
Abstract
Cyanophycin or cyanophycin granule peptide is a protein that results from non-ribosomal protein synthesis in microorganisms such as cyanobacteria. The amino acids in cyanophycin can be used as a feedstock in the production of a wide range of chemicals such as acrylonitrile, polyacrylic acid, 1,4-butanediamine, and urea. In this study, an auxotrophic mutant (Rhizopus oryzae M16) of the filamentous fungus R. oryzae 99-880 was selected to express cyanophycin synthetase encoding genes. These genes originated from Synechocystis sp. strain PCC6803, Anabaena sp. strain PCC7120, and a codon optimized version of latter gene. The genes were under control of the pyruvate decarboxylase promoter and terminator elements of R. oryzae. Transformants were generated by the biolistic transformation method. In only two transformants both expressing the cyanophycin synthetase encoding gene from Synechocystis sp. strain PCC6803 was a specific enzyme activity detected of 1.5 mU/mg protein. In one of these transformants was both water-soluble and insoluble cyanophycin detected. The water-soluble fraction formed the major fraction and accounted for 0.5% of the dry weight. The water-insoluble CGP was produced in trace amounts. The amino acid composition of the water-soluble form was determined and constitutes of equimolar amounts of arginine and aspartic acid.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
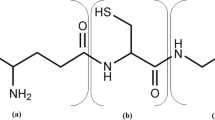
Cyanophycin or cyanophycin granule peptide (CGP) was first discovered in 1887 by Borzi while examining cyanobacteria with a microscope (Borzi 1887). Later, it was discovered that all groups of cyanobacteria produce CGP (Oppermann-Sanio and Steinbuchel 2002) as well as many heterotrophic bacteria (Krehenbrink et al. 2002). CGP is a polypeptide that results from non-ribosomal protein synthesis by a single enzyme, cyanophycin synthetase (CphA) (Ziegler et al. 1998). The molecule consists of an α-amino-α-carboxy linked aspartic acid backbone to which arginine residues are linked to the β-carboxyl group of each aspartic acid (Simon and Weathers 1976) (Fig. 1). The non-ribosomal synthesis results in a polydisperse size distribution. In addition, the molecular mass depends on the CGP producing organism (Krehenbrink et al. 2002); in cyanobacteria, the molecular mass ranges from 25 to 100 kDa (Hai et al. 1999; Lang et al. 1972; Simon 1971, 1973). At neutral pH and under physiological ionic strength, CGP is in general water insoluble, although a CGP form was described that is water soluble at neutral pH (Ziegler et al. 2002). The water-insoluble CGP can become water soluble at extreme pH values (below 3 or above 9) or under high ionic strength (Fuser and Steinbuchel 2005; Simon 1971).

Schematic overview of a CGP molecule. Schematic representation of a CGP molecule with an aspartic acid backbone and arginine side chains (Simon and Weathers 1976)
CGP has potentially many industrial applications, it can be used for the production of poly(l-aspartic acid) (PAA) generated by β-cleavage of the side chains. PAA can be used as substitute for non-degradable poly(anionic) molecules such as polyacrylic acid (Sanders et al. 2007). CGP can also be applied for the production of anti-scalants, dispersing agents, or bulk chemicals such as acrylonitrile (Könst et al. 2009), urea, and 1,4-butanediamine (Könst et al. 2010; Schwamborn 1998; Scott et al. 2007).
Currently, the application of CGP is hampered by the lack of a cost-effective production process in cyanobacteria as a result of the low yield and strict growth requirements such as light and complex media. In order to increase the production efficiency, cphA-encoding genes from various biological sources were expressed in Escherichia coli (Berg et al. 2000; Frey et al. 2002; Krehenbrink et al. 2002; Ziegler et al. 2002; Ziegler et al. 1998; Hai et al. 1999; Aboulmagd et al. 2000) and other commercially relevant bacteria such as Corynebacterium glutamicum, Ralstonia eutropha, and Pseudomonas putida (Aboulmagd et al. 2001; Voss et al. 2004; Diniz et al. 2006). The maximal yield in these bacteria varied strongly with a maximum yield of 35% (w/w). In eukaryotic microorganisms like Saccharomyces cerevisiae (Bröker et al. 2008) and Pichia pastoris (Bröker et al. 2010), the maximal CGP yield dropped to 15% w/w).
