmictec.2010.09.017
Chuenchor W, Pengthaisong S, Robinson RC, Yuvaniyama J, Svasti J, Cairns JRK (2011) The structural basis of oligosaccharide binding by rice BGlu1 β-glucosidase. J Struct Biol 173(1):169–179. doi:10.1016/j.jsb.2010.09.021
Cornish-Bowden A (2012) Fundamentals of enzyme kinetics, 4th edn. Wiley-VCH, Weinheim
Feng HY, Drone J, Hoffmann L, Tran V, Tellier C, Rabiller C, Dion M (2005) Converting a β-glycosidase into a β-transglycosidase by directed evolution. J Biol Chem 280(44):37088–37097. doi:10.1074/jbc.M502873200
Frutuoso MA, Marana SR (2013) A single amino acid residue determines the ratio of hydrolysis to transglycosylation catalyzed by β-glucosidases. Prot Pept Lett 20(1):102–106
Giacomini C, Irazoqui G, Gonzalez P, Batista-Viera F, Brena BM (2002) Enzymatic synthesis of galactosyl-xylose by Aspergillus oryzae β-galactosidase. J Mol Catal B-Enzym 19:159–165. doi:10.1016/S1381-1177(02)00163-7
Goderis H, Ampe G, Feyton M, Fouwe B, Guffens W, Van Cauwenbergh S, Tobback P (1987) Lipase-catalyzed ester exchange reactions in organic media with controlled humidity. Biotech Bioeng 30:258–266
Greenspan L (1977) Humidity fixed points of binary saturated aqueous solutions. J Res Natl Bureau Standards A Phys Chem 81A(1):89–96
Hansson T, Andersson M, Wehtje E, Adlercreutz P (2001) Influence of water activity on the competition between β-glycosidase-catalysed transglycosylation and hydrolysis in aqueous hexanol. Enzym Microb Technol 29(8–9):527–534. doi:10.1016/s0141-0229(01)00421-5
Hermans J, Berendsen HJC, Vangunsteren WF, Postma JPM (1984) A consistent empirical potential for water-protein interactions. Biopolymers 23(8):1513–1518. doi:10.1002/bip.360230807
Honda Y, Fushinobu S, Hidaka M, Wakagi T, Shoun H, Taniguchi H, Kitaoka M (2008) Alternative strategy for converting an inverting glycoside hydrolase into a glycosynthase. Glycobiol 18(4):325–330. doi:10.1093/glycob/cwn011
Johansson P, Brumer H, Baumann MJ, Kallas AM, Henriksson H, Denman SE, Teeri TT, Jones TA (2004) Crystal structures of a poplar xyloglucan endotransglycosylase reveal details of transglycosylation acceptor binding. Plant Cell 16(4):874–886. doi:10.1105/tpc.020065
Joshi MD, Sidhu G, Nielsen JE, Brayer GD, Withers SG, McIntosh LP (2001) Dissecting the electrostatic interactions and pH-dependent activity of a family 11 glycosidase. Biochem 40(34):10115–10139. doi:10.1021/bi0105429
Joshi MD, Sidhu G, Pot I, Brayer GD, Withers SG, McIntosh LP (2000) Hydrogen bonding and catalysis: a novel explanation for how a single amino acid substitution can change the pH optimum of a glycosidase. J Mol Biol 299(1):255–279. doi:10.1006/jmbi.2000.3722
Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL (2001) Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B 105(28):6474–6487. doi:10.1021/jp003919d
Kaper T, Leemhuis H, Uitdehaag JCM, van der Veen BA, Dijkstra BW, van der Maarel MJEC, Dijkhuizen L (2007) Identification of acceptor substrate binding subsites +2 and +3 in the amylomaltase from Thermus thermophilus HB8. Biochem 46(17):5261–5269. doi:10.1021/bi602408j
Kelly RM, Leemhuis H, Rozeboom HJ, van Oosterwijk N, Dijkstra BW, Dijkhuizen L (2008) Elimination of competing hydrolysis and coupling side reactions of a cyclodextrin glucanotransferase by directed evolution. Biochem J 413(3):517–525. doi:10.1042/BJ20080353
Kone FM, Le Bechec M, Sine JP, Dion M, Tellier C (2009) Digital screening methodology for the directed evolution of transglycosidases. Prot Eng, Design Sel: PEDS 22(1):37–44. doi:10.1093/protein/gzn065
Koshland DE (1953) Stereochemistry and the mechanism of enzymatic reactions. Biol Rev 28(4):416–436. doi:10.1111/j.1469-185X.1953.tb01386.x
Krivov GG, Shapovalov MV, Dunbrack RL (2009) Improved prediction of protein side-chain conformations with SCWRL4. Prot-Struct Func Bioinf 77(4):778–795. doi:10.1002/prot.22488
