
Abstract
Clostridium perfringens is a cause for increasing concern due to its responsibility for severe infections both in humans and animals, especially poultry. To find new control strategies to treat C. perfringens infection, we investigated the activity and delivery of a bacteriophage endolysin. We identified a new endolysin, designated CP25L, which shows similarity to an N-acetylmuramoyl-l-alanine amidase domain and is distinct from other C. perfringens endolysins whose activity has been demonstrated in vitro. The cp25l gene was cloned and expressed in Escherichia coli, and the gene product demonstrated lytic activity against all 25 C. perfringens strains tested. The probiotic strain Lactobacillus johnsonii FI9785 was engineered to deliver the endolysin to the gastrointestinal tract. The integration of the nisRK two-component regulatory system from the Lactococcus lactis nisin A biosynthesis operon into the chromosome of L. johnsonii allowed constitutive expression of the endolysin under the control of the nisA promoter (P nisA ), while the use of a signal peptide (SLPmod) led to successful secretion of the active endolysin to the surrounding media. The high specificity and activity of the endolysin suggest that it may be developed as an effective tool to enhance the control of C. perfringens by L. johnsonii in the gastrointestinal tract.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Clostridium perfringens is a rod-shaped, Gram-positive, anaerobic spore-forming bacterium with the ability to produce potent toxins. According to the pattern of toxin expression, C. perfringens strains are classified into five toxinotypes: A, B, C, D, and E (Miyamoto et al. 2012; Petit et al. 1999). The interest in this microorganism is increasing because it is responsible for a range of debilitating conditions, including food poisoning, gas gangrene, necrotic enteritis (NE) both in humans and animals, and non-food-borne gastrointestinal infections (Keyburn et al. 2008; Lindstrom et al. 2011). C. perfringens associated food poisoning is classed among the most common gastrointestinal diseases in industrialized countries (Lindstrom et al. 2011; Scallan et al. 2011). One of the most frequently occurring infections caused by C. perfringens type A is NE in poultry; in countries where the use of antimicrobial growth promoters in feed has been abolished, the incidence of NE has increased, and the productivity of the poultry industry has been reduced (Lee et al. 2011; Van Immerseel et al. 2009). A range of different alternatives to the conventional antimicrobial growth promoter approach are being investigated to reduce the numbers of these pathogens in the food chain and improve poultry health. In addition to vaccination (Lee et al. 2011) or the use of natural antimicrobials (Timbermont et al. 2010), there are a number of examples of using probiotic microbes (Gil de los Santos et al. 2012; Kizerwetter-Swida and Binek 2009; La Ragione et al. 2004) or lytic enzymes (Liu et al. 2010; Zhang et al. 2010) to control NE in poultry. Bacteriophages and their endolysins present a further opportunity to reduce C. perfringens without disturbing the balance of the microbiota in the gastrointestinal (GI) tract.
Bacteriophage therapy is a rapidly growing area of research (Lu et al. 2011; O'Flaherty et al. 2009), and tests in poultry have demonstrated the potential of bacteriophages to control levels of C. perfringens, Salmonella, and Campylobacter (Atterbury et al. 2007; El-Shibiny et al. 2009; Miller et al. 2010). However, the narrow host range of bacteriophages and the rapid development of resistance can limit their application as control agents. An alternative approach is to develop bacteriophage proteins as novel antimicrobial agents. At the end of their lytic cycle, most dsDNA bacteriophages release their progeny by lysis of the host cell via the activity of two proteins—the holin and the endolysin. The holins are small hydrophobic proteins which disrupt the cell membrane to produce holes, allowing the endolysin to access the cell wall (Catalao et al. 2012). The endolysins target bonds in the peptidoglycan, resulting in the destruction of the cell and the release of newly produced virions. Although evolved to act from inside the cell, endolysins which act on Gram-positive bacteria are also effective when applied exogenously, giving them the potential to be used as targeted antibacterial agents (Fischetti 2010).
Although there are several variations in endolysin architecture, the majority of endolysins from bacteriophages infecting Gram-positive bacteria consist of two functional regions: the enzymatically active region contains one or more domains providing the catalytic activity of the enzyme (e.g., muramidase, glucosaminidase, endopeptidase, or amidase), attacking specific bonds in the peptidoglycan, while the cell wall binding region allows the endolysin to bind to specific ligands found in the cell wall of the host bacterium and so provides specificity (Schmelcher et al. 2012). Properties such as this specificity for their host pathogen, leading to minimal disturbance of the normal flora, the low risk of bacterial resistance, and the ability to kill colonizing pathogens on mucosal surfaces make them ideal anti-infective agents (Fischetti 2010; O’Flaherty et al. 2009). Recent studies demonstrated the efficiency of endolysins in killing or controlling pathogenic bacteria in animal models (Fenton et al. 2010; Grandgirard et al. 2008; Gupta et al. 2011; Yoong et al. 2006); they have also shown efficacy against biofilms (Meng et al. 2011), as disinfectants (Hoopes et al. 2009) and as the basis of detection systems for bacteria (Kretzer et al. 2007). Several bacteriophage endolysins which produce lytic activity against C. perfringens cells have been described (Nariya et al. 2011; Schmitz et al. 2011; Simmons et al. 2010; Zimmer et al. 2002). A number have also been identified by sequencing and genome mining, producing a rich resource of information for such enzymes (Morales et al. 2012; Oakley et al. 2011; Schmitz et al. 2011; Volozhantsev et al. 2012; Volozhantsev et al. 2011).