Filamentous fungi of Rhizopus oryzae spp. have great potential in biotechnological applications. This is due to their ability to utilize a range of simple carbon substrates such as d-glucose, d-xylose, sucrose, and lactose (Vially et al. 2010). Next to simple carbohydrates, R. oryzae spp. can also grow on agricultural waste streams (Abedinifar et al. 2009; Bulut et al. 2004; Guo et al. 2010; Xu et al. 2010; Yao et al. 2010; Yen and Lee 2010; Yu and Hang 1989). Using these carbon sources, it can produce ethanol and organic acids like l-(+)-lactic, fumaric and l-(+)-malic acid (Lockwood et al. 1936; Magnuson and Lasure 2004). These organic acids have wide applications in the food and feed industry. In addition, these compounds can be applied as feedstock in order to produce renewable resources like plastics, fibers, solvents, and oxygenated chemicals (Datta and Henry 2006; Engel et al. 2008; Goldberg et al. 2006; Roshchin 2010). The biotechnological potential of R. oryzae spp. further increased by the publication of the genome sequence of strain 99-880 in 2004 and with the development of transformation systems based on uracil auxotrophy (Skory and Ibrahim 2007).
To investigate the potential for the production of CGP in a fungal expression system, we have expressed the cphA-encoding genes from Synechocystis sp. strain PCC6803, Anabaena sp. strain PCC7120 and its codon-optimized version in the auxotrophic mutant R. oryzae M16 derived from R. oryzae 99-880.
Materials and methods
Strains, media, growth conditions, and methods
One Shot® Mach1™ T1 Phage-Resistant E. coli (Invitrogen Carlsbad, CA) was used for plasmid maintenance and propagation. The cells were grown in Luria-Bertani (LB) medium containing 50 μg/ml ampicillin or kanamycin at 37°C with agitation at 250 rpm. In this study, R. oryzae 99-880 (Fungal Genetics Stock Center FGSC 9543) for which the genome sequence is known and the from this strain-derived orotate phosphoribosyltransferase (pyrF) auxotrophic mutant M16 (Skory and Ibrahim 2007) were used. The auxotrophic mutant M16 was a kind gift of Dr. C. D. Skory of the USDA, Peoria, IL. Spores were obtained by cultivation on synthetic Rhizopus (RZ) medium (Skory 2000) containing 1.5% (w/v) agar and 2% d-glucose. For growth of R. oryzae M16, the medium was supplemented with 0.5 mg/ml uracil. The plates were incubated for 4 days at 30°C. The spores were harvested using a saline Tween-80 solution [0.9% (w/v) NaCl, 0.005% (v/v) Tween-80]. Details for the strains and plasmids are listed in Table 1. All experiments were performed with biomass obtained from liquid cultures unless stated otherwise. Biomass of R. oryzae transformants and wild-type was generated by cultivation in shake flasks containing 100 ml RZ medium using 100 g/l of D-glucose as a carbon source, inoculated with 106 spores per milliter. The cultures were incubated for 72 h at 30°C with constant agitation in an orbital shaker at 200 rpm. To maintain a stable pH, 10 g/l CaCO3 was added after 18 h. When the CaCO3 was almost dissipated, fresh CaCO3 was added.
DNA techniques
The cphA 7120 gene originating from Anabaena sp. strain PCC7120 was cloned from plasmid pBBR1MCS-2::cphA 7120 (Voss et al. 2004). The cphA 6803 gene originating from Synechocystis sp. strain PCC6803 was cloned from plasmid pMa/c5-914::cphA 6803 (Frey et al. 2002). Both plasmids were a kind gift from Prof. Dr. A. Steinbüchel of the WWU Münster, Germany. The genes were cloned by PCR amplification using Phusion DNA polymerase (Finnzymes, Espoo, Finland). The oligonucleotide primers used for PCR amplification were 7120FS and 7120RS for cphA 7120 and 6803FS and 6803RP for cphA 6803 (Table 2). The correct size of the DNA fragments was verified by agarose gel electrophoresis using 0.8% agarose with 0.5 times Tris–acetate–EDTA (TAE) buffer. The amplicons were ligated into the plasmid pJET1.2/blunt using the CloneJET™ PCR Cloning Kit (Fermentas International Inc, Burlington, Canada) according to the manufacturer's instructions. Codon-optimized versions of both genes were ordered from DNA 2.0 (Menlo Park, CA). Codon optimization was performed based on the codon table of Kazusa DNA Research Institute (Kisarazu, Japan). Electro transformation of E. coli was performed with the Gene pulser II using cuvettes with a 0.2-cm gap (Bio-Rad, Hercules, CA). Isolation of plasmid DNA was performed using a GeneJETTM plasmid miniprep kit (Fermentas International Inc, Burlington, Canada) according to the manufacturer’s instructions. The plasmid pJet::cphA 7120 and pJet::cphA 6803 were digested using the restriction enzymes SphI and PacI (New England Biolabs, Ipswich, MA), subsequently ligated into the SphI and PacI sites of plasmid pPdcExPyrF. Plasmid pPdcExPyrF was a kind gift of Dr. C. D. Skory of the USDA, Peoria, IL. The nucleotide sequences of all fragments were verified by DNA sequence analysis (Baseclear BV, Leiden, The Netherlands).
