
Abstract
Functional metagenomics, the study of the collective genome of a microbial community by expressing it in a foreign host, is an emerging field in biotechnology. Over the past years, the possibility of novel product discovery through metagenomics has developed rapidly. Thus, metagenomics has been heralded as a promising mining strategy of resources for the biotechnological and pharmaceutical industry. However, in spite of innovative work in the field of functional genomics in recent years, yields from function-based metagenomics studies still fall short of producing significant amounts of new products that are valuable for biotechnological processes. Thus, a new set of strategies is required with respect to fostering gene expression in comparison to the traditional work. These new strategies should address a major issue, that is, how to successfully express a set of unknown genes of unknown origin in a foreign host in high throughput. This article is an opinionating review of functional metagenomic screening of natural microbial communities, with a focus on the optimization of new product discovery. It first summarizes current major bottlenecks in functional metagenomics and then provides an overview of the general metagenomic assessment strategies, with a focus on the challenges that are met in the screening for, and selection of, target genes in metagenomic libraries. To identify possible screening limitations, strategies to achieve optimal gene expression are reviewed, examining the molecular events all the way from the transcription level through to the secretion of the target gene product.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
One of the major hurdles in microbial ecology is the inability to culture most of the microbial diversity present in ecosystems under laboratory conditions. The observed divergence between the numbers of bacterial cells forming colonies on plates and the cell count obtained by microscopic examination is known as “the great plate count anomaly” (Staley and Konopka 1985). In fact, only a fraction of the microbial diversity present in most ecosystems (1–5%) can be accessed through standard cultivation techniques (Curtis and Sloan 2004; Nichols 2007; Staley and Konopka 1985; Torsvik and Øvreås 2002). Thus, we can only speculate about the environmental importance and economical value of the majority of organisms that have remained unexplored so far. To access and explore this hitherto unexplored microbiota, the genetic material of the collective cells from an environmental sample can be directly extracted. This microbial community DNA, also known as the metagenome, can be further analyzed using modern technologies such as screens of constructed expression libraries and direct high-throughput sequencing. The molecular analysis strategies used to examine microbial metagenomes have been denoted as metagenomics techniques. Metagenomics has come a long way since the term was first introduced by Handelsman et al. (1998). Recently, the enormous potential of metagenomics to promote both bioexploration and our understanding of ecosystems has become clear (Hil and Fenical 2010; Imhoff et al. 2011; Lefevre et al. 2008; Mocali and Benedetti 2010; Riesenfeld et al. 2004; Singh and Macdonald 2010; Warnecke and Hess 2009). Clearly, the screening of metagenomic libraries allows one to study genes and functions from previously inaccessible microbes, opening up exiting new possibilities for the development of novel products (Fernández-Arrojo et al. 2010; Singh et al. 2008; Uchiyama and Miyazaki 2009;Warnecke et al. 2007).
Screens of metagenomic libraries have been performed by two fundamentally different strategies, i.e., using (1) a function-based approach and (2) a sequence-based approach (Kakirde et al. 2010; Schloss and Handelsman 2003). In the first strategy, screening is based on the detection of expression of target genes in the cloning host. In the second one, the focus is on the detection of target genetic sequences, for instance, by hybridization or PCR screening. An alternative is offered by direct sequencing.
Despite the potential for mining of genetic novelty, the yields from function-based metagenomic studies often fall short of yielding products with sufficient novelty for biotechnological processes (Beloqui et al 2008; Hil and Fenical 2010; Singh and Macdonald 2010). One key reason for this is likely an often low level of gene expression in the library host (Van Elsas et al. 2008a). Alternatively, the screening method may have too low sensitivity to make gene expression easily detectable (Gabor et al. 2004). An additional caveat is the frequent rediscovery of already known functions, which limits the success of the metagenomics approach (Binga et al. 2008). The first two limitations appear to exacerbate the apparent inaccessibility of the extant genetic diversity through functional metagenomics (Lefevre et al. 2008). Considering this, we here pose the question “is there such a thing as a great screen anomaly”? If so, what strategies could be developed to solve this problem? Bluntly speaking, the central question underlying the success of metagenomics-based explorations of natural microbial communities is: “How to express a large number of genes of unknown origin at high throughput and successfully screen for specific functions?”
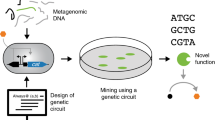
This review aims to discuss the major bottlenecks that pertain to function-based metagenomics of the microbiota in natural systems for bioexploration. By reviewing the status of functional metagenomics, an overview will be given of the most important aspects of currently employed exploration of such microbial systems, and strategies for future improvements are given. Figure 1 depicts the general outline of microbial metagenomics.

Schematic overview of the major function-based metagenomic assessment strategies discussed in this article
Sample selection and pretreatments
Metagenomic libraries have already been constructed from a broad range of environments to access the genetic potential of the microbial communities present. The studies have included soil (Brennerova et al. 2009; Fan et al. 2011; Jiang et al. 2011; Lämmle et al 2007; Van Elsas et al 2008a), sediment (Jeon et al. 2009; Parsley et al. 2010; Zanaroli et al. 2010), freshwater (Wexler et al. 2005), marine environments (Breitbart et al. 2002; Martin-Cuadrado et al. 2007; Venter et al. 2004), and the guts of animals (Bao et al. 2011; Li et al. 2008; Wang et al. 2011). Also, extreme environments such as the Arctic (Jeon et al. 2009), glacial ice (Simon et al. 2009), acidic (Morohoshi et al. 2011; Tyson et al. 2004), and hypersaline environments (Ferrer et al. 2005) as well as a hyperthermal pond (Rhee et al. 2005) have been addressed by metagenomics-based studies. Extreme environments are of obvious interest in the search for novel enzymatic activities and properties.
Clearly, the success of metagenomics exploration of microbial communities will be dependent on the make-up of these in each environment, as well as on the specifics of the environment being investigated. For instance, in cases where particular catabolic functions are sought, screening based on the utilization of specific substrates has been proposed (Brennerova et al. 2009; De Vasconcellos et al. 2010; Tirawongsaroj et al. 2008).
To enhance the chances of finding useful target functions, ecological enhancement (also called habitat biasing) has been proposed in order to manipulate the local microbial community prior to the extraction of the metagenomic DNA. Thus, the prevalence of the target functions in the total extracted metagenome is increased in situ, and so is the target gene hit rate. In practical terms, an environmental sample is biased towards specific groups of organisms by adding substrates or modifying its physicochemical conditions (Van Elsas et al. 2008b). This then results in an enrichment of target functions in the resulting metagenome. As an example, such an experiment has been set up in order to attempt to bias soil microbial communities towards organisms that use chitin as a carbon source under conditions of native versus high pH (Kielak et al., manuscript in review). An advantage of this strategy is its low cost and effort, together with the generally low-tech procedures. However, a side effect of ecological enhancement is that organisms that depend on the activities of the target microbes can also proliferate, thus resulting in a potential “false” enrichment and reduction of the (optimized) target gene hit rate. However, by fine-tuning the selective criteria applied, this problem can be minimized.
In another approach, specific functions/activities within a microbial community can be targeted to increase their activity/expression. Thus, stable isotope probing (SIP) has been applied as a method to selectively target functions involved in an ecological process, thereby making the underlying genes accessible (Cebron et al. 2007, Dumont et al. 2006). SIP allows one to distinguish the metabolically active members of a microbial community from the inactive ones using the addition of a substrate labeled with a stable isotope (13C or 15N) to the environmental sample. If sufficient isotope has been incorporated into the DNA of the active microorganisms, this labeled (“heavy”) DNA can be separated from unlabeled (“light”) DNA by density gradient ultracentrifugation and further analyzed. The method thus enables the establishment of a direct link between function and identity (Chen and Murrell 2010; Cupples 2011; Dumont and Murrell 2005; Radajewski et al. 2003; Uhlik et al. 2009). Depending on the type of labeled substrate used, one can additionally bias the sample in much the same way as in ecological enhancement, targeting specific active ecotypes within a sample. However, a major drawback of SIP remains the fact that unnaturally high concentrations of labeled substrate may be required, next to too extended incubation times, in order to attain sufficient yields of labeled DNA in the active organisms. The former may result in growth inhibition, whereas the latter might accrue an accumulation of the label in the “wrong” trophic classes. An additional practical disadvantage is the prohibitively high cost of labeled substrate. Another problem of SIP is technical as the differentiation between labeled and unlabeled DNA may be difficult: unlabeled high G–C% DNA may have a density profile that approaches that of labeled low G–C% DNA (Buckley et al. 2007).