Lactobacillus johnsonii FI9785 is a poultry-isolated strain which has been shown to act as a competitive exclusion agent against C. perfringens in chickens (La Ragione et al. 2004). As well as acting as probiotics in their own right, lactic acid bacteria have also shown promise as delivery systems for the secretion of biologically active interleukins or peptidoglycan hydrolases (Braat et al. 2006; Fernandez et al. 2009; Turner et al. 2007). The promoter of the structural gene encoding the lantibiotic nisin A (P nisA ) is induced by nisin A via signal transduction using the two component regulatory system NisRK (Kuipers et al. 1995). This system has been exploited to develop gene expression systems in Lactococcus lactis, Leuconostoc lactis, and Lactobacillus helveticus (de Ruyter et al. 1996; Kleerebezem et al. 1997), and in this study, we have used the NisRK system to express the endolysin CP25L, originating from a prophage of C. perfringens (Gervasi et al. 2013), in a probiotic strain of L. johnsonii which was engineered for delivery to the GI tract.
Materials and methods
Bacterial strains and growth conditions
C. perfringens strains (listed in Table 1) were obtained from the NCTC (HPA, London, UK), or from in-house culture collections (IFR, Norwich, UK). C. perfringens 5416–97 is a type A strain (Grant et al. 2008). Strains were maintained in Robertson’s cooked-meat medium (SGL) at room temperature and were grown anaerobically at 37 °C in brain heart infusion broth (BHI, Oxoid) supplemented with vitamin K [50 μg/l], hemin [5 mg/l], resazurin [1 mg/l], and l-cysteine [0.5 g/l]), termed BHI + C. Escherichia coli strains were grown in L broth with shaking at 37 °C, and L. johnsonii strain FI9785 (NCIMB 41621) (La Ragione et al. 2004) and derivatives were grown in MRS broth (Oxoid) at 37 °C. Commensal, environmental, and clostridial strains (listed in Tables 1 and 2) were obtained from IFR culture collections, the DSMZ (Braunschweig, Germany), or the NCIMB (Aberdeen, UK) and were grown as recommended by DSMZ or in BHI + C.
Endolysin CP25L analysis and subcloning
The endolysin sequence is available in the vB_CpeS-CP51 genome nucleotide sequence, accession number KC237729. Endolysin amino acid similarities and conserved domains were determined using Blastp (Altschul et al. 1997) and the NCBI non-redundant protein sequences database. Amino acid alignments were performed using the ClustalW algorithm in Vector NTI (Invitrogen) and visualized with GeneDoc (Nicholas et al. 1997).
Genomic DNA was extracted from cells of C. perfringens 5416–97 (grown to mid-exponential phase) using the Genomic DNA extraction kit with a Genomic Tip 20/G column (Qiagen) as described by the manufacturer, with the addition of 50 U mutanolysin (Sigma) to aid cell lysis. The putative endolysin gene cp25l was amplified from genomic DNA using Phusion DNA polymerase (Finnzymes). To facilitate cloning into the NdeI and XhoI sites of expression vector pET15b (Novagen), primers were designed to incorporate an NdeI site at the initiating methionine codon CP25L_NDE (5’-GAATGTCATATGTATATAAATCAATCA-3’, altered nucleotides underlined throughout) and a XhoI site downstream of the stop codon CP25L_XHO (5’-ACTCGAGGTGGGATAATTCCTACC-3’). Due to the presence of an internal NdeI site, internal primers were designed to create a 1 bp substitution (T771C) without changing the resulting amino acid sequence. Primer CP25L_GTG (5’-TATTGCATGTGGTAGTCAAAAGAT-3’) was used in conjunction with CP25L_NDE, and primer CP25L_CAC (5’-TTGACTACCACATGCAATATAAAAATG-3’) was paired with CP25L_XHO, then the two products were spliced and amplified using overlap extension polymerase chain reaction (Horton et al. 1989) with primers CP25L_NDE and CP25L_XHO. The spliced product was restricted with NdeI and XhoI (New England Biolabs) and cloned into restricted pET15b that had been dephosphorylated with Antarctic Phosphatase (New England Biolabs), using Fastlink ligase (Epicentre). Ligation products were transformed into chemically competent E. coli TOP10 cells (Invitrogen), and transformants were selected with ampicillin (100 μg/ml). After confirmation by sequencing, construct pET15b-cp25l and the empty control vector pET15b were transformed for expression into chemically competent E. coli BL21(DE3) cells (Invitrogen).
An endolysin expression and secretion construct was produced in pUK200 (Wegmann et al. 1999) placing the endolysin coding sequence downstream of the signal peptide SLPmod (Fernandez et al. 2009), and a 6xHis tag, all under the control of the promoter P nisA (pUK200-slpmod-6xHis-cp25l). Ligation products were transformed into electrocompetent E. coli MC1022, selected with chloramphenicol (15 μg/ml) and confirmed by sequencing. To provide the two-component regulatory system required for PnisA-controlled expression, the nisRK genes from L. lactis FI5876 (Dodd et al. 1992) were inserted into the chromosome of L. johnsonii FI9785 using the thermosensitive pG + host9 vector (Maguin et al. 1996). An integration region was amplified from L. johnsonii FI9785 genomic DNA, and the resulting 1,033-bp product was cloned into the blunt-ended SpeI site of pG + host9 (creating plasmid pFI2657). A 2.4-kb region encoding nisRK was amplified from the genome of L. lactis FI5876 using the primer pair nisRK_F (5’-CCCGGGAGAATCTTAAAGAGTCTAGGG-3’) and nisRK_R (5’-AAAAAGTAATCCTTAGAGATTAC-3’) and cloned into a blunt-ended BstEII site located within the integration region of pFI2657, creating pFI2652. Subsequently, L. johnsonii FI9785 was transformed with pFI2652, and the nisRK sequences were integrated into the chromosome by gene replacement (Maguin et al. 1996) using 30 °C as the permissive temperature and 42 °C as the non-permissive temperature, to create the nisRK-carrying strain FI10744. The lysin expression construct and the control vector pUK200 were transformed into electrocompetent L. johnsonii FI10744 as described (Horn et al. 2005), and positive transformants were selected using chloramphenicol (7.5 μg/ml) to give the endolysin delivery strain FI10744-L and the vector control FI10744-V.