Transformation of R. oryzae and stability assay of transformants
Transformation of R. oryzae M16 spores was achieved by particle bombardment. M5 tungsten particles (Bio-Rad, Hercules, CA) were coated with plasmid DNA according to the manufacturer's instructions. The particles were delivered by the PDS-1000/He Biolistic Particle Delivery System (Bio-Rad, Hercules, CA), having a distance between the rupture disc and the particles of 1.6 cm. The distance between the particles and the spores was 6 cm. Spores (105) of R. oryzae M16 were plated on RZ medium and bombarded. The pressure was set with a rupture disc at 1,100 psi. To allow the formation of biomass and spores, the plates were incubated at 30°C for 5 to 7 days. After sporulation, the spores were harvested, and serial dilutions were made that were used to inoculate RZ plates with one spore. This process was repeated to ensure single progeny. In order to determine the stability of the transformants, serial dilutions of the spores grown from the glycerol stocks were made ranging from 8 to 8 × 109 spores per plate. The spores were plated on RZ medium containing 5-fluoroorotic acid (5-FOA) and 5-FOA complemented with 0.5 mg/ml uracil.
Transcript analysis
To determine the presence of cphA mRNA in the transformants, liquid cultures were grown as described in the previous section. Mycelium was harvested after 72 h, frozen in liquid nitrogen, and ground using a Braun micro-dismembrator (Braun, Melsungen, Germany). RNA was isolated and cDNA was generated as described by Oliveira et al. (2008). The cDNA served as the template for quantitative real-time PCR (qPCR). Primers were designed with the Primer3 program to have a specific melting temperature of 60 ± 1°C, GC content of 50 ± 5%, and amplicon sizes between 139 and 150 bp. The primers used were 7120qF, 7120qR, 7120coqF, 7120coqR 6803qF, and 6803qR. Two reference genes were used; as an external reference gene kanamycin, the primers were kanqF and kanqR (Table 2) and as an internal reference gene pyruvate decarboxylase was used with the primers PDCqF and PDCqR (Table 2). The PCR mixes were pipetted with the CAS-1200 robot (Corbett Life Science, Sydney, Australia). Reaction mixtures for real-time PCR had a total volume of 16 μl and contained 4 μl cDNA (2.5 ng/μl), 1.2 μl of each primer (1.2 μM), and 10 μl ABsolute QPCR SYBR Green Mix (ABgene, Epsom, UK). The Rotor-Gene 3000 (Corbett Life Science) was used for thermal cycling and real-time detection of the DNA. The melting analysis feature was used to determine primer–dimer formation, and the comparative quantitation feature was used to determine take-off and amplification values. Relative expression was calculated by the Pfaffl method (Pfaffl 2001). Normalization was performed on the basis of the added kanamycin external transcript proportional to the total RNA used for cDNA synthesis. Additionally, normalization was performed on the basis of pyruvate decarboxylase (PDC).
Cyanophycin synthetase activity assay
CphA activity was determined in cell-free extracts by a scintillation assay performed as described by Aboulmagd et al. (2000) using L-[2,3,4,5-3H] arginine monohydrochloride (GE Healthcare, Piscataway, NJ) with a specific activity of 1.59 TBq/mmol. The cell-free extract was obtained by centrifugation of a suspension of 100 mg grinded mycelium in 500 μl 20 mM Tris/HCl buffer pH 7.5 (4ºC, 16,100 g). To determine the effect of protease activity, 12.5 μl protease inhibitor cocktail (P8215) (Sigma-Aldrich, St. Louis, MO) was added to the cell-free extract. The activity was measured by adding the reaction mixture in a 1 to 10 ratio to Ultima gold scintillation liquid (Perkin-Elmer Life Sciences, Boston, MA) and scintillation counting in a model 1600 TR Tri-Carb liquid scintillation counter (Packard Instrument Company, Meriden, CT). To determine the specific activity, the protein concentrations in the cell-free extract were determined with a Bradford protein assay (Bio-Rad, Hercules, CA). The protein standards were prepared with bovine serum albumin. All experiments were performed in triplicate. The specific enzyme activity was expressed in units per milligram, which represents the incorporation rate of l-arginine in nanomoles per minute per milligram of total protein in the cell-free extract.