Despite such limitations, SIP is a very valuable tool to reduce sample complexity and increase the hit rates of particular target genes (Chen and Murrell 2010). It is especially practical in the search for target metabolic genes for biotechnical applications.
DNA extraction and processing
Extraction of microbial community DNA for use in metagenomic library construction can be roughly divided into two strategies: (1) “direct extraction”—the microbial community DNA is directly isolated from the sample and (2) “indirect extraction”—the microbial cells are first isolated from the sample prior to cell lysis (Robe et al. 2003; Van Elsas et al. 2008a). Both methods have their own specific advantages and biases. Four key parameters that define the suitability of the DNA extracted by each method for subsequent metagenomics analysis have been identified: yield, purity, fragment size, and representativeness. Unfortunately, in practice, these factors often stand in negative relation to one another. Enhancing one will often have a negative effect on other factors. As a matter of example, these extraction trade-offs may result in either low-yield extracts containing large DNA fragment sizes versus high-yield small-fragment DNA. A low average fragment size obviously impedes the subsequent analysis of larger operons, for which larger insert libraries are needed (Williamson et al. 2011).
A recent study (Delmont et al. 2011) showed that the apparent functional diversity present in an ecosystem is not severely affected by the DNA extraction method, possibly reflecting the high functional redundancy in most natural microbial communities. However, the inevitable biases inherent to any DNA extraction method can lead to unrepresentative (biased) microbial community DNA. This caveat also impacts our strategies to explore the rare biosphere. An interesting new method for separating DNA from highly contaminated samples, called synchronous coefficient of drag alteration, applies a rotating dipole and quadruple electric field in an aqueous gel by which DNA is concentrated at a focal point while contaminants are pushed outwards (Pel et al. 2009).
In particular cases (Neufeld et al. 2008), e.g., after indirect extraction of DNA from a sample or in a SIP experiment, DNA yields may be low, indicating the need for pre-amplification to allow metagenomic library construction. Regular PCR amplification is often not suitable due to the requirement for specific annealing sites of the primers. A suitable technique is offered by multiple displacement amplification (MDA). MDA is based on the use of phi29 DNA polymerase and random hexamer primers and results in high-fidelity replication, in a random fashion, of the different DNA fragments present in the sample. Although under debate, the method was shown to work without biases due to primer specificity (Binga et al. 2008; Blanco and Salas 1985; Blanco et al. 1989; Nelson et al. 2002). However, MDA can also yield chimeric artifacts (Neufeld et al. 2008; Simon et al. 2009). Neufeld et al. (2008) clearly showed the potential of SIP combined with MDA. They incubated a marine microbial community with in situ concentrations of labeled substrate (methanol) and subsequently performed MDA on the labeled DNA for construction of a fosmid library. They found that the amplified DNA was very representative of the sample.
Metagenomic library construction and gene expression
Construction of a metagenomic library should be accompanied by the careful selection of the appropriate average DNA fragment size. Moreover, suitable vectors and expression hosts should be selected. It is vital to understand that, for most natural ecosystems, complete coverage of the extant diversity cannot be achieved, and so most libraries will consist of fragmentary randomly sampled genes from an overall DNA pool. Only the most abundant fraction of the gene pool will be present in the library, and hence the extracted DNA pool is a sub-selection of the complete metagenome. The extracted DNA pool is further biased by factors such as the effect of sampling, cell separation, lysis intensity, and DNA size variation. Delmont et al. (2011) recently showed that up to 80% increase in genetic diversity is achievable by diversifying the extraction factors (meaning, adding extra extraction modules) in comparison to the most effective single extraction strategy. Simple stochasticity thus dictates that the prevalence of genes from the dominant biosphere will greatly exceed that of genes from the rare biosphere. Therefore, the selection of the vector/host system is ideally guided by prior knowledge about the prevalence and distribution of different bacterial types in the sampled habitat.
Once a metagenomic library has been constructed, screens need to be performed in high throughput to uncover the genes of interest. The two common strategies, i.e., (1) functional and (2) sequence-based (genetic) screening, have been widely applied. It is obvious that functional screening provides a very straightforward way towards the objective. Thus, the target genes in metagenomic libraries are expressed in a relevant experimental setup in order to visualize (detect) them and confirm their assignment to the function. In contrast, genetic screening is dependent on prior knowledge of expected gene sequences or motifs. Direct hybridization of PCR-based screenings has been the method of choice; however, current high-throughput sequencing has opened up the way to employ direct sequencing-based analyses.
In most metagenomics studies performed thus far, Escherichia coli has been used as the cloning host as an extended genetic toolkit is available for this host. Depending on the size of the DNA fragment that needs to be inserted, different vectors have been employed. For small fragments, plasmids <15 kb, for larger fragments cosmids (15–40 kb), fosmids (25–45 kb), and/or bacterial artificial chromosomes (BACs) (100–200 kb) have been successfully used (Angelov et al. 2009; Kakirde et al. 2011; Uchiyama and Miyazaki 2009; Van Elsas et al. 2008a).
In order to eliminate the limitations generated by using E. coli as a single host, shuttle vectors and non-E. coli host systems have been developed. Bacterial strains from genera like Burkholderia, Bacillus, Sphingomonas, Streptomyces, and Pseudomonas have thus been reported as alternative hosts (Courtois et al. 2003; Eyers et al. 2004; Martinez et al. 2004; Van Elsas et al. 2008a).
When expressing the metagenomic library material in a host organism, two strategies can be applied: (1) single-host expression and (2) multi-host expression. Although most functional expression screens have been conducted with a single host, in recent years a shift to multi-host gene expression has been taking place. This is due to the idea that a substantial part of the transformed genes cannot be successfully expressed in a single organism and that the use of multiple hosts either sequentially or in parallel offers great advantages.
Possible causes of lack of gene expression
A central issue concerning the detectable expression of genes of metagenomes in suitable hosts is, thus, the inability to detectably express a major fraction of the target genes. This might be due to a plethora of factors, such as codon usage differences, improper promoter recognition, lack of proper initiation factors, ribosomal entry, improper protein folding, absence of essential co-factors, accelerated enzymatic breakdown of the gene product, inclusion body formation, toxicity of the gene product, or the inability of the host to secrete the gene expression product. To what degree these different factors contribute to the inability to detect the expression of genes in a metagenomic library will differ per host/gene combination. This makes the question as to what percentage of genes within a library can be expressed by an available host very difficult to answer. What we do know is that codon usage is a particularly important factor in the successful expression of foreign genes (Kudla et al. 2009). Most organisms have a preference for specific codons when generating proteins or encoding signals for initiation or termination of translation. The preferred codons are referred to as “optimal” codons. However, the nature of such codons varies between species (Goodarzi et al. 2008). The occurrence of the resulting “codon dialects” between different species is termed codon usage bias (CUB). This phenomenon is particularly important regarding the expression of foreign genes in a metagenomics host, as is done in functional metagenome screens. Kudla et al. (2009) clearly showed the effect of codon bias by synthesizing and expressing 154 genes encoding the green fluorescent protein (GFP) with randomly introduced silent mutations in the third base position. The resulting expression levels varied 250-fold across all variants, clearly illustrating the dramatic effect that CUB has on gene expression. Besides overall codon usage, also the preference for start codons can vary greatly across bacterial species (Villegas and Kropinski 2008). Furthermore, CUB has been shown to be important in translation (Sørensen et al. 1989), protein folding (Zalucki et al. 2009), and secretion (Power et al. 2004; Zalucki and Jennings 2007).