Protein expression, analysis, and partial purification
Crude protein extracts were produced from IPTG-induced E. coli BL21(DE3) cells containing pET15b-cp25l or pET15b in NP buffer (20 mM sodium phosphate buffer pH 6.5) as previously described (Mayer et al. 2008). His-CP25L was purified using the nickel-nitrilotriacetic acid (Ni-NTA) Fast Start kit (Qiagen). For expression in L. johnsonii, cells were grown to mid-exponential phase then incubated for 2 h with or without nisin (10 ng/ml). Cells were harvested by centrifugation for 10 min at 2,500×g and 4 °C, and the cell pellet was frozen while the supernatant was concentrated 20-fold using Amicon columns (Ultra-4 ultracell-30 k, Millipore) or 166-fold by shaking for 5 min after the addition of an equal volume of 100 % ethanol pre-cooled to −80 °C, followed by centrifugation for 30 min at 10,000×g and 4 °C. The resulting pellet was freeze-dried prior to resuspension in Ni-NTA elution buffer (EB, 50 mM sodium phosphate, 300 mM NaCl, 250 mM imidazole pH 8). Crude cell extracts were produced in EB by bead beating as with E. coli. Protein quantification, electrophoresis, and Western blot analysis with a His Tag® monoclonal antibody (Novagen) were all performed as described previously (Mayer et al. 2008).
Lysis assays
The lysis of C. perfringens NCTC 3110 cells resuspended in phosphate-buffered saline (PBS) was assessed by turbidity reduction assay as previously described (Mayer et al. 2008) in duplicate 300-μl assays comprising 270 μl resuspended cells and 30 μl crude or Ni-NTA purified endolysin extract or the appropriate buffer controls. Cells for lysis assays were harvested by centrifugation, flash-frozen in liquid nitrogen, and stored at −20 °C until use. Hen egg white lysozyme (Sigma) was used as a positive control at 500 U per 300 μl assay. The activity in different media was assessed by replacing the PBS with BHI + C or chemostat medium, a complex medium used for in vitro fermentations (CM: peptone water 2 g/l (Oxoid), yeast extract 2 g/l (Oxoid), NaCl 0.1 g/l, K2HPO4 0.04 g/l, KH2PO4 0.04 g/l, MgSO4.7H2O 0.01 g/l, CaCl2.6H2O 0.01 g/l, NaHCO3 2 g/l, Tween 80 2 ml, hemin 0.02 g/l dissolved in 400 μL 1 M NaOH, vitamin K1 10 μl of 5 % v/v aqueous solution, cysteine HCl 0.5 g/l, bile salts 0.5 g/l, and 1 % glucose all from Sigma (Mandalari et al. 2007)), which was adjusted to pH 6 to approximate colon conditions. Activity against different species was assessed using 1 μg of Ni-NTA purified protein during a 1-h incubation. The endolysin activity of crude protein extracts and concentrated culture supernatants from the engineered L. johnsonii strains was measured by plate assay as described previously (Stentz et al. 2010). Briefly, C. perfringens cells were grown to stationary phase in 100 ml BHI broth, autoclaved, pelleted by centrifugation then resuspended in 1 ml of PBS, and added to 100 ml of PBS with 1 % agar at 55 °C. After pouring into plates, small holes were punched into the seeded agar, and 10 μl of protein extract (equivalent to 30 μg total crude protein) or 20 μl of concentrated supernatants was loaded into the wells, and the plates were then incubated for 24 h at 37 °C. To demonstrate lytic activity from growing colonies, C. perfringens cells were processed in the same way then incorporated into plates containing MRS agar with 7.5 μg/ml chloramphenicol. Overnight cultures of FI10744-L and FI10744-V were subsequently streaked onto these plates, and the plates were incubated for 1–3 days at 37 °C.
C. perfringens NCTC 3110 viability assays were performed by adding 166 μg of Ni-NTA purified CP25L protein to 5 ml of pre-reduced BHI + C under anaerobic conditions. The protein was added to the media either immediately before C. perfringens inoculation (20 μl of a fresh overnight culture) or during the early exponential phase of growth (optical density [OD600] ca. 0.3). To determine C. perfringens viability, samples were taken every 2 h for 8 h and serially diluted in PBS before being plated onto BHI agar and incubated for 24 to 48 h at 37 °C. Results were calculated as the total number of bacteria from triplicate counts.