Kuriki T, Kaneko H, Yanase M, Takata H, Shimada J, Handa S, Takada T, Umeyama H, Okada S (1996) Controlling substrate preference and transglycosylation activity of neopullulanase by manipulating steric constraint and hydrophobicity in active center. J Biol Chem 271(29):17321–17329
Kurosu J, Sato T, Yoshida K, Tsugane T, Shimura S, Kirimura K, Kino K, Usami S (2002) Enzymatic synthesis of α-arbutin by α-anomer-selective-glucosylation of hydroquinone using lyophilized cells of Xanthomonas campestris WU-9701. J Biosci Bioeng 93(3):328–330. doi:10.1263/Jbb.93.328
Laemmli UK (1970) Cleavage of structural proteins during assembly of head of bacteriophage-T4. Nature 227(5259):680. doi:10.1038/227680a0
Larsbrink J, Izumi A, Hemsworth GR, Davies GJ, Brumer H (2012) Structural enzymology of Cellvibrio japonicus Agd31B protein reveals α-transglucosylase activity in glycoside hydrolase family 31. J Biol Chem 287(52):43288–43299. doi:10.1074/jbc.M112.416511
Lu Y, Yang HT, Hu HY, Wang Y, Rao ZH, Jin C (2009) Mutation of Trp137 to glutamate completely removes transglycosyl activity associated with the Aspergillus fumigatus AfChiB1. Glycoconj J 26(5):525–534. doi:10.1007/s10719-008-9203-z
Luang S, Cho J-I, Mahong B, Opassiri R, Akiyama T, Phasai K, Komvongsa J, Sasaki N, Hua Y-L, Matsuba Y, Ozeki Y, Jeon J-S, Cairns JRK (2013) Rice Os9BGlu31 is a transglucosidase with the capacity to equilibrate phenylpropanoid, flavonoid, and phytohormone glycoconjugates. J Biol Chem 288(14):10111–10123. doi:10.1074/jbc.M112.423533
Lundemo P, Adlercreutz P, Karlsson EN (2013) Improved transferase/hydrolase ratio through rational design of a family 1 β-glucosidase from Thermotoga neapolitana. Appl Environ Microb 79(11):3400–3405. doi:10.1128/Aem.00359-13
Lundemo P, Karlsson EN, Adlercreutz P (2014) Preparation of two glycoside hydrolases for use in micro-aqueous media. J Mol Catal B-Enzym 108:1–6. doi:10.1016/j.molcatb.2014.06.009
Mackenzie LF, Wang QP, Warren RAJ, Withers SG (1998) ) Glycosynthases: mutant glycosidases for oligosaccharide synthesis. J Am Chem Soc 120(22):5583–5584. doi:10.1021/Ja980833d
Mala S, Dvorakova H, Hrabal R, Kralova B (1999) Towards regioselective synthesis of oligosaccharides by use of α-glucosidases with different substrate specificity. Carbohyd Res 322(3–4):209–218. doi:10.1016/S0008-6215(99)00222-0
Malet C, Planas A (1998) From β-glucanase to β-glucansynthase: glycosyl transfer to α-glycosyl fluorides catalyzed by a mutant endoglucanase lacking its catalytic nucleophile. FEBS Lett 440(1–2):208–212. doi:10.1016/S0014-5793(98)01448-3
McIntosh LP, Hand G, Johnson PE, Joshi MD, Korner M, Plesniak LA, Ziser L, Wakarchuk WW, Withers SG (1996) The pKa of the general acid/base carboxyl group of a glycosidase cycles during catalysis: a 13C-NMR study of Bacillus circulans xylanase. Biochem 35(31):9958–9966. doi:10.1021/bi9613234
Mladenoska I, Grey CE, Winkelhausen E, Kuzmanova S, Adlercreutz P (2007) Competition between transglycosylation and hydrolysis in almond β-glucosidase-catalyzed conversion of p-nitrophenyl-β-D-glucoside in monophasic water/alcohol mixtures. Biocatal Biotransform 25(5):382–385. doi:10.1080/10242420701510494
Mladenoska I, Winkelhausen E, Kuzmanova S (2008) Transgalactosylation/hydrolysis ratios of various β-galactosidases catalyzing alkyl-β-galactoside synthesis in single-phased alcohol media. Food Technol Biotech 46(3):311–316
Oikawa T, Tsukagawa Y, Chino M, Soda K (2001) Increased transglycosylation activity of Rhodotorula glutinis endo-β-glucanase in media containing organic solvent. Biosci Biotech Bioch 65(8):1889–1892. doi:10.1271/Bbb.65.1889
Olsson MHM, Sondergaard CR, Rostkowski M, Jensen JH (2011) PROPKA3: consistent treatment of internal and surface residues in empirical pK (a) predictions. J Chem Theory Comput 7(2):525–537. doi:10.1021/ct100578z
Osanjo G, Dion M, Drone J, Solleux C, Tran V, Rabiller C, Tellier C (2007) Directed evolution of the α-L-fucosidase from Thermotoga maritima into an α-L-transfucosidase. Biochem 46(4):1022–1033. doi:10.1021/bi061444w