Gabor et al. (2004) quantified the probability of detection of particular genes by random expression cloning on a theoretical basis using 32 prokaryotic genomes (belonging to Euryarchaeota, Crenarchaeota, Firmicutes, Actinobacteria, and Proteobacteria). Three theoretical modes of expression were examined: i.e. (1) independent expression with the ribosomal binding site (RBS) and promoter provided by the insert, (2) expression by transcriptional fusion with the RBS on the insert, and (3) expression by translational fusion with both RBS and promoter on the vector. The latter option was considered to be irrelevant due to its low chance of expression in a real-life experiment. About 40% of the extant enzymatic activities may be accessible by random cloning in E. coli, with a range of 7–73% between the five taxa examined. However, this study was based on purely theoretical bioinformatics considerations and did not take into account key factors that play defined roles in successful gene expression, such as the presence of co-factors, protein folding, and/or secretion.
One way to more successfully express genes in metagenomic library hosts may be to engineer the host expression machinery on the basis of the expected prevalence of genes from source hosts. Thus, it would be interesting to tinker with the host’s transcription and translation systems, thereby increasing the recognition of the foreign RBS predicted to be prevalent in the metagenome (Bernstein et al. 2007). Moreover, one could boost the co-expression of chaperone proteins to promote proper protein folding, whereas enhancement of secretion of the target gene product is another possibility (Ferrer et al. 2004; Jhamb et al. 2008). Not only the host but also the vector can be engineered to maximize the rate or frequency of gene expression. An example is the use of dual-orientation promoters on the vector, which may effectively increase the rate of successful gene expression (Lämmle et al. 2007). However, such promoters are probably most useful in small-fragment libraries where native promoters may not be present in the insert.
Single-host systems
Most single-host metagenomic expression systems rely on E. coli. This organism is readily used regarding gene expression assays based on many different vectors. Because of its status as the most well-known model host, there is ample knowledge about different useful gene expression strategies. In fact, a wide variety of expression systems is available for use in E. coli and many genetic constructs have been assayed this way. There is also a broad range of strains capable of efficient replication of such vectors, which are either single- or multi-copy, and confer low-frequency recombination and protection against lytic phages (Sørensen and Mortensen 2005).
Multiple-host systems
An alternative to the single-host strategy, which increases the rate of gene expression, is the use of multiple hosts, either sequentially or in parallel. The use of multiple hosts diversifies the available expression machinery, thus increasing the chance of successful gene expression. At the same time, the effect of gene product toxicity and enzymatic breakdown can be overcome. To express genes from metagenomes in multiple hosts, shuttle vectors with broad host range are of use. An example of an advanced vector design (combining key characteristics) for broad-host-range screenings was recently presented by Aakvik et al. (2009). Fosmid libraries constructed using broad-host-range fosmid and BAC vector pRS44 were successfully transferred into Pseudomonas fluorescens and Xanthomonas campestris. The main features of this vector are (1) inducible copy number for controlled gene expression, which minimizes possible gene product toxicity but allows high-level gene expression for effective detection in screenings, (2) the ability to stably hold inserts of up to 200 kb, and (3) a high capacity to be efficiently transferred to a wide range of hosts (Aakvik et al. 2009). A nice example of a metagenomic study in which broad-host-range vectors were used was provided by Craig et al. (2010). Metagenomic libraries derived from soil were constructed in an IncP1-α broad-host-range cosmid vector using six selected proteobacterial host strains, i.e., Agrobacterium tumefaciens, Burkholderia graminis, Caulobacter vibrioides, E. coli, Pseudomonas putida, and Ralstonia metallidurans. Library screenings were conducted on the basis of three types of phenotypic traits: antibiosis, pigmentation, and colony morphology. Remarkably, a high diversity of expression profiles between the different hosts was found, with little overlap (Craig et al. 2010). This illustrates the fact that the same metagenomic library can yield totally different expression data, purely based on the expression host used. Furthermore, the still rather low frequencies of clones with desired genes indicated the need for more robust screening methods to lower detection thresholds. Another broad-host-range study (Martinez et al. 2004), which targeted novel drugs, had already underlined the need for multiple-host gene expression. Parallel screenings of metagenomic libraries in multiple hosts yielded diverse expression profiles of antibiotic- producing genes between hosts (Martinez et al. 2004).
On the basis of the foregoing, we conclude that an investment in the development of more sophisticated host–vector systems on the basis of a broad range of host organisms is needed. In particular, the development of host–vector systems with environmentally prevalent strains from phyla that are relatively incompatible with the E. coli expression machinery (like Acidobacteria and Verrucomicrobia) holds great potential to increase the rates of expression of genes from metagenomes.
Future developments
Metagenomic approaches are increasingly being assisted by massive (directly obtained) sequence information. To bypass the difficulties of gene expression, it should be possible, on the basis of such information, to “translate” a whole coding sequence to the expression signal and optimal codon usage typical for E. coli. As an example, Bayer et al. (2009) codon-optimized 89 genes with possible relation to methyl halide transferases for expression in E. coli and obtained an impressive result. That is, 94% of the predicted genes were expressed and showed methyl halide transferase activity. This example clearly indicates the high potential of codon optimization strategies, in this case, evidenced in E. coli.
Functional screening
It is the ability to detect, isolate, and characterize expressed genes in a metagenomic library which determines the success of any function-based metagenomic assessment. A broad array of screening methods can be used (summarized in Fig. 1). Among these, three general detection strategies are distinguished (Simon and Daniel 2009):
-
(1)
Phenotypic insert detection (PID), where the expression of a particular trait is used to identify positive clones
-
(2)
Modulated detection (MD), a strategy that relies on the production of a gene product that is necessary for growth under selective conditions
-
(3)
Substrate induction, a strategy that is based on the induced expression of cloned genes via a specific substrate
Although a distinction is made between these three detection strategies, categorizing them into separate groups would be incorrect. Often, a combination of them is used in order to perform and optimize the screening. For instance, phenotypic detection through a GFP reporter gene can be combined with substrate-induced expression of the insert gene (Uchiyama et al. 2005).
PID
PID is the most commonly used approach for the functional screening of metagenomic libraries. The screen is based on the detection of specific phenotypic traits. The intensity (level) of gene expression is an important issue here since faint expression signals can be easily missed in high-throughput screenings. A possible aid is offered by microfluidic approaches using nanoliter volumes. These can offer increasing sensitivity of the assay since less gene product is required to yield a detectable phenotype (Taupp et al. 2011).
Specific phenotypic traits may be detected in multiple ways. The first way is based on direct expression, for instance, by detecting pigmentation or colony morphology (Brady 2007), both of which may directly result from the expressed inserted gene (Craig et al. 2010; LeCleir et al. 2007). Another way is the (indirect) reaction or interaction of an added substance with the expressed gene product or a product that is a consequence of this expression. Lastly, detection can be based on co-expression of a reporter gene which is linked to the target gene in the library. A high diversity of methods has been developed based on these three strategies, of which the most prominent ones will be discussed below.
Direct detection
Visual detection is a phenotypic screening method that is relatively straightforward and “low-tech”. However, it is also quite labor-intensive. This screening method works by positive clones displaying a trait (as the result of the expression of a library gene) which is directly observable. Examples of such observable traits are colony pigmentation, irregular colony morphology, or halo formation on plate overlays. Coupling this direct detection method with high-throughput technologies, such as that offered by 384-well plates, colony picking robots and microplate readers, not only shortens processing time but also enhances the reliability and comparability of screenings performed on different clones. A disadvantage of this method is its rather low resolution or sensitivity. For example, if expression is low in a certain positive clone, a phenotypic trait might not be readily detectable, resulting in an incorrect rejection of “sub-threshold” positive clones. Furthermore, the method does not allow direction to be given to the screen. In a recent study (Craig et al. 2010), clones were screened in high-throughput in multiple hosts based on the three phenotypic traits mentioned above. These traits were chosen based on the fact that they are commonly associated with small-molecule production (Craig et al. 2010). This illustrates the fact that these methods are more suited to a broad-range exploration of the metagenome for pleiotrophic traits than to directed searches for a specific pathway or metabolite.