Isolation and analysis of cyanophycin
Water-soluble and water-insoluble CGP was extracted using a modified method of Ziegler et al. (2002). Mycelium was harvested and disrupted as described in a previous section and 1 g (wet weight) was resuspended in 10 ml 20 mM Tris/HCl buffer pH 7.5. The suspension was centrifuged for 20 min at 4,600 g and 4°C. The cell-free extract was separated from the pellet, and water-soluble CGP was extracted from the cell-free extract. The soluble fraction was incubation at 65°C for 20 min and centrifuged for 20 min at 4,600 g at 4°C. The supernatant was incubated overnight with proteinase K after which the proteins were precipitated with ice-cold ethanol, washed with acetone, and air-dried.
Water-insoluble CGP was isolated from the biomass pellet obtained during the isolation of water-soluble CGP. This pellet was resuspended in 0.1 M HCl until the CaCO3 was dissipated and the pH of the sample was 1. The sample was centrifuged for 20 min at 4,600 g at 4°C. The supernatant was neutralized with 0.1 M NaOH and re-centrifuged. The pellet was washed twice with demineralized water and dried. At each step, the dry weight of the removed material was gravimetrically determined to calculate the weight percentage of the accumulated CGP. The molecular mass of the CGP was determined by SDS-PAGE analysis using an 11.5% (w/v) polyacrylamide gel as described by Laemmli (1970) with 50 μg of dried protein per slot. The protein marker used was a precision plus protein standard from Bio-Rad (Bio-Rad, Hercules, CA). The gels were stained with Coomassie Brilliant Blue R-250. To determine the amino acid composition, the isolated protein was hydrolyzed in 6 M HCl with 1% w/v of phenol at 110°C for 24 h under nitrogen atmosphere. The individual amino acids were derivatized with OPA-reagent (o-phthaldialdehyde) and FMOC (9-fluorenylmethoxycarbonyl chloride) (Sigma-Aldrich, St. Louis, MO). The amino acid analysis was done using the Dionex rapid separation liquid chromatography (RSLC) system (Dionex Corporation, Sunnyvale, CA) with an Acquity UPLC® BEH C18 reversed phase column (Waters, Milford, MA) using an Ultimate 3000 variable wavelength detector (Dionex Corporation, Sunnyvale, CA).
Results
Transformant stability
The stability of transformants generated in this study for the pyrF phenotype was determined with the aid of 5-fluoro-orotic acid (5-FOA) and uracil selection. If the selection marker is lost, pyrF transformants are unable to metabolize 5-FOA, thereby preventing cell death, and uracil in the plates would facilitate cell growth. None of the transformants or the wild type were able to grow on the plates containing 5-FOA and uracil. This shows that all generated transformants were stable for the pyrF phenotype.
CphA expression in R. oryzae transformants
The cphA-encoding genes from Anabaena sp. strain PCC7120 (cphA 7120) and Synechocystis sp. strain PCC6803 (cphA 6803) were selected for expression in R. oryzae 99-880 on basis of their close codon usage. The GC difference in the first three bases for cphA 7120 was 3%, 3%, and 13% for cphA 6803, the difference was 2%, 2%, and 2%. To further increase the efficiency of gene translation, codon-optimized genes were designed and cloned by DNA 2.0 (Menlo Park, CA). All the genes were cloned into the R. oryzae expression vector pPdcExPyrF (Table 1). It was impossible to obtain a vector containing the codon-optimized gene of cphA 6803, despite the fact that several E. coli strains were used as a host for plasmid propagation. The transformation of R. oryzae spores with the expression vectors was accomplished by the biolistic transformation method. In total, 40, 14, and 38 transformants were isolated with the unmodified cphA 7120, the codon optimized cphA 7120 and the cphA 6803-encoding genes, respectively (Table 3). The total number of transformants isolated directly after the transformation with the codon-optimized gene was 60, yet many failed to grow or sporulate in the isolation process for single progeny. All generated transformants were grown in liquid RZ medium for 72 h after which the mycelium was harvested for RNA extraction and protein analysis. The transformants were screened with quantitative real-time PCR (qPCR) for the presence and the amount of the specific cphA transcript. Not all isolated transformants expressed the cphA-encoding 8 out of 40 for cphA7120, 1 out of 14 for codon optimized cphA 7120, and 14 out of 38 for cphA 6803 (Table 3). The transcript levels for the cphA 7120-encoding genes represented 0.01% to 0.4% of the PDC transcript. For the transformants, expressing the cphA 6803-encoding gene was much higher, ranging from 0.8% to 39.5% of the PDC transcript. In the wild-type strain, the apparent transcript level of the cphA-encoding genes represented 1‰ of the PDC gene; this was considered to be an a-specific transcript.