Results
Bioinformatic analysis of the CP25L sequence
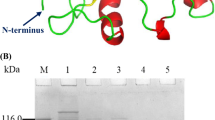
The cp25l gene was identified previously within the genome of a mitomycin C-inducible prophage of C. perfringens 5416–97, vB_CpeS-CP51 (Gervasi et al. 2013). Blastp analysis of CP25L identified two regions with similarity to conserved domains separated by a central section (Fig. 1). In the N-terminal part of the protein, there is an area with similarity to domains associated with cell wall binding and lysis, including the peptidoglycan recognition protein (PGRP, cd06583, amino acids 19–135, E value 1.24e-23), lysozyme (PHA00447, amino acids 22–134, E value 3.62e-25) and amidase_2 (N-acetylmuramoyl-l-alanine amidase, pfam01510, amino acids 20–128, E value 2.37e-22). An area with similarity to a bacterial SH3 domain (SH3_3, pfam08239, amino acids 322–375, E value 4.14e-09) is located at the C terminus. Alignments of this protein with other C. perfringens endolysins whose activity has been demonstrated experimentally revealed that there are only very low levels of amino acid conservation with the amidase_3 domain-containing lysins ply3626 (Zimmer et al. 2002) (28 % consensus/14.7 % identity), plyCP26F, or plyCP39O (Simmons et al. 2010) (14.1 %/7.4 % and 13.5 %/7.7 %, respectively), or with the glycosyl hydrolase_25 domain-containing Pms (Nariya et al. 2011) (26.3 %/16.3 %) or PlyCM (Schmitz et al. 2011) (25.5 %/15.5 %). The conservation with Pms and PlyCM is highest in the region of the SH3_3 hypothetical domain near the C terminus, with 44.8 % consensus/29.3 % identity between this 54 amino acid region in CP25L and the corresponding regions in Pms and PlyCM. The region of the amidase_2 domain also showed some homology to predicted endolysins from podoviruses of C. perfringens (Volozhantsev et al. 2012); for example, the CP25L sequence shows 66.9 % consensus/57.4 % identity over the first 135 amino acids covering the amidase_2 domain with the predicted lysin from ΦZP2. Interestingly, when CP25L was analyzed by Blastp, the sequences showing the highest similarity had the amino acid conservation concentrated in the center of the sequence, between the enzymatic and SH3 domains in an area without homology to a functional domain (Fig. 1). These sequences were all annotated as putative amidases from C. perfringens sequenced genomes in the NCBI databases, and it is possible that conservation of this area is relevant to the target cell wall.

Alignment of the amino acid sequence of CP25L compared with putative N-acetylmuramoyl-l-alanine amidases from C. perfringens genomes identified by Blastp analysis. Coloring of letters indicates degree of amino acid conservation between sequences (black, 100 %, dark grey 80 %, light grey 60 %); lines above CP25L sequence indicate areas of homology with the PGRP and amidase_2 domains (black line) or the SH3_3 bacterial domain (grey)
Expression and activity of CP25L
CP25L was expressed in E. coli as a His-tagged protein, giving a product of c. 45 kD which was visible in crude extracts and bound a His-tag antibody (data not shown). Crude protein extracts showed lytic activity when incubated with C. perfringens cells (58.5 ± 1.8 % drop in OD600 over 4 min when using 33 μg/ml protein), while lytic activity of the crude protein extracts from empty vector control samples was minimal over this period and similar to buffer controls; lysis of cells which had not been frozen after harvest was slower (12.4 ± 0.1 %) but still resulted in complete lysis within 10 min (data not shown).
Ni-NTA columns were used to purify His-tagged CP25L from E. coli cells to assess lytic activity and host range of the endolysin (Fig. 2a). Strong lytic activity was shown by an effective drop in the OD600 reading within 6 min of addition of 3, 33, or 100 μg/ml of Ni-NTA-purified protein to frozen cells of C. perfringens (Fig. 2b). The speed of cell lysis obtained with 3 μg/ml of protein was similar to that obtained with 33 μg/ml, the OD600 dropping from c. 0.65 to c. 0.13 in less than 5 min. Lower concentrations gave slower lysis—0.3 μg/ml gave a 9.1 ± 0.1 % drop in OD600 over 4 min compared with 79.5 ± 1.4 % using 3 μg/ml, while 0.03 μg/ml failed to produce appreciable lysis. However, lysis from 100 μg/ml was slightly slower than that from 33 μg/ml; this phenomenon of reduced activity with higher concentrations has been observed previously (Mayer et al. 2010) and may be associated with precipitation of the protein at high concentrations. As noted with other clostridial species (Mayer et al. 2008; Mayer et al. 2012), slow gradual lysis of cells was also commonly observed in buffer controls.

Expression of CP25L in E. coli. a SDS-PAGE analysis of products from Ni-NTA column purification. Lanes 1, SeeBlue Plus2 marker, 2, cell lysate, 3, column flow through, 4–5 column washes, 6–8 successive 1 ml column eluates. Lanes contain 3.25 μl samples. b, c Lytic activity of Ni-NTA-purified protein on C. perfringens showing the effect of protein concentration (b) and lysis medium (c). b Closed square, 100 μg/ml; open square, 33 μg/ml; small closed square, 3 μg/ml; closed triangle, 0.3 μg/ml; and open triangle, 0.03 μg/ml of CP25L compared with buffer control (EB, closed circle). c Cells were resuspended in PBS (closed circle), BHI + C, (closed triangle), or CM (closed square) and incubated with 33 μg/ml CP25L (closed symbols) or EB (open symbols). Results are the mean of duplicate assays ± standard deviation
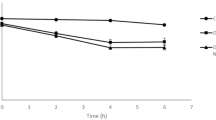
The Ni-NTA-purified CP25L was stable at 4 °C for several months, and its activity was not affected by a 30-min incubation at 37 °C or 45 °C; however, treatment at 65 °C for 30 min, or 42 °C overnight, or a pasteurization treatment at 72 °C for 20 s all abolished lytic activity (data not shown). Stability at room temperature was monitored over 3 weeks, the activity remaining largely unchanged after 8 days compared with a sample stored at 4 °C, but, after 15 and 22 days storage at room temperature, the endolysin activity had reduced to c. 83 % and c. 62 % respectively, compared with the protein samples stored at 4 °C (Fig. 3).

CP25L stability. Endolysin activity was measured in turbidity reduction assays using 33 μg/ml Ni-NTA-purified protein. Results represent the activity of samples stored at room temperature as a percentage of the activity of endolysin stored at 4 °C assayed at the same time; activity was calculated as the percent decrease in OD600 over 4 min of linear lysis using the mean of duplicate assays ± standard deviation
Lysis assays were performed to examine the activity of CP25L when cells were suspended in broth media such as BHI + C or CM (Fig. 2c). Lytic activity was observed when cells were resuspended in BHI + C but was reduced in comparison to PBS; however, the lysis in CM, a complex medium designed to simulate GI tract conditions, was more effective. When tested under anaerobic conditions in BHI + C growth medium, which allows rapid growth of C. perfringens, the addition of endolysin to C. perfringens culture at the point of cell inoculation resulted in a dramatic 5-log reduction in viable cell counts compared with the culture not exposed to endolysin (Fig. 4a). However, on continued culturing, the pathogen was able to recover its population density. When the lysin was added to the culture during the log phase, it failed to reduce the bacterial count (Fig. 4b).