Indicator medium
The use of indicator medium constitutes a direct way to detect particular small molecules, chemical reactions, or metabolic, catabolic, or antibiotic capabilities of a clone. It is a popular detection method given its suitability for high-throughput application, as well as its amenability to many experiments, from broad screenings of diverse gene products to the isolation of very specific metabolic capabilities (De Vasconcellos et al. 2010; Fan et al. 2011; Morohoshi et al. 2011; Tirawongsaroj et al. 2008). Furthermore, the relative sensitivity of this method allows it to detect changes in, e.g., pH at moderate expression levels, especially when combined with droplet-based microfluidics, where detectable concentrations are easily reached due to the small volume in use.
A nice example of the use of indicator medium in screenings is the isolation of novel metallo-proteases from metagenomic libraries using milk-infused plates. The screen was based on detection of proteolytic activity in the E. coli clones, which confer the ability to hydrolyze milk proteins. The library clones were incubated on skimmed milk-containing agar plates and proteolytic activity was detected by the formation of clear haloes on the plates (Waschkowitz et al. 2009).
The use of indicator media holds great promise as the successful expression of foreign genes in the host can be readily monitored in high throughput. Relying on the successful expression of foreign genes for detection might yield low amounts of positive hits yet give the guarantee that the gene is functional in the metagenome host. However, a problem is that the target enzymes are only expressed intracellularly in the metagenome host and the cell membrane might not necessarily be permeable to the indicator substances present in the medium. Hence, secretion of the gene product is necessary for detection. Moreover, other problems can arise, such as enzymatic breakdown or intracellular product accumulation (Sørensen and Mortensen 2005), which can result in toxicity. Forced cell lysis may hold the solution, as it may bring the indicator substance into contact with the intracellular target proteins (Bao et al. 2011). However, care should be taken to not denature the expressed proteins by the lysing agents since otherwise protein activity as well as its interaction with the indicator substrate might be lost. Furthermore, mechanical cell disruption can be time-consuming and labor-intensive. A possible way of avoiding this is to use an autolytic vector. Li et al. (2007) developed a UV-inducible autolytic vector for use in high-throughput screenings. An SRRz lysis gene cassete was inserted downstream of a UV-inducible promotor in E. coli. Cell lysis efficiency was tested by expressing β-galactosidase in E. coli prior to UV induction. After UV-induced lysis, extracellular (supernatant) β-galactosidase activity was compared to the total of intracellular (pellet) and extracellular β-galactosidase activity to quantify lysis efficiency. A lysis effiencency of 60% or more was observed at a temperature of 30 °C. This is comparable to conventional lysozyme treatment. However, at 37 °C, the lysis rate was less consistent. Thus, use of such an autolytic vector might provide a simple alternative to existing lysis techniques (Li et al. 2007).
In addition to the beta-galactosidase-based screen, several other chromogenic and fluorogenic reporter techniques were proven to be efficient in the identification of the activities of enzymes encoded by the inserted gene(s). LeCleir et al. (2007) thus showed the presence of chitinolytic enzymes in an estuarine metagenomic library by cleavage of fluorogenic analogs of chitin.
MD
MD does not rely on the direct detection of an expressed gene, but it uses a predesigned expression route. By modulating the expression host and/or vector systems, selection and detection of inserted genes can be manipulated, for instance by the coexpression of reporter genes or heterologous complementation. This results in more specific screenings and standardized detectable signals.
Reporter genes
The use of reporter genes is a suitable method for high-throughput screenings. The lacZ gene encoding beta-galactosidase (resulting in colony coloring upon growth on X-Gal-containing medium) is frequently used as a reporter gene. In an experiment to screen for metagenomic clones containing genes that interfere with quorum sensing (QS), this reporter gene was used. Screening was achieved by measuring the potential degradation of the QS signaling molecules. An A. tumefaciens strain containing a traI-lacZ gene fusion was used in the screening. By inducing the traI gene with the QS signal molecule homoserine lactone 3-oxo-C 8 -HSL, lacZ is activated. This results in beta-galactosidase production, yielding blue colonies. If, however, 3-oxo-C 8 -HSL is broken down by the host, lacZ induction is inhibited and no blue color appears. This would be an indication of a quorum sensing-inhibitory or degradation activity of the clone. The experiment yielded 438 positive clones showing QS inhibition (Schipper et al. 2009).
A great advantage of this approach is that detection of positive clones does not rely on successful expression of a gene product downstream of transcription. Nor does a possibly faint expression of the inserted gene hamper detection, as it might in other detection methods. This can be of great benefit when searching for genes that might be hard to express in the host.
Heterologous complementation (HC)
Heterologous complementation (HC) relies on exploring foreign genes to achieve genetic complementation in the host, resulting in the expression of a gene product that is vital for growth under selective conditions. The technique allows for great selectivity and, thereby, a screen can be precisely directed to search for specific genes (Kellner et al. 2011; Simon et al. 2009). An example is presented by Simon et al. (2009) who screened metagenomic plasmid and fosmid libraries derived from glacial ice for DNA polymerase encoding genes. This was achieved by using an E. coli strain that carries a cold-sensitive mutation in the 5′–3′ exonuclease domain of DNA polymerase I, which is lethal at temperatures below 20 °C. By growing the clones on antibiotic-containing plates compatible with vector resistance, at a temperature of 18 °C, positive clones can be identified that complemented the lethal mutation. Using this approach, 17 plasmids and 1 fosmid with the desired phenotype were retrieved from the clone library. Sequence analysis of nine positive clones indicated that indeed DNA polymerase genes had been isolated from the library. Taking into account the conserved nature of DNA polymerase genes and the degree of homology to known DNA polymerase genes, this result led to the conclusion that the genes had been recovered from as-yet-unexplored microorganisms (Simon et al. 2009).
Another example of HC is provided by a study that attempted to isolate novel lysine racemase genes from a metagenome (Chen et al. 2009). An E. coli strain carrying a lysine auxotrophy mutation was used to screen for the aforementioned genes in a metagenomic library derived from garden soil. Clones were grown on d-lysine-supplemented medium. Since only successful recombinant clones would be able to catabolize d-lysine, unsuccessful clones would starve on the medium. Using this method, a positive clone was identified and sequenced. To confirm that the inserted gene was derived from the metagenome and not from digested DNA fragments during library construction, primers based on the detected lyr (lysine racemase) gene were designed. These primers then successfully amplified the lyr gene directly from the metagenomic DNA by PCR (Chen et al. 2009).
Induction by substrate
Induction of gene expression by substrate is particularly practical for the detection of catabolic genes. Expression of catabolic genes is often induced by substrates and/or metabolites of catalytic enzymes. The regulatory elements of these catabolic genes are generally situated close to the genes themselves. These elements have been shown to work in host organisms like E. coli.
Substrate-induced gene expression
Uchiyama et al. (2005) developed the so-called substrate-induced gene expression (SIGEX) system for use as a screening method for particular catabolic genes. To make this method high-throughput-compatible, an operon-trap GFP expression vector was used, resulting in co-expressed GFP upon substrate-induced expression of any responsive inserted gene. This GFP expression subsequently enabled the separation of positive clones by fluorescence-assisted cell sorting (FACS). In the study, metagenomic genes from groundwater were found that could be induced by benzoate and naphthalene. This yielded 58 benzoate- and four naphthalene-positive clones (Uchiyama et al. 2005). By changing the substrate, the scope of the screen can be adjusted and different catabolic genes can be targeted. Possibly unknown gene functions might even be deduced from the inducing substrate that is used. However, induction of expression by other effectors than the substrate used can result in the detection of false positives.
Metabolite-regulated expression
Metabolite-regulated expression constitutes a similar technique to the above one (Williamson et al. 2005). It aims to detect biologically active small molecules by an intracellular luxI–luxR biosensor system. In this system, gene expression will be induced or inhibited by quorum sensing. After a certain concentration of gene product is met, induction of expression of a transcriptional activator will take place by binding of luxR, which activates luxI; subsequent expression of a reporter gene will ensue. The successful expression of the reporter gene allows high-throughput screening by FACS. An advantage of this system is that it does not rely on secretion of a gene product like screens with indicator organisms do (a common technique to identify antibiotics). Instead this technique screens intracellularly for expressed small molecules (Williamson et al. 2005).