CphA activity in R. oryzae transformants
Transformants in which a cphA transcript was detected were further analyzed for CphA activity as determined with the scintillation assay. In two transformants, both expressing the cphA 6803 gene, specific enzyme activity was detected. These transformants were named transformant cphA 6803# 1and transformant cphA 6803#2. Both transformants had a specific enzyme activity of 1.5 mU/mg proteins, which was much higher than the wild-type in which an activity of 0.27 mU/mg protein was measured (Table 4). To determine if protease activity was the cause of the low specific enzyme activity, a broad-spectrum protease inhibitor cocktail was added to fresh cell-free extract. This did not lead to an increase in the enzyme activity (data not shown).
Determination of the presence of CGP
CGP was extracted using the protocols for water-soluble and insoluble CGP. Only in transformant cphA 6803#1 water-soluble and insoluble CGP was detected by SDS-PAGE analysis (Fig. 2). This transformant accumulated mainly water-soluble CGP to a maximal amount of 0.5% dry weight of the biomass (Table 4). The water-insoluble CGP was accumulated in such small quantities that it was impossible to accurately determine the dry weight percentage. The protein fractions were analyzed on SDS-PAGE gel. Both forms of CGP appeared to be poly-disperse in molecular mass. The molecular mass of the water-insoluble CGP ranged from 25 to 37 kDa, whereas the molecular mass of the water-soluble CGP ranged between 10 and 20 kDa (Fig. 2).

Water-soluble and insoluble CGP extracted from R. oryzae transformants SDS-PAGE analysis of the CGP accumulated in transformants of R. oryzae M 16 expressing cphA-encoding gene from Synechocystis sp. Strain PCC6803. a The water-soluble CGP samples. b The water-insoluble samples. Per lane, 50 μg sample of each of the different transformants was loaded. a Lane 1; 20 μg CGP from S. cerevisiae G175 expressing the cphA-encoding gene from Synechocystis sp. strain PCC6308 (Bröker et al. 2008), lane 2; R. oryzae 99-880, lane 3; R. oryzae M16 transformant cphA 6803 #2, 4; R. oryzae transformant M16 cphA 6803 #1; 5, protein marker. b Lane 1; 20 μg CGP from S. cerevisiae G175 expressing the cphA-encoding gene from Synechocystis sp. strain PCC6308 (Bröker et al. 2008); lane 2; protein marker, lane 3; R. oryzae 99-880, lane 4; R. oryzae M16 expressing cphA 6803 #1
Amino acid composition of CGP
The amino acid composition of the isolated CGP was determined by rapid separation liquid chromatography (RSLC). The samples were first hydrolyzed by acid hydrolysis, and as a result, it was not possible to discriminate between aspartic acid and asparagine, nor between glutamic acid and glutamine. The amino acid composition of the water-soluble CGP comprised of equimolar amounts of arginine and aspartic acid/asparagine. These fractions represented 70.4 mol% of the total protein fraction. The remainder of the sample consisted primarily of leucine, glycine, serine, and lysine, with trace amounts of other amino acids. A sample from the wild-type strain extracted by the same method was also analyzed as a reference sample. In this sample, the combined fractions of aspartic acid/asparagine and arginine represented only 16.4 mol% of the total protein present in the sample. The most abundant amino acid fractions were glutamic acid/glutamine, aspartic acid/asparagine and alanine representing 11.4, 11.2, and 10.8 mol%, respectively. Lysine represented 7.2 mol% of the protein fraction in the wild-type sample and only 5.7 mol% in the CGP sample. The amino acid composition of the water-insoluble CGP was not accurately determined due to the low CGP content in the sample.