Viability assays under anaerobic conditions to examine the effect of 33 μg/ml Ni-NTA-purified CP25L (black bars) on the viability of C. perfringens in comparison with the buffer control (white bars). Endolysin was added to the media either at point of inoculation (0 h) (a), or during the early exponential phase (OD600 0.3, 3.5 h) (b)
The host range of CP25L was tested using frozen cells of C. perfringens strains and a variety of clostridial and non-clostridial species. All of the 25 C. perfringens strains tested were sensitive to the endolysin, but the speed of lysis varied greatly (Table 1). For some strains, the drop in turbidity was immediate, while with others there was a lag of up to c. 20 min before lysis was detected. Sensitivity to the positive control lysozyme was also variable, with some strains being insensitive while others exhibited rapid lysis. There was no apparent relationship between sensitivity to the CP25L lysin and to lysozyme. In addition, CP25L also showed activity against Clostridium bifermentans, Clostridium acetobutylicum, Clostridium beijerinckii, Bacillus subtilis, and Bacillus cereus cells (Table 1) but failed to cause lysis of a range of other bacteria (data not shown, strains listed in Table 2).
L. johnsonii as an endolysin delivery vehicle
The cp25l gene was cloned into an expression and delivery system which combined expression from the P nisA , in conjunction with the chromosomally located signal transduction nisRK genes, with a signal peptide to secrete the lysin. To confirm lysin expression in this host, crude protein extracts from L. johnsonii were used in turbidity reduction assays (Fig. 5). This showed firstly that extracts from L. johnsonii carrying the lysin expression construct (FI10744-L) had a lytic activity that was absent from extracts of the empty vector control (FI10744-V) and secondly that lytic activity from cells induced with nisin was equivalent to that from uninduced cells. This demonstrates that the nisRK-P nisA system produced in L. johnsonii FI10744-L gives constitutive expression without the requirement for nisin induction. Secretion of the endolysin was assessed using L. johnsonii culture supernatants (Fig. 6a). The lytic activity associated with the concentrated supernatant was demonstrated in samples prepared from cultures of FI10744-L grown either with or without nisin induction, showing that the SLPmod signal peptide is effective for lysin secretion. Constitutive endolysin expression and secretion was further demonstrated by growing colonies of L. johnsonii on plates incorporating autoclaved C. perfringens cells, with clear development of zones of lysis around colonies expressing the lysin (Fig. 6b).

Endolysin expression in L. johnsonii. Frozen cells of C. perfringens were incubated with 33 μg/ml crude protein extracts from L. johnsonii FI10744-L (filled square) or FI10744-V (filled triangle), or EB (open circle). Filled symbols were from cultures induced with nisin; open symbols had no exposure to nisin. Results are the mean of duplicate assays ± standard deviation

Endolysin delivery from L. johnsonii. a Plate assay showing lysis from crude protein extracts (1–4) and concentrated supernatants (5–7, 20× concentrated; 9–12, 166× concentrated) from L. johnsonii FI10744-L induced with nisin (1, 5, 9) or uninduced (3, 7, 11), L. johnsonii FI10744-V induced with nisin (2, 6, 10) or uninduced (4, 12); 8, EB control. b Lytic zones produced by growing colonies of FI10744-L (L) compared with control FI10744-V (V) on media incorporating autoclaved C. perfringens
Discussion
C. perfringens is widely distributed in the intestine of animals, especially poultry, and can be pathogenic to the host. It is responsible both for severe infections and important economic losses (Lee et al. 2011; Scallan et al. 2011); targeting the pathogen in poultry will have impacts not just on poultry welfare and production but also on carriage in the food chain and the incidence of human infections. In this study, we have examined the ability of a novel endolysin to kill C. perfringens cells and demonstrated endolysin delivery by a probiotic strain of L. johnsonii.
The gene for CP25L is located within a prophage which can be released as a bacteriophage from the C. perfringens chromosome (Gervasi et al. 2013). In common with three other endolysins which have been demonstrated to show activity against C. perfringens, it is predicted to be an amidase (Simmons et al. 2010; Zimmer et al. 2002). A number of prophage lysin-like enzymes found in the genomes of C. perfringens contain an amidase domain, and the presence of an SH3 domain is also very prevalent in these proteins (Schmitz et al. 2011). In CP25L, the predicted SH3_3 domain is present in the C-terminal region commonly associated with endolysin specificity (Fischetti 2010; Schmelcher et al. 2012). The C-terminal and the central region between the amidase domain and the SH3_3 domain showed high similarity to other putative amidases from C. perfringens, and it will be interesting to investigate whether these regions are associated with recognition of the C. perfringens cell wall.
When expressed from E. coli, CP25L showed a potent lytic activity against C. perfringens NCTC 3110, a type B strain and was also active against all 24 other C. perfringens strains tested. However, activity varied widely between strains, even between two strains of the same type A (NCTC 8238 and 8239). Although sensitivity to the lysozyme positive control also varied to a lesser extent between strains, there was no relationship between the speed of lysis with lysozyme and that with CP25L. This large difference in activity between strains was also found with the amidase endolysins Ply3626, PlyCP39O, and PlyCP26F and the glucosyl hydrolase endolysins SM101 and PlyCM (Nariya et al. 2011; Schmitz et al. 2011; Simmons et al. 2010; Zimmer et al. 2002), although CP25L shows a slightly broader host range than most of these lysins. The sensitivity of other selected clostridial and Bacillus species might indicate shared binding targets in the cell walls of these organisms.