Major hurdles and future prospects in functional metagenomics
Functional screening will remain an essential element to be developed further in metagenomics aimed at mining natural microbial communities for new products for biotechnology. Two main hurdles can still be identified, i.e., the successful expression of genes in metagenomic clone libraries and the subsequent screening and selection of genes of interest from expressed inserts.
The metagenomic expression paradigm
Two facets are important in the expression of foreign genes in a metagenomics library host, i.e., the nature of the DNA insert and that of the expression machinery (consisting of a vector and host with tunable genetic circuitry). In “traditional” foreign gene expression by genetic modification using known source and host genetic backgrounds, the two facets have become relatively well known. Thus, we have learned that the host expression machinery needs to be chosen and tuned to the requirements of the specific genetic insert. Often, the insert was first codon-optimized for the expression host to maximize the chance of successful gene expression. In contrast, in functional metagenomics a new expression paradigm is encountered, in which very little is known of the genetic insert prior to gene expression. Hence, prior knowledge on the estimated prevalence of source genes in the metagenomic library can be extremely helpful and may be required. On the basis of such knowledge, suitable host/vector systems can be selected. And, in addition to that, the host expression machinery might be tuned to a broad range of inserts, with varying fragment types, origins and sizes. It seems to be the relatively narrow-range expression machinery present in E. coli which is preventing an effective match on the individual insert level, leading to the current expression bottlenecks that are often encountered in functional metagenomic screenings. Broader expression systems might overcome this bottleneck. There is still a need for the development of additional expression hosts from less studied phyla and of robust shuttle and induction systems for these hosts. But even with a broadly applicable expression system, trans-acting processes together with the shear randomness of insertions into the vectors used will continue to limit our ability to optimize the rate of expression of the target genes present in the metagenome. In Fig. 2, an overview of the different expression stages that are met in functional metagenomic screens is provided.

Schematic overview of the expression process from induction all the way to secretion of the light of troubleshooting during a metagenomic library screening
One obvious strategy would be to scale up functional screenings by the use of multiple hosts and screening methods in parallel (e.g. Craig et al. 2010). This enables one to increase the chance to successfully express and detect inserted genes. However, to prevent such an experiment to become costly and time-consuming, high-throughput, potentially microfluidic, technologies are required. This might be combined with cell lysis procedures, either chemically or by the use of an autolytic vector, to minimize biases by enzymatic breakdown of the gene product, toxicity or secretion problems.
The alternative to the upscaling of metagenomic screenings is to restrict the scope of the study by narrowing down the insert gene source diversity so that the expression machinery can be more specifically engineered to suit the experimental demands. To confine the scope of a metagenomics screen, ecological enhancement can be very useful as it decreases library complexity by an increase of the prevalence of genes of preselected and dominating target microbes. One could also think of preselecting genetic material on the basis of G–C content by ultracentrifugation (Holben 2011), thereby providing a better match between the sampled metagenome and the expression host. Heterologous complementation is also a very suitable detection method in such focused screens because of its specificity and independence of successful insert expression.
A more radical approach would be to tune the selected target genes in the metagenome library to match the most convenient expression machinery of the host. This would require sequencing of a metagenomic library to identify target genes and subsequent optimization of the codons and signal sequences of these to suit the expression host. Finally, optimized sequences would be synthesized, inserted, and functionally screened in the relevant host.
Computational, sequence-based, and high-throughput technologies may allow us to codon-optimize and synthesize a complete metagenomic operon for functional screening. The possibilities are enormous, like accessing ever rarer genes, as well as increasing positive hit rates. However enticing this idea may be, as well as its potential possibilities, currently only selections of existing libraries are realistic candidates for such experiments.
New technologies useful in functional screenings
The recent growth of the use of “library based” approaches in metagenomics-based mining is directly related to the rapid technological developments in molecular biotechnology. Screening methodologies have evolved in the light of the necessity to understand complex ecological and biochemical interactions in different environments. The original screenings, based on the isolation of mutant (transformed) cells of host organisms on culture media, were considered to provide insufficient power or throughput (Link et al. 2007; Shuman 2003). Thus, screening systems able to rapidly identify the presence and activity of enzymes, effects of mutations, or interaction and changes among microbial community members were desired. Microarray (chip)-based technologies coupled with microfluidic devices, cell compartmentalization, flow cytometry, and cell sorting have been proposed as promising new technologies (Link et al. 2007; Ottesen et al. 2006; Tracy et al. 2010). It is important to pinpoint the major roles fluorescence-based assays are playing in single-cell analyses. Improved enzyme detection, resulting from the discovery of new genes, isolation of proteins with high affinity, as well as phylogenetic analyses were reported (Aharoni et al. 2006; Feng et al. 2007; Melkko et al. 2007). Large metagenome libraries have been screened via single-cell fluorescent assays and high-throughput flow cytometry in the quest for novel catalytic activities (Link et al. 2006; Santoro et al. 2002; Varadarajan et al. 2005). These screening methods offer higher levels of quantification and the possibility to detect multiple traits in one assay. Considering the evolution of screening approaches, from Sydney Brenner’s affirmation “just toothpicks and logic” to higher-level technologies (which have become available at low cost), increases in the rates of discovery of new bio-engineered molecules can be predicted.
The hunt for novelty
The increasing human impact on the environment in the last century has urged developments in our food, waste treatment, agriculture, and biomedical industries. In this respect, explorations of diverse habitats have increased with the increasing demand for new enzymes, antibiotics, and other active biomolecules as well as biofuels (Ferrer et al. 2009; Lorenz and Eck 2005; Schloss and Handelsman 2003). A recently developed database—denominated MetaBioMe (Sharma et al. 2010)—offers access to 510 “commercially useful” enzymes (CUEs) by linking protein databases with data from metagenomic and bacterial genomic datasets. These CUEs have been classified into nine broad application categories, namely: agriculture, biosensor, biotechnology, energy, environment, food and nutrition, medical, other industries, and miscellaneous. Among these, biotechnology, food and nutrition, medicine, and biodegradation of toxic compounds are considered to be of utmost importance. So far, one of the most frequently targeted habitats for finding genetic “novelty” was soil. The cryptic microbial treasures of different types of soil and sediments, including those in extreme conditions (e.g., low pH, high temperature, high salt concentration), have promised the presence of an enormous reservoir of different enzymes. Of late, such environments are certainly underexplored. However, metagenomics-derived products have already found their way to the biotechnology market (Table 1), although their metagenomic origin is not always revealed by the manufacturer. Moreover, it is often protected by patents.
Conclusion
High-throughput technology has been often associated with increasing the success of function-based metagenomic screens. However, high-throughput is more of a way of compensating for the often low hit rates in metagenomics screens than a true improvement of methods. The upscaling of a functional metagenomics screen by adopting a high-throughput strategy using existing screening techniques may indeed increase the chance of identifying target genes in a metagenome, and indeed there is the rightful expectation that these high-throughput screens will become more effective by the use of microfluidic strategies, substrates with higher sensitivity, smartly designed induction systems, and easily detectable reporter genes. Collectively, such improvements may result in the lowering of detection thresholds and saving of costs.
Depending on the purpose of the application, there is still ample room for improvement of expression strategies, such as careful host (range) selection, co-expression of chaperones, or codon optimization. It is the latter strategy that holds great potential for the future by screening of sequence databases to identify genes of interest, after which the targeted genes are codon-optimized and synthesized before being expressed in the target host. Key in overcoming expression bottlenecks is certainly a more intricate understanding of the complex aspects involved in expression of foreign genes (Fig. 3). Thus, identification of crucial hurdles involved in the inability to express genes of any metagenome should be considered as the spearhead that allows us to move forward. Ultimately, a more directed approach in improving existing gene expression systems should be envisaged.