Discussion
CGP production in R. oryzae
The goal of this study was to produce the polypeptide CGP in the filamentous fungus R. oryzae. The transcript levels of the different cphA-encoding genes varied strongly in the transformants and in only two transformants expressing cphA 6803 was specific enzyme activity detected. The specific activity of CphA in the two transformants was 1.5 mU/mg protein. This is about 1,000 times lower than described for other eukaryotic microorganisms. In S. cerevisiae (Bröker et al. 2008) and P. pastoris (Bröker et al. 2010) both expressing cphA 6308, the CphA activity was 0.91 and 2.01 U/mg, respectively. In addition, no correlation was detected between transcript levels and the measured enzyme activity for the R. oryzae transformants. This in contrast to the results described in a study by Mertens et al. (2006) with R. oryzae NRRL395 using the same promoter. Here, a clear correlation was observed between transcript levels and Green Fluorescence Protein (GFP) accumulation. Furthermore, CGP only accumulated in one of the two transformants which displayed enzyme activity.
There are several possible explanations for the low enzyme activity and CGP accumulation in the cphA transformants of R. oryzae. One option is a putative instability of the introduced construct. Kroll et al. (2011) reported a much lower CGP production by engineered E. coli cells grown on mineral medium compared to those grown in complex media. It was hypothesized that this was due to the instability of the introduced plasmid. Apparently, the production of CGP in mineral medium, also applied in our study, is a strong metabolic burden resulting in an unstable genotype. A comparable phenomenon was observed in CGP producing transformants of S. cerevisiae. Here, a loss in enzyme activity and CGP accumulation occurred in transformants after several cultivation rounds, whereas the gene itself remained present in the cells (Dr. A. Bröker, personal communication). The isolation of single progeny transformants and biomass generation with R. oryzae in this study requires three consecutive sporulation events and are equivalent to many cultivation rounds. A loss comparable to that of S. cerevisiae's enzyme activity after prolonged cultivation can explain the absence or low amounts of active enzyme and CGP. Another option can be a putative toxicity of the cphA-encoding gene products. The putative toxicity can also be a reason why there is a discrepancy between the mRNA levels and enzyme activity. Yet the discrepancy can also be a result of the direct inactivation of the CphA protein by, e.g., proteolytic activity.
Currently, a limited number of heterologous genes were successfully expressed in R. oryzae. These are genes coding for GFP (Mertens et al. 2006) and lactate dehydrogenase A from R. oryzae NRRL395 in R. oryzae in 99-880 (Skory and Ibrahim 2007). These genes are either small or originate from closely related organisms. The cphA 6803-encoding gene from cyanobacteria is phylogenetically more distant and with a size of approximately 3 kbp significantly larger than the previously expressed genes. This demonstrated that larger heterologous genes can also be successfully expressed in R. oryzae.
References
Abedinifar S, Karimi K, Khanahmadi M, Taherzadeh MJ (2009) Ethanol production by Mucor indicus and Rhizopus oryzae from rice straw by separate hydrolysis and fermentation. Biomass and Bioenerg 33:828–833
Aboulmagd E, Oppermann-Sanio FB, Steinbuchel A (2000) Molecular characterization of the cyanophycin synthetase from Synechocystis sp strain PCC6308. Arch Microbiol 174:297–306
Aboulmagd E, Voss I, Oppermann-Sanio FB, Steinbuchel A (2001) Heterologous expression of cyanophycin synthetase and cyanophycin synthesis in the industrial relevant bacteria Corynebacterium glutamicum and Ralstonia eutropha and in Pseudomonas putida. Biomacromolecules 2:1338–1342
Berg H, Ziegler K, Piotukh K, Baier K, Lockau W, Volkmer-Engert R (2000) Biosynthesis of the cyanobacterial reserve polymer multi-l-arginyl-poly-l-aspartic acid (cyanophycin)—mechanism of the cyanophycin synthetase reaction studied with synthetic primers. Eur J Biochem 267:5561–5570