The ability to lyse cells in complex media at physiologically relevant pH values suggests that the endolysin might be able to function in GI tract conditions, without causing collateral damage to the normal microflora. However, as demonstrated in small cultures, despite initial evidence of control, the C. perfringens population was able to recover from endolysin treatment in a medium which favored rapid growth, while addition to a rapidly growing culture was ineffective. It is possible that the lysin remains attached to the cell walls of lysed cells, leading to a depletion in concentration and allowing rapidly growing cells to outstrip the lytic activity. Control might be more effective under less optimal growth conditions, such as in the competitive environment of the GI tract, and the lysin was shown to function in a complex medium that permits the growth of gut bacteria in simulated colon conditions with pH 6 and incubation temperature of 37 °C. However, survival of the lysin and contact with the C. perfringens target are further hurdles to overcome in the GI tract. Together with the finding that the lytic activity is not stable after prolonged incubation at 42 °C, these results indicate that a continuous high production of protein prior to infection would be required for pathogen control. This would be a feasible proposition in an environment where the endolysin was delivered to the gut via a colonizing probiotic.
L. johnsonii FI9785 was previously shown to reduce colonization and shedding of C. perfringens in chickens after a single oral dose (La Ragione et al. 2004), making it an ideal tool for preventative treatment. The use of a delivery system incorporating the signal peptide SLPmod, which has previously been demonstrated to be effective in the delivery of interleukin-12 to the mouse gut by L. lactis (Fernandez et al. 2009), gave successful production and secretion of a functional enzyme, as demonstrated by the lysis assays. Furthermore, the combination of the nisin A promoter and the chromosomally located signal transduction genes gave constitutive production without the need for nisin induction, which is of great potential benefit for use in animals. Production in L. johnsonii was lower than that in E. coli, and this is an area which may benefit from improvement. Another possibility is co-delivery or co-application with other antimicrobials—the combination of a phage endolysin with nisin achieved a strong synergistic effect against Staphylococcus aureus (Garcia et al. 2010), and the ability of lactobacilli to produce a range of bacteriocins, in addition to their other probiotic properties (Lebeer et al. 2008), make them valuable candidates as vehicles to deliver specific antimicrobials to gut communities. This preliminary study indicates that either large numbers of probiotic bacteria combined with high delivery rates and high activity of one or more antimicrobial or some method of attracting the bacterium to its target will be necessary for effective control.
The probiotic L. johnsonii has the capability to exclude C. perfringens from the GI tract of poultry birds. In this study, we have demonstrated its ability to express a biologically active anti-C. perfringens endolysin, indicating that the engineered strain has the potential to be an even more effective control agent for this pathogen. Future work will focus on the efficacy of the CP25L-secreting L. johnsonii FI10744-L strain in controlling C. perfringens in a GI tract environment, on its own or in co-application with other antimicrobials.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Atterbury RJ, Van Bergen MA, Ortiz F, Lovell MA, Harris JA, De Boer A, Wagenaar JA, Allen VM, Barrow PA (2007) Bacteriophage therapy to reduce Salmonella colonization of broiler chickens. Appl Environ Microbiol 73:4543–4549
Braat H, Rottiers P, Hommes DW, Huyghebaert N, Remaut E, Remon JP, van Deventer SJ, Neirynck S, Peppelenbosch MP, Steidler L (2006) A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn's disease. Clin Gastroenterol Hepatol 4:754–759
Catalao MJ, Gil F, Moniz-Pereira J, Sao-Jose C, Pimentel M (2012) Diversity in bacterial lysis systems: bacteriophages show the way. FEMS Microbiol Rev. doi:10.1111/1574-6976.12006
de Ruyter PG, Kuipers OP, de Vos WM (1996) Controlled gene expression systems for Lactococcus lactis with the food-grade inducer nisin. Appl Environ Microbiol 62:3662–3667
Dodd HM, Horn N, Hao Z, Gasson MJ (1992) A lactococcal expression system for engineered nisins. Appl Environ Microbiol 58:3683–3693
El-Shibiny A, Scott A, Timms A, Metawea Y, Connerton P, Connerton I (2009) Application of a group II Campylobacter bacteriophage to reduce strains of Campylobacter jejuni and Campylobacter coli colonizing broiler chickens. J Food Prot 72:733–740
Fenton M, Casey PG, Hill C, Gahan CG, Ross RP, McAuliffe O, O'Mahony J, Maher F, Coffey A (2010) The truncated phage lysin CHAP(k) eliminates Staphylococcus aureus in the nares of mice. Bioeng Bugs 1:404–407
Fernandez A, Horn N, Wegmann U, Nicoletti C, Gasson MJ, Narbad A (2009) Enhanced secretion of biologically active murine interleukin-12 by Lactococcus lactis. Appl Environ Microbiol 75:869–871
Fischetti VA (2010) Bacteriophage endolysins: a novel anti-infective to control Gram-positive pathogens. Int J Med Microbiol 300:357–362
Garcia P, Martinez B, Rodriguez L, Rodriguez A (2010) Synergy between the phage endolysin LysH5 and nisin to kill Staphylococcus aureus in pasteurized milk. Int J Food Microbiol 141:151–155