The metagenomic expression bottleneck
However, when metagenomic strategies are designed to yield more optimal functional screenings, there might be a risk of being trapped in an overdesigned experimental setup, which leaves insufficient room for the discovery of the “real” instead of the “similar” unknown biosphere. This holds especially true when selecting target genes from sequenced metagenomic libraries. Following this, caution should be taken when weighing the advantages and disadvantages of different DNA processing strategies for library construction.
To conclude, we posit that “the great screen anomaly” is a current reality; however, the term “great expression inability” might more appropriately describe current obstacles that hamper greater screening efficiencies. To what extent this anomaly will persist (as its predecessor “the great plate count anomaly” has) remains uncertain. Considerable fine-tuning of methods is clearly still needed to make functional screening representative of the environmental diversity and to boost its efficiency in assigning genes to function. Nevertheless, considering the pace at which innovative technologies evolve in this area, “the great screen anomaly” probably awaits a very unsure future with respect to its existence in the next decennium.
References
Aakvik T, Degnes KF, Dahlsrud R, Schmidt F, Dam R, Yu L, Völker U, Ellingsen TE, Valla S (2009) A plasmid RK2-based broad-host-range cloning vector useful for transfer of metagenomic libraries to a variety of bacterial species. FEMS Microbial Lett 296(2):149–158
Aharoni A, Thieme K, Chiu CP, Buchini S, Lairson LL, Chen H, Strynadka NC, Wakarchuk WW, Withers SG (2006) High-throughput screening methodology for the direct evolution of glycosyltransferases. Nat Methods 3(8):609–614
Angelov A, Mientus M, Liebl S, Liebl W (2009) A two-host fosmid system for functional screening of (meta)genomic libraries from extreme thermophiles. Syst Appl Microbiol 32(3):177–185
Bao L, Huang Q, Chang L, Zhou J, Lu H (2011) Screening and characterization of a cellulase with endocellulase and exocellulase activity from yak rumen metagenome. J Mol Catal B: Enzym 73:104–110
Bayer TS, Widmaier DM, Temme K, Mirsky EA, Santi DV, Voigt CA (2009) Synthesis of methyl halides from biomass using engineered microbes. J Am Chem Soc 131(18):6508–6515
Beloqui A, de Maria PD, Golyshin PN, Ferrer M (2008) Recent trends in industrial microbiology. Curr Opin Microbiol 11(3):240–248
Bernstein JR, Butler T, Shen CR, Liao JC (2007) Directed evolution of ribosomal protein S1 for enhanced translational efficienty of high CG Rhodopseudomonas palustris DNA in Escherichia coli. J Biol Chem 282(26):18929–18936
Binga EK, Lasken RS, Neufeld JD (2008) Something from (almost) nothing: the impact of multiple displacement amplification on microbial ecology. ISME J 2(3):233–241
Blanco L, Salas M (1985) Characterization of a 3′–5′ exonuclease activity in the phage phi29-encoded DNA polymerase. Nucl Acids Res 13(4):1239–1249
Blanco L, Bernad A, Lazaro J, Martin G, Garmendia C, Sales M (1989) Highly efficient DNA synthesis by the phage phi 29 DNA polymerase. Symmetrical mode of DNA replication. J Biol Chem 264(15):8935–8940
Brady SF (2007) Construction of soil environmental DNA cosmid libraries and screening for clones that produce biologically active small molecules. Nat Protocols 2(5):1297–1305
Breitbart M, Salamon P, Andresen B, Mahaffy JM, Segall AM, Mead D, Azam F, Rohwer F (2002) Genomic analysis of uncultured marine viral communities. Proc Natl Acad Sci U S A 99(22):14250–14255
Brennerova MV, Josefiova J, Brenner V, Pieper DH, Junca H (2009) Metagenomics reveals diversity and abundance of meta-cleavage pathways in microbial communities from soil highly contaminated with jet fuel under air-sparging bioremediation. Environ Microbiol 11(9):2216–2227
Buckley DH, Huangyutitham V, Hsu S-F, Nelson TA (2007) Stable isotope probing with 15N achieved by disentangling the effects of genome G+C content and isotope enrichment on DNA density. Appl Environ Microbiol 73(10):3189–3195
Cebron A, Bodrossy L, Chen Y, Singer AC, Thompson IP, Prosser JI, Murrell JC (2007) Identity of active methanotrophs in landfill cover soil as revealed by DNA-stable isotope probing. FEMS Microbiol Ecol 62(1):12–23
Chen Y, Murrell JC (2010) When metagenomics meets stable-isotope probing: progress and perspectives. Trends Microbiol 18(4):157–163
Chen IC, Lin WD, Hsu SK, Thiruvengadam V, Hsu WH (2009) Isolation and characterization of a novel lysine racemase from a soil metagenomic library. Appl Environ Microbiol 75(15):5161–5166
Courtois S, Cappellano CM, Ball M, Francou FX, Normand P, Helynck G, Martinez A, Kolvek SJ, Hopke J, Osburne MS, August PR, Nalin R, Guerineau M, Jeannin P, Simonet P, Pernodet JL (2003) Recombinant environmental libraries provide access to microbial diversity for drug discovery from natural products. Appl Environ Microbiol 69(1):49–55
Craig JW, Chang FY, Kim JH, Obiajulu SC, Brady SF (2010) Expanding small-molecule functional metagenomics through parallel screening of broad-host-range cosmid environmental DNA libraries in diverse Proteobacteria. Appl Environ Microbiol 76(5):1633–1641
Cupples AM (2011) The use of nucleic acid based stable isotope probing to identify the microorganisms responsible for anaerobic benzene and toluene biodegradation. J Microbiol Methods 85(2):83–91
Curtis TP, Sloan WT (2004) Prokaryotic diversity and its limits: microbial community structure in nature and implications for microbial ecology. Curr Opin Microbiol 7:221–226
De Vasconcellos SP, Angolini CFF, Garcia INS, Dellagnezze BM, da Silva CC, Marsaioli AJ, Neto EVD, de Oliveira VM (2010) Screening for hydrocarbon biodegraders in a metagenomic clone library derived from Brazilian petroleum reservoirs. Org Geochem 41(7):675–681
Delmont TO, Robe P, Clark I, Simonet P, Vogel TM (2011) Metagenomic comparison of direct and indirect soil DNA extraction approaches. J Microbiol Methods 86(3):397–400
Dumont MG, Murrell JC (2005) Stable isotope probing—linking microbial identity to function. Nat Rev Microbiol 3(6):499–504
Dumont MG, Neufeld JD, Murrell JC (2006) Isotopes as tools for microbial ecologists. Curr Opin Biotechnol 17(1):57–58
Eyers L, George I, Schuler L, Stenuit B, Agathos SN, El Fantroussi S (2004) Environmental genomics: exploring the unmined richness of microbes to degrade xenobiotics. Appl Microbiol Biotechnol 66(2):123–130
Fan X, Liu X, Wang K, Wang S, Huang R, Liu Y (2011) Highly soluble expression and molecular characterization of an organic solvent-stable and thermotolerant lipase originating from the metagenome. J Mol Catal B: Enzym 72(3–4):319–326
Feng Y, Duan CJ, Pang H, Mo XC, Wu CF, Yu Y, Hu YL, Wei J, Tang JL, Feng JX (2007) Cloning and identification of novel cellulase genes from uncultured microorganisms in rabbit cecum and characterization of the expressed cellulases. Appl Microbiol Biotechnol 75(2):319–328
Fernández-Arrojo L, Guazzaroni ME, López-Cortés N, Beloqui A, Ferrer M (2010) Metagenomic era for biocatalyst identification. Curr Opin Biotechnol 21(6):725–733
Ferrer M, Chernikova TN, Timmis KN, Golyshin PN (2004) Expression of a temperature-sensitive esterase in a novel chaperone-based Escherichia coli strain. Appl Environ Microbiol 70(8):4499–4504