Borzi A (1887) Le communicazioni intracellulari delle Nostochinee. Malpighia 1:28–74
Bröker A, Oppermann-Sanio FB, Reichelt R, Steinbuchel A (2008) Synthesis and accumulation of cyanophycin in transgenic strains of Saccharomyces cerevisiae. Appl Environ Microbiol 74:3410–3418
Bröker A, Witthoff S, Krause JP, Steinbuchel A (2010) Establishment of cyanophycin biosynthesis in Pichia pastoris and optimization by use of engineered cyanophycin synthetases. Appl Environ Microbiol 76:1062–1070
Bulut S, Elibol M, Ozer D (2004) Effect of different carbon sources on l(+)-lactic acid production by Rhizopus oryzae. Biochem Eng J 21:33–37
Datta R, Henry M (2006) Lactic acid: recent advances in products, processes and technologies—a review. J Chem Technol Biot 81:1119–1129
Diniz SC, Voss I, Steinbuchel A (2006) Optimization of cyanophycin production in recombinant strains of Pseudomonas putida and Ralstonia eutropha employing elementary mode analysis and statistical experimental design. Biotechnol Bioeng 93:698–717
Engel CAR, Straathof AJJ, Zijlmans TW, van Gulik WM, van der Wielen LAM (2008) Fumaric acid production by fermentation. Appl Environ Microbiol 78:379–389
Frey KM, Oppermann-Sanio FB, Schmidt H, Steinbuchel A (2002) Technical-scale production of cyanophycin with recombinant strains of Escherichia coli. Appl Environ Microbiol 68:3377–3384
Fuser G, Steinbuchel A (2005) Investigations on the solubility behavior of cyanophycin. Solubility of cyanophycin in solutions of simple inorganic salts. Biomacromolecules 6:1367–1374
Goldberg I, Rokem JS, Pines O (2006) Organic acids: old metabolites, new themes. J Chem Tech Biotechnol 81:1601–1611
Guo Y, Yan Q, Jiang Z, Teng C, Wang X (2010) Efficient production of lactic acid from sucrose and corncob hydrolysate by a newly isolated Rhizopus oryzae GY18. J Ind Microbiol Biotechnol 37:1137–1143
Hai T, Oppermann-Sanio FB, Steinbuchel A (1999) Purification and characterization of cyanophycin and cyanophycin synthetase from the thermophilic Synechococcus sp MA19. Fems Microbiol Lett 181:229–236
Könst PM, Franssen MCR, Scott EL, Sanders JPM (2009) A study on the applicability of l-aspartate alpha-decarboxylase in the biobased production of nitrogen containing chemicals. Green Chem 11:1646–1652
Könst PM, Turras PMCCD, Franssen MCR, Scott EL, Sanders JPM (2010) Stabilized and immobilized Bacillus subtilis arginase for the biobased production of nitrogen-containing chemicals. Adv Synth Catal 352:1493–1502
Krehenbrink M, Oppermann-Sanio FB, Steinbuchel A (2002) Evaluation of non-cyanobacterial genome sequences for occurrence of genes encoding proteins homologous to cyanophycin synthetase and cloning of an active cyanophycin synthetase from Acinetobacter sp. strain DSM 587. Arch Microbiol 177:371–380
Kroll J, Klinter S, Steinbuchel A (2011) A novel plasmid addiction system for large-scale production of cyanophycin in Escherichia coli using mineral salts medium. Appl Microbiol Biotechnol 89:593–604
Laemmli UK (1970) Cleavage of structural proteins during assembly of head of bacteriophage-T4. Nature 227:680–685
Lang NJ, Simon RD, Wolk CP (1972) Correspondence of cyanophycin granules with structured granules in Anabaena cylindrica. Archiv Fur Mikrobiologie 83:313–320
Lockwood LB, Ward GE, May OE (1936) The physiology of Rhizopus oryzae. journal of agricultural research 53:849–857
Magnuson J, Lasure LL (2004) Advances in fungal biotechnology for industry, agriculture and medicin. Organic acid production by filamentous fungi. Kluwer Academic/Plenum Publishers, New York, NY, pp 307–340
Mertens JA, Skory CD, Ibrahim AS (2006) Plasmids for expression of heterologous proteins in Rhizopus oryzae. Arch Microbiol 186:41–50
Oliveira JM, van der Veen D, de Graaff LH, Qin L (2008) Efficient cloning system for construction of gene silencing vectors in Aspergillus niger. Appl Microbiol Biotechnol 80:917–924
Oppermann-Sanio FB, Steinbuchel A (2002) Occurrence, functions and biosynthesis of polyamides in microorganisms and biotechnological production. Naturwissenschaften 89:11–22