Gervasi T, Lo Curto R, Narbad A, Mayer MJ (2013) Complete genome sequence of PhiCP51, a temperate bacteriophage of Clostridium perfringens. Arch Virol. doi:10.1007/s00705-013-1647-1
Gil de los Santos JR, Storch OB, Fernandes CG, Gil-Turnes C (2012) Evaluation in broilers of the probiotic properties of Pichia pastoris and a recombinant P. pastoris containing the Clostridium perfringens alpha toxin gene. Vet Microbiol 156:448–451
Grandgirard D, Loeffler JM, Fischetti VA, Leib SL (2008) Phage lytic enzyme Cpl-1 for antibacterial therapy in experimental pneumococcal meningitis. J Infect Dis 197:1519–1522
Grant KA, Kenyon S, Nwafor I, Plowman J, Ohai C, Halford-Maw R, Peck MW, McLauchlin J (2008) The identification and characterization of Clostridium perfringens by real-time PCR, location of enterotoxin gene, and heat resistance. Foodborne Pathog Dis 5:629–639
Gupta R, Prasad Y (2011) P-27/HP endolysin as antibacterial agent for antibiotic resistant Staphylococcus aureus of human infections. Curr Microbiol 63:39–45
Hoopes JT, Stark CJ, Kim HA, Sussman DJ, Donovan DM, Nelson DC (2009) Use of a bacteriophage lysin, PlyC, as an enzyme disinfectant against Streptococcus equi. Appl Environ Microbiol 75:1388–1394
Horn N, Wegmann U, Narbad A, Gasson MJ (2005) Characterisation of a novel plasmid p9785S from Lactobacillus johnsonii FI9785. Plasmid 54:176–183
Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68
Keyburn AL, Boyce JD, Vaz P, Bannam TL, Ford ME, Parker D, Di Rubbo A, Rood JI, Moore RJ (2008) NetB, a new toxin that is associated with avian necrotic enteritis caused by Clostridium perfringens. PLoS Pathog 4:e26
Kizerwetter-Swida M, Binek M (2009) Protective effect of potentially probiotic Lactobacillus strain on infection with pathogenic bacteria in chickens. Pol J Vet Sci 12:15–20
Kleerebezem M, Beerthuyzen MM, Vaughan EE, de Vos WM, Kuipers OP (1997) Controlled gene expression systems for lactic acid bacteria: transferable nisin-inducible expression cassettes for Lactococcus, Leuconostoc, and Lactobacillus spp. Appl Environ Microbiol 63:4581–4584
Kretzer JW, Lehmann R, Schmelcher M, Banz M, Kim KP, Korn C, Loessner MJ (2007) Use of high-affinity cell wall-binding domains of bacteriophage endolysins for immobilization and separation of bacterial cells. Appl Environ Microbiol 73:1992–2000
Kuipers OP, Beerthuyzen MM, de Ruyter PG, Luesink EJ, de Vos WM (1995) Autoregulation of nisin biosynthesis in Lactococcus lactis by signal transduction. J Biol Chem 270:27299–27304
La Ragione RM, Narbad A, Gasson MJ, Woodward MJ (2004) In vivo characterization of Lactobacillus johnsonii FI9785 for use as a defined competitive exclusion agent against bacterial pathogens in poultry. Lett Appl Microbiol 38:197–205
Lebeer S, Vanderleyden J, De Keersmaecker SC (2008) Genes and molecules of lactobacilli supporting probiotic action. Microbiol Mol Biol Rev 72:728–764
Lee KW, Lillehoj HS, Jeong W, Jeoung HY, An DJ (2011) Avian necrotic enteritis: experimental models, host immunity, pathogenesis, risk factors, and vaccine development. Poult Sci 90:1381–1390
Lindstrom M, Heikinheimo A, Lahti P, Korkeala H (2011) Novel insights into the epidemiology of Clostridium perfringens type A food poisoning. Food Microbiol 28:192–198
Liu D, Guo Y, Wang Z, Yuan J (2010) Exogenous lysozyme influences Clostridium perfringens colonization and intestinal barrier function in broiler chickens. Avian Pathol 39:17–24
Lu TK, Koeris MS (2011) The next generation of bacteriophage therapy. Curr Opin Microbiol 14:524–531
Maguin E, Prevost H, Ehrlich SD, Gruss A (1996) Efficient insertional mutagenesis in lactococci and other gram-positive bacteria. J Bacteriol 178:931–935
Mandalari G, Nueno Palop C, Tuohy K, Gibson GR, Bennett RN, Waldron KW, Bisignano G, Narbad A, Faulds CB (2007) In vitro evaluation of the prebiotic activity of a pectic oligosaccharide-rich extract enzymatically derived from bergamot peel. Appl Microbiol Biotechnol 73:1173–1179
Mayer MJ, Narbad A, Gasson MJ (2008) Molecular characterization of a Clostridium difficile bacteriophage and its cloned biologically active endolysin. J Bacteriol 190:6734–6740
Mayer MJ, Payne J, Gasson MJ, Narbad A (2010) Genomic sequence and characterization of the virulent bacteriophage ΦCTP1 from Clostridium tyrobutyricum and heterologous expression of its endolysin. Appl Environ Microbiol 76:5415–5422
Mayer MJ, Gasson MJ, Narbad A (2012) Genomic sequence of bacteriophage ATCC 8074-B1 and activity of its endolysin and engineered variants against Clostridium sporogenes. Appl Environ Microbiol 78:3685–3692
Meng X, Shi Y, Ji W, Zhang J, Wang H, Lu C, Sun J, Yan Y (2011) Application of a bacteriophage lysin to disrupt biofilms formed by the animal pathogen Streptococcus suis. Appl Environ Microbiol 77:8272–8279
Miller RW, Skinner EJ, Sulakvelidze A, Mathis GF, Hofacre CL (2010) Bacteriophage therapy for control of necrotic enteritis of broiler chickens experimentally infected with Clostridium perfringens. Avian Dis 54:33–40
Miyamoto K, Li J, McClane BA (2012) Enterotoxigenic Clostridium perfringens: detection and identification. Microbes Environ 27:343–349
Morales CA, Oakley BB, Garrish JK, Siragusa GR, Ard MB, Seal BS (2012) Complete genome sequence of the podoviral bacteriophage PhiCP24R, which is virulent for Clostridium perfringens. Arch Virol 157:769–772
Nariya H, Miyata S, Tamai E, Sekiya H, Maki J, Okabe A (2011) Identification and characterization of a putative endolysin encoded by episomal phage phiSM101 of Clostridium perfringens. Appl Microbiol Biotechnol 90:1973–1979
Nicholas KB, H.B. N, Deerfield DW (1997) GeneDoc: analysis and visualization of genetic variation. EMBNEW.NEWS 4:14.