Ferrer M, Golyshina OV, Chernikova TN, Khachane AN, Martins Dos Santos VA, Yakimov MM, Timmis KN, Golyshin PN (2005) Microbial enzymes mined from the Urania deep-sea hypersaline anoxic basin. Chem Biol 12(8):895–904
Ferrer M, Beloqui A, Timmis KN, Golyshin PN (2009) Metagenomics for mining new genetic resources of microbial communities. J Mol Microbiol Biotechnol 16(1–2):109–123
Gabor EM, Alkema WB, Janssen DB (2004) Quantifying the accessibility of the metagenome by random expression cloning techniques. Environ Microbiol 6(9):879–886
Goodarzi H, Torabi N, Najafabadi HS, Archetti M (2008) Amino acid and codon usage profiles: adaptive changes in the frequentcy of amino acids and codons. Gene 407(1–2):30–41
Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM (1998) Molecular biological access to the chemisrty of unknown soil microbes, a new frontier fornatural products. Chem Biol 5(10):245–249
Hil RT, Fenical W (2010) Pharmaceuticals from marine natural products: surge or ebb? Curr Opin Biotechnol 21(6):777–779
Holben WE (2011) GC fractionation allows comparative total microbial community analysis, enhances diversity assessment, and facilitates detection of minority populations of minority populations of bacteria. In: Bruijn FJ (ed) Handbook of molecular microbial ecology I: metagenomics and complementary approaches. Wiley, Hoboken, pp 183–196
Imhoff JF, Labes A, Wiese J (2011) Bio-mining the microbial treasures of the ocean: new natural products. Biotechnol Adv 29(5):468–482
Jeon JH, Kim JT, Kang SG, Lee JH, Kim SJ (2009) Characterization and its potential application of two esterases derived from the arctic. Mar Biotechnol 11(3):307–316
Jhamb K, Jawed A, Sahoo DK (2008) Immobilized chaperones: a productive alternative to refolding of bacterial inclusion body proteins. Process Biochem 43(6):587–597
Jiang C, Li SX, Luo FF, Jin K, Wang Q, Hao ZY, Wu LL, Zhao GC, Ma GF, Shen PH, Tang XL, Wu B (2011) Biochemical characterization of two novel β-glucosodase genes by metagenome expression cloning. Bioresour Technol 102(3):3272–3278
Kakirde KS, Parsley LC, Liles MR (2010) Size does matter: application-driven approaches for soil metagenomics. Soil Biol Biochem 42(11):1911–1923
Kakirde KS, Wild J, Godiska R, Mead DA, Wiggings AG, Goodman RM, Szybalski W, Liles MR (2011) Gram negative shuttle BAC vector for heterologous expression of metagenomic libraries. Gene 475(2):57–62
Kellner H, Luis P, Portetelle D, Vandenbol M (2011) Screening of a soil metatranscriptomic library by functional complementation of Saccharomyces cerevisiae mutants. Microbiol Res 166(5):360–368
Kudla G, Murray AW, Tollervey D, Plotkin JB (2009) Coding-sequence determinants of gene expression in Escherichia coli. Science 324(5924):255–258
Lämmle K, Zipper H, Breuer M, Hauer B, Buta C, Brunner H, Rupp S (2007) Identification of novel enzymes with different hydrolytic activities by metagenome expression cloning. J Biotechnol 127(4):575–592
LeCleir GR, Buchan A, Maurer J, Moran MA, Hollibaugh JT (2007) Comparison of chitinolytic enzymes from an alkaline, hypersaline lake and an estuary. Environ Microbiol 9(1):197–205
Lefevre F, Robe P, Jarrin C, Ginolhac A, Zago C, Auriol D, Vogel TM, Simonet P, Nalin R (2008) Drug from hidden bugs: their discovery via untapped resources. Res Microbiol 159(3):153–161
Li S, Xu LH, Hua H, Ren CA, Lin ZL (2007) A set of UV-inducible autolytic vectors for high throughput screening. J Biotechnol 127(4):647–652
Li M, Wang BH, Zhang MH, Rantalainen M, Wang SY, Zhou HK, Zhang Y, Shen J, Pang XY, Zhang ML, Wei H, Chen Y, Lu HF, Zuo J, Su MM, Qiu YP, Jia W, Xiao CN, Smith LM, Yang SL, Holmes E, Tang HR, Zhao GP, Nicholson JK, Li LJ, Zhao LP (2008) Symbiotic gut microbes modulate human phenotypes. Proc Natl Acad Sci U S A 105(6):2117–2122
Link AJ, Vink MKS, Agard NJ, Prescher JA, Bertozzi CR, Tirrell DA (2006) Discovery of aminoacyl-tRNA synthetase activity through cell-surface display of noncanonical amino acids. Proc Natl Acad Sci U S A 103:10180–10185
Link AJ, Jeon KJ, Georgiu G (2007) Beyond toothpicks: new methods for isolating mutant bacteria. Nat Rev Microbiol 5:680–688
Lorenz P, Eck J (2005) Metagenomics and industrial applications. Nat Rev Microbiol 3:510–516
Martin-Cuadrado AB, López-García P, Alba JC, Moreira D, Monticelli L, Strittmatter A, Gottschalk G, Rodríguez-Valera F (2007) Metagenomic of the deep Mediterranean, a warm bathypelagic habitat. PLoS One 2(9):e914
Martinez A, Kolvek SJ, Yip CLT, Hopke J, Brown KA, MacNeil IA (2004) Genetically modified bacterial strains and novel bacterial artificial chromosome shuttle vectors for constructing environmental libraries and detecting heterologous natural products in multiple expression hosts. Appl Environ Microbiol 70(4):2452–2463
Melkko S, Zhang Y, Dumelin CE, Scheuerman J, Neri D (2007) Isolation of high-affinity trypsin inhibitors from a DNA-encoded chemical library. Angew Chem 119:4755–4758
Mocali S, Benedetti A (2010) Exploring research frontiers in microbiology: the challenge of metagenomics in soil microbiology. Res Microbiol 161(6):497–505
Morohoshi T, Oikawa M, Sato S, Kikuchi N, Kato N, Ikeda T (2011) Isolation and characterization of a novel lipases from a metagenomic library of the microbial community in the pitcher fluid of the carnivorous plant Nepenthes hybrida. J Biosci Bioeng 112(4):315–320
Nelson JR, Cai YC, Giesler TL, Farchaus JW, Sundaram ST, Ortiz-Rivera M, Hosta LP, Hewitt PL, Mamone JA, Palaniappan C, Fuller CW (2002) TempliPhi, phi29 DNA polymerase based rolling circle amplification of templates for DNA sequencing. Biotechniques 32:44–47
Neufeld JD, Chen Y, Dumont MG, Murrell JC (2008) Marine methylotrophs revealed by stable-isotope probing, multiple displacement amplification and metagenomics. Environ Microbiol 10(6):1526–1535
Nichols D (2007) Cultivation gives context to microbial ecologist. FEMS Microbiol Ecol 60(3):351–357
Ottesen EA, Hong JW, Quake SR, Leadbetter JR (2006) Microfluidic digital PCR enables multigene analysis of individual environmental bacteria. Science 314(5804):1464–1467
Parsley LC, Consuegra EJ, Kakirde KS, Land AM, Harper WF, Liles MR (2010) Identification of diverse antimicrobial resistance determinants carried on bacterial, plasmid, or viral metagenomes from an activated sludge microbial assemblage. Appl Environ Microbiol 76(11):3753–3757
Pel J, Broemeling D, Mai L, Poon H, Tropini G, Holt RA Warren R, Marziali A (2009) Nonlinear electrophoretic response yields a unique parameter for separation of biomolecules. Proc Natl Acad Sci U S A 106(35):14796–14801
Power PM, Jones RA, Beacham IR, Bucholtz C, Jennings MP (2004) Whole genome analysis reveals a high incidence of non-optimal codons in secretory signal sequences of Escherichia coli. Biochem Biophs Res Commun 322(3):1038–1044
Radajewski S, McDonald IR, Murrel JC (2003) Stable-isotope probing of nucleic acids: a window to the function of uncultured microorganisms. Curr Opin Biotechnol 14(3):296–302
Rhee JK, Ahn DG, Kim YG, Oh JW (2005) New thermophilic and thermostable esterase with sequence similarity to the hormone-sensitive lipase family, cloned from a metagenomic library. Appl Environ Microbiol 71(2):817–825