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:36–45
Roshchin AI (2010) Arylmaleic anhydrides via Heck arylation of fumaric acid. Tetrahedron Lett 51:3633–3635
Sanders J, Scott E, Weusthuis R, Mooibroek H (2007) Bio-refinery as the bio-inspired process to bulk chemicals. Macromol Biosci 7:105–117
Schwamborn M (1998) Chemical synthesis of polyaspartates: a biodegradable alternative to currently used polycarboxylate homo- and copolymers. Polym Degrad Stabil 59:39–45
Scott E, Peter F, Sanders J (2007) Biomass in the manufacture of industrial products—the use of proteins and amino acids. Appl Microbiol Biotechnol 75:751–762
Simon RD (1971) Cyanophycin granules from blue-green alga Anabaena cylindrica—reserve material consisting of copolymers of aspartic acid and arginine. P Natl Acad Sci USA 68:265-&
Simon RD (1973) Measurement of cyanophycin granule polypeptide contained in blue-green alga Anabaena cylindrica. J Bacteriol 114:1213–1216
Simon RD, Weathers P (1976) Determination of the structure of the novel polypeptide containing aspartic acid and arginine which is found in Cyanobacteria. Biochim Biophys Acta 420:165–176
Skory CD (2000) Isolation and expression of lactate dehydrogenase genes from Rhizopus oryzae. Appl Environ Microbiol 66:2343–2348
Skory CD, Ibrahim AS (2007) Native and modified lactate dehydrogenase expression in a fumaric acid producing isolate Rhizopus oryzae 99-880. Curr Genet 52:23–33
Vially G, Marchal R, Guilbert N (2010) l(+) Lactate production from carbohydrates and lignocellulosic materials by Rhizopus oryzae UMIP 4.77. World J Microb Biot 26:607–614
Voss I, Diniz SC, Aboulmagd E, Steinbuchel A (2004) Identification of the Anabaena sp. strain PCC7120 cyanophycin synthetase as suitable enzyme for production of cyanophycin in gram-negative bacteria like Pseudomonas putida and Ralstonia eutropha. Biomacromolecules 5:1588–1595
Xu Q, Li S, Fu Y, Tai C, Huang H (2010) Two-stage utilization of corn straw by Rhizopus oryzae for fumaric acid production. Bioresource Technol 101:6262–6264
Yao W, Wu X, Zhu J, Miller C, Sun B (2010) Bioconversion of the nutrients in dairy manure for l-(+)-lactic acid production by Rhizopus oryzae. Am Sco Agri Biotechnol Eng 2:1079–1090
Yen HW, Lee YC (2010) Production of l-(+)-lactic acid from raw sweet potato powders by Rhizopus oryzae immobilized in sodium alginate capsules. Appl Biochem Biotech 162:607–615
Yu RC, Hang YD (1989) Kinetics of direct fermentation of agricultural commodities to l-(+)-lactic acid by Rhizopus oryzae. Biotechnol Lett 11:597–600
Ziegler K, Deutzmann R, Lockau W (2002) Cyanophycin synthetase-like enzymes of non-cyanobacterial eubacteria: characterization of the polymer produced by a recombinant synthetase of Desulfitobacterium hafniense. Z Naturforsch [C] 57:522–529
Ziegler K, Diener A, Herpin C, Richter R, Deutzmann R, Lockau W (1998) Molecular characterization of cyanophycin synthetase, the enzyme catalyzing the biosynthesis of the cyanobacterial reserve material multi-l-arginyl-poly-l-aspartate (cyanophycin). Eur J Biochem 254:154–159
Acknowledgements
We would like to thank Dr. C. D. Skory for providing plasmid pPdcExPyrF and R. oryzae M16 and Prof. Dr. A. Steinbüchel for providing a sample of CGP and plasmids pBBR1MCS-2::cphA 7120 and pMa/c5-914::cphA 6803. We thank A. van Zeeland for performing RSLC analysis of the amino acid composition of the accumulated CGP and Dr. A. Bröker for the discussion on gene stability. This project was supported by a grant (EOSLT02034) provided by SenterNovem (Utrecht, the Netherlands).
Competing interests
The authors declare that they have no competing interests.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Meussen, B.J., Weusthuis, R.A., Sanders, J.P.M. et al. Production of cyanophycin in Rhizopus oryzae through the expression of a cyanophycin synthetase encoding gene. Appl Microbiol Biotechnol 93, 1167–1174 (2012). https://doi.org/10.1007/s00253-011-3604-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3604-9