O'Flaherty S, Ross RP, Coffey A (2009) Bacteriophage and their lysins for elimination of infectious bacteria. FEMS Microbiol Rev 33:801–819
Oakley BB, Talundzic E, Morales CA, Hiett KL, Siragusa GR, Volozhantsev NV, Seal BS (2011) Comparative genomics of four closely related Clostridium perfringens bacteriophages reveals variable evolution among core genes with therapeutic potential. BMC Genomics 12:282
Petit L, Gibert M, Popoff MR (1999) Clostridium perfringens: toxinotype and genotype. Trends Microbiol 7:104–110
Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, Roy SL, Jones JL, Griffin PM (2011) Foodborne illness acquired in the United States—major pathogens. Emerg Infect Dis 17:7–15
Schmelcher M, Donovan DM, Loessner MJ (2012) Bacteriophage endolysins as novel antimicrobials. Future Microbiol 7:1147–1171
Schmitz JE, Ossiprandi MC, Rumah KR, Fischetti VA (2011) Lytic enzyme discovery through multigenomic sequence analysis in Clostridium perfringens. Appl Microbiol Biotechnol 89:1783–1795
Simmons M, Donovan DM, Siragusa GR, Seal BS (2010) Recombinant expression of two bacteriophage proteins that lyse Clostridium perfringens and share identical sequences in the C-terminal cell wall binding domain of the molecules but are dissimilar in their N-terminal active domains. J Agric Food Chem 58:10330–10337
Stentz R, Bongaerts RJ, Gunning AP, Gasson M, Shearman C (2010) Controlled release of protein from viable Lactococcus lactis cells. Appl Environ Microbiol 76:3026–3031
Timbermont L, Lanckriet A, Dewulf J, Nollet N, Schwarzer K, Haesebrouck F, Ducatelle R, Van Immerseel F (2010) Control of Clostridium perfringens-induced necrotic enteritis in broilers by target-released butyric acid, fatty acids and essential oils. Avian Pathol 39:117–121
Turner MS, Waldherr F, Loessner MJ, Giffard PM (2007) Antimicrobial activity of lysostaphin and a Listeria monocytogenes bacteriophage endolysin produced and secreted by lactic acid bacteria. Syst Appl Microbiol 30:58–67
Van Immerseel F, Rood JI, Moore RJ, Titball RW (2009) Rethinking our understanding of the pathogenesis of necrotic enteritis in chickens. Trends Microbiol 17:32–36
Volozhantsev NV, Oakley BB, Morales CA, Verevkin VV, Bannov VA, Krasilnikova VM, Popova AV, Zhilenkov EL, Garrish JK, Schegg KM, Woolsey R, Quilici DR, Line JE, Hiett KL, Siragusa GR, Svetoch EA, Seal BS (2012) Molecular characterization of podoviral bacteriophages virulent for Clostridium perfringens and their comparison with members of the Picovirinae. PLoS One 7:e38283
Volozhantsev NV, Verevkin VV, Bannov VA, Krasilnikova VM, Myakinina VP, Zhilenkov EL, Svetoch EA, Stern NJ, Oakley BB, Seal BS (2011) The genome sequence and proteome of bacteriophage PhiCPV1 virulent for Clostridium perfringens. Virus Res 155:433–439
Wegmann U, Klein JR, Drumm I, Kuipers OP, Henrich B (1999) Introduction of peptidase genes from Lactobacillus delbrueckii subsp. lactis into Lactococcus lactis and controlled expression. Appl Environ Microbiol 65:4729–4733
Yoong P, Schuch R, Nelson D, Fischetti VA (2006) PlyPH, a bacteriolytic enzyme with a broad pH range of activity and lytic action against Bacillus anthracis. J Bacteriol 188:2711–2714
Zhang G, Mathis GF, Hofacre CL, Yaghmaee P, Holley RA, Duranc TD (2010) Effect of a radiant energy-treated lysozyme antimicrobial blend on the control of clostridial necrotic enteritis in broiler chickens. Avian Dis 54:1298–1300
Zimmer M, Vukov N, Scherer S, Loessner MJ (2002) The murein hydrolase of the bacteriophage phi3626 dual lysis system is active against all tested Clostridium perfringens strains. Appl Environ Microbiol 68:5311–5317
Acknowledgements
The authors are grateful to Kerry I’Anson, Mike Peck, Clare Aldus, Sandra Stringer, and Lee Kellingray (IFR); Dr. M. Brett (Health Protection England, London); and Prof. R. M. La Ragione (AHVLA, UK) for the provision of strains, and Dr. E. Maguin (INRA) for provision of the pG + host9 vector. This research was funded by the Biotechnology and Biological Sciences Research Council Core Science Grants IFR/08/1 and BB/J004529/1, EU-funded project TORNADO (FP7-KBBE-222720) and by the University of Messina. The authors declare they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Gervasi, T., Horn, N., Wegmann, U. et al. Expression and delivery of an endolysin to combat Clostridium perfringens . Appl Microbiol Biotechnol 98, 2495–2505 (2014). https://doi.org/10.1007/s00253-013-5128-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-5128-y