Riesenfeld CS, Schloss PD, Handelsman J (2004) Metagenomics, genomic analysis of microbial communities. Annu Rev Genet 38:525–552
Robe P, Nalin R, Capellano C, Vogel TA, Simonet P (2003) Extraction of DNA from soil. Eur J Soil Biol 39:183–190
Santoro SW, Wang L, Herberich B, King DS, Schultz PG (2002) An efficient system for the evolution of aminoacyl-tRNA synthetase specificity. Nat Biotechnol 20:1044–1048
Schipper C, Hornung C, Bijtenhoorn P, Quitschau M, Grond S, Streit W (2009) Metagenome-derived clones encoding two novel lactonase family proteins involved in biofilm inhibition in Pseudomonas aeruginosa. Appl Environ Microbiol 75(1):224–233
Schloss PD, Handelsman J (2003) Biotechnological prospects from metagenomics. Curr Opin Biotechnol 14(3):303–310
Sharma VK, Kumar N, Prakash T, Taylor TD (2010) MetaBioME: a database to explore commercially useful enzymes in metagenomic datasets. Nucleic Acids Res 38:468–472
Shuman HA (2003) Just toothpicks and logic: how some labs succeed at solving complex problems. J Bacteriol 185(2):387–390
Simon C, Daniel R (2009) Achievements and new knowledge unraveled by metagenomic approaches. Appl Microbiol Biotechnol 85(2):265–276
Simon C, Herath J, Rockstroh S, Daniel R (2009) Rapid identification of genes encoding DNA polymerases by function-based screening of metagenomic libraries derived from glacial ice. Appl Environ Microbiol 75(9):2964–2968
Singh BK, Macdonald CA (2010) Drug discovery from uncultivable microorganisms. Drug Discov Today 15(17–18):792–799
Singh B, Gautam SK, Verma V, Kumar M, Singh B (2008) Metagenomics in animal gastrointestinal ecosystem: potential biotechnological prospects. Anaerobe 14(3):138–144
Sørensen HP, Mortensen KK (2005) Advanced genetic strategies for recombinant protein expression in Escherichia coli. J Biotechnol 115(2):113–128
Sørensen MA, Kurland CG, Pedersen S (1989) Codon usage determines translation rate in Escherichia coli. J Mol Biol 207(2):365–377
Staley JT, Konopka A (1985) Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu Rev Microbiol 39:321–346
Taupp M, Mewis K, Hallam SJ (2011) The art and design of functional metagenomic screens. Curr Opin Biotechnol 22(3):1–8
Tirawongsaroj P, Sriprang R, Harnpicharnchai P, Thongaram T, Champreda V, Tanapongpipat S, Pootanakit K, Eurwilaichitr L (2008) Novel thermophilic and thermostable lipolytic enzymes from a Thailand hot spring metagenomic library. J Biotechnol 133(1):42–49
Torsvik V, Øvreås L (2002) Microbial diversity and function in soil: from genes to ecosystems. Curr Opin Microbiol 5(3):240–245
Tracy BP, Gaida SM, Papoutsakis ET (2010) Flow cytometry for bacteria: enabling metabolic engineering, synthetic biology and the elucidation of complex phenotypes. Curr Opin Biotechnol 21:85–99
Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF (2004) Community structure and metabolism through reconstruction of the microbial genomes from the environment. Nature 428(6978):37–43
Uchiyama T, Miyazaki K (2009) Functional metagenomics for enzyme discovery: challenges to efficient screening. Curr Opin Biotechnol 20(6):616–622
Uchiyama T, Abe T, Ikemura T, Wantanabe K (2005) Substrate-induced gene-expression screening of environmental metagenome libraries for isolation of catabolic genes. Nat Biotechnol 23(1):1038–1048
Uhlik O, Jecna K, Leigh MB, Mackova M, Macek T (2009) DNA-based stable isotope probing: a link between community structure and function. Sci Total Environ 407(12):3611–3619
Van Elsas JD, Speksnijder AJ, van Overbeek LS (2008a) A procedure for the metagenomics exploration of disease-suppressive soils. J Microbiol Methods 75:515–522
Van Elsas DJ, Costa R, Jansson J, Sjöling S, Bailey M, Nalin R, Vogel TM, van Overbeek L (2008b) The metagenomics of disease-suppressive soils—experiences from the METACONTROL project. Trends Biotechnol 26(11):591–601
Varadarajan N, Gam J, Olsen MJ, Georgiou G, Iverson BL (2005) Engineering of protease variants exhibiting high catalytic activity and exquisite substrate selectivity. Proc Natl Acad Sci U S A 102(19):6855–6860
Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu DY, Paulsen I, Nelson KE, Nelson W, Fouts DE, Levy S, Knap AH, Lomas MW, Nealson K, White O, Peterson J, Hoffman J, Parsons R, Baden-Tillson H, Pfannkoch C, Rogers YH, Smith HO (2004) Environmental genome shotgun sequencing of the Sargasso Sea. Science 304(5667):66–74
Villegas A, Kropinski AM (2008) An analysis of initiation codon utilization in the domain Bacteria—concerns about the quality of bacterial genome annotation. Microbiology 154(9):2559–2561
Wang GZ, Luo HY, Wang YR, Huang HQ, Shi PJ, Yang PL, Meng K, Bai YG, Yao B (2011) A novel cold-active xylanase gene from the environmental DNA of goat rumen contents: direct cloning, expression and enzyme characterization. Biores Technol 102(3):3330–3336
Warnecke F, Hess M (2009) A perspective: metatranscriptomics as a tool for the discovery of novel biocatalysts. J Biotechnol 142(1):91–95
Warnecke F, Luginbuhl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT, Cayouette M, McHardy AC, Djordjevic G, Aboushadi N, Sorek R, Tringe SG, Podar M, Martin HG, Kunin V, Dalevi D, Madejska J, Kirton E, Platt D, Szeto E, Salamov A, Barry K, Mikhailova N, Kyrpides NC, Ottesen EA, Zhang XN, Hernandez M, Murillo C, Acosta LG, Rigoutsos I, Tamayo G, Green BD, Chang C, Rubin EM, Mathur EJ, Robertson DE, Hugenholtz P, Leadbetter JR (2007) Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450(7169):560–565
Waschkowitz T, Rockstroh S, Daniel R (2009) Isolation and characterization of metalloproteases with a novel domain stucture by contruction and screening of metagenomic libraries. Appl Environ Microbiol 75(8):2506–2516
Wexler M, Bond PL, Richardson DJ, Johnston AWB (2005) A wide host-range metagenomic library from a waste water treatment plant yields a novel alcohol/aldehyde dehydrogenase. Environ Microbiol 7(12):1917–1926
Williamson LL, Borlee BR, Schloss PD, Guan CH, Allen HK, Handelsman J (2005) Intracellular screen to identify metagenomic clones that induce or inhibit a quorum-sensing biosensor. Appl Environ Microbiol 71(10):6335–6344
Williamson KE, Kan J, Polson SW, Williamson SJ (2011) Optimizing the indirect extraction of prokaryotic DNA from soils. Soil Biol Biochem 43(4):736–748
Zalucki YM, Jennings MP (2007) Experimental confirmation of a key role for non-optimal codons in protein export. Biochem Biophys Res Commun 355(1):143–148
Zalucki YM, Beacham IR, Jennings MP (2009) Biased codon usage in signal peptides: a role in protein export. Trends Microbiol 17(4):146–150
Zanaroli G, Balloi A, Negroni A, Daffonchio D, Young L, Fava F (2010) Characterization of the microbial community from the marine sediment of the Venice lagoon capable of reductive dechlorination of coplanar polychlorinated bephenyls (PCBs). J Hazard Mater 178(1–3):417–426
Acknowledgments
This work was funded by the EU Metaexplore project (KBBE-222625).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Ekkers, D.M., Cretoiu, M.S., Kielak, A.M. et al. The great screen anomaly—a new frontier in product discovery through functional metagenomics. Appl Microbiol Biotechnol 93, 1005–1020 (2012). https://doi.org/10.1007/s00253-011-3804-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3804-3