Perez-Sanchez M, Cabrera AC, Garcia-Martin H, Sinisterra JV, Garcia JI, Hernaiz MJ (2011) Improved synthesis of disaccharides with Escherichia coli β-galactosidase using bio-solvents derived from glycerol. Tetrahedron 67(40):7708–7712. doi:10.1016/j.tet.2011.08.009
Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29(7):845–854. doi:10.1093/bioinformatics/btt055
Ribeirao M, Pereira-Chioccola VL, Eichinger D, Rodrigues MM, Schenkman S (1997) Temperature differences for trans-glycosylation and hydrolysis reaction reveal an acceptor binding site in the catalytic mechanism of Trypanosoma cruzi trans-sialidase. Glycobiol 7(8):1237–1246. doi:10.1093/glycob/7.8.1237
Ribeiro MH (2011) Naringinases: occurrence, characteristics, and applications. Appl Microbiol Biot 90(6):1883–1895. doi:10.1007/s00253-011-3176-8
Roitner M, Schalkhammer T, Pittner F (1984) Characterization of naringinase from Aspergillus niger. Monatsh Chem 115(10):1255–1267
Rojas-Cervellera V, Ardevol A, Boero M, Planas A, Rovira C (2013) Formation of a covalent glycosyl-enzyme species in a retaining glycosyltransferase. Chem-Eur J 19(42):14018–14023. doi:10.1002/chem.201302898
Saitoh H, Takagaki K, Majima M, Nakamura T, Matsuki A, Kasai M, Narita H, Endo M (1995) Enzymatic reconstruction of glycosaminoglycan oligosaccharide chains using the transglycosylation reaction of bovine testicular hyaluronidase. J Biol Chem 270(8):3741–3747
Seidle HF, Huber RE (2005) Transglucosidic reactions of the Aspergillus niger family 3 β-glucosidase: qualitative and quantitative analyses and evidence that the transglucosidic rate is independent of pH. Arch Biochem Biophys 436(2):254–264. doi:10.1016/j.abb.2005.02.017
Soares CM, Teixeira VH, Baptista AM (2003) Protein structure and dynamics in nonaqueous solvents: insights from molecular dynamics simulation studies. Biophys J 84(3):1628–1641
Taira T, Fujiwara M, Dennhart N, Hayashi H, Onaga S, Ohnuma T, Letzel T, Sakuda S, Fukamizo T (2010) Transglycosylation reaction catalyzed by a class V chitinase from cycad, Cycas revoluta: a study involving site-directed mutagenesis, HPLC, and real-time ESI-MS. BBA-Proteins Proteom 1804(4):668–675. doi:10.1016/j.bbapap.2009.10.015
Teze D, Hendrickx J, Czjzek M, Ropartz D, Sanejouand YH, Tran V, Tellier C, Dion M (2014) Semi-rational approach for converting a GH1 β-glycosidase into a β-transglycosidase. Prot Eng Des Sel 27(1):13–19. doi:10.1093/protein/gzt057
Turner C, Turner P, Jacobson G, Almgren K, Waldeback M, Sjoberg P, Karlsson EN, Markides KE (2006) Subcritical water extraction and β-glucosidase-catalyzed hydrolysis of quercetin glycosides in onion waste. Green Chem 8(11):949–959
Uitdehaag JCM, Mosi R, Kalk KH, van der Veen BA, Dijkhuizen L, Withers SG, Dijkstra BW (1999) X-ray structures along the reaction pathway of cyclodextrin glycosyltransferase elucidate catalysis in the α-amylase family. Nat Struct Biol 6(5):432–436. doi:10.1038/8235
van Rantwijk F, Oosterom MWV, Sheldon RA (1999) Glycosidase-catalysed synthesis of alkyl glycosides. J Mol Catal B-Enzym 6(6):511–532. doi:10.1016/S1381-1177(99)00042-9
Yamamoto I, Muto N, Nagata E, Nakamura T, Suzuki Y (1990) Formation of a stable L-ascorbic-acid α-glucoside by mammalian α-glucosidase-catalyzed transglucosylation. Biochim Biophys Acta 1035(1):44–50. doi:10.1016/0304-4165(90)90171-R
Acknowledgments
This work was supported by the Swedish Research Council Formas (Grant no. 213-2012-820).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical statement
This work was supported by the Swedish Research Council Formas (Grant no. 213–2012-820). This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(PDF 447 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lundemo, P., Karlsson, E.N. & Adlercreutz, P. Eliminating hydrolytic activity without affecting the transglycosylation of a GH1 β-glucosidase. Appl Microbiol Biotechnol 101, 1121–1131 (2017). https://doi.org/10.1007/s00253-016-7833-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7833-9