
Abstract
Polyketides are important bioactive natural products biosynthesized by bacteria, fungi, and plants. The enzymes that synthesize polyketides are collectively referred to as polyketide synthases (PKSs). Because many of the natural hosts that produce polyketides are difficult to culture or manipulate, establishing a universal heterologous host that is genetically tractable has become an important goal toward the engineered biosynthesis of polyketides and analogues. Here, we summarize the recent progresses in engineering Escherichia coli as a heterologous host for reconstituting PKSs of different types. Our increased understanding of PKS enzymology and structural biology, combined with new tools in protein engineering, metabolic engineering, and synthetic biology, has firmly established E. coli as a powerful host for producing polyketides.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polyketides constitute an important family of structurally diverse natural products, which include many clinically useful drugs such as the antibiotics erythromycin and tetracycline, anticancer epothilone, and the anti-hypercholesterolemia lovastatin. The pharmaceutical values of many polyketides have led to intense efforts in recent decades toward understanding and engineering the corresponding biosynthetic pathways. These concerted efforts from several disciplines, such as biochemistry, structural biology, genetics, and metabolic engineering, have enabled many examples of rational and combinatorial biosynthesis of “unnatural” natural products, as well as enzymatic tools that can be used in the semisynthesis of natural product-derived targets (Baltz 2006; Weissman and Leadlay 2005; Olano et al. 2009).
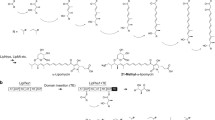
Polyketides are synthesized by a family of multifunctional enzymes known as polyketide synthases (PKSs). PKSs assemble the core structures (or aglycons) of polyketides via the sequential Claisen-like condensations of extender units derived from caboxylated acyl-CoA precursors, in a head-to-tail fashion (Staunton and Weissman 2001). Based on the structures of the polyketide products, as well as biochemical features of the PKSs, PKSs are currently classified into types I, II, and III subgroups. Type I PKSs are megasynthases in which catalytic domains are typically found in a single polypeptide. A modular type I PKS, such as the 6-deoxyerythronolide B synthase (DEBS) (Donadio and Katz 1992), consists of multiple modules and each module catalyzes one round of chain elongation and modification. Linear juxtaposition of modules facilitates unidirectional transfer of the growing polyketide from the upstream to the downstream modules in assembly line-like fashion (Cane et al. 1998). An iterative type I PKS, such as the lovastatin nonaketide synthase (LovB) (Kennedy et al. 1999), is a monomodule megasynthase in which a single set of catalytic domains are used repeatedly in a highly programmed fashion. Type II PKSs (also known as bacterial aromatic PKSs) are composed of mostly dissociated, monofunctional enzymes that function repeatedly in the synthesis of a poly-β-ketone backbone (Hertweck et al. 2007). Type II PKSs are involved in the synthesis of aromatic polyketides, such as the aglycons of actinorhodin (McDaniel et al. 1993) and daunorubicin (Hutchinson 1997). In both type I and II PKSs, the minimal PKS components that are required to perform one round of decarboxylative condensation consist of a β-ketoacylsynthase (KS), an acyltransferase (AT), and a phosphopantethienylated acyl carrier protein (ACP) (Fig. 1). The elongated polyketide product synthesized by the minimal PKS can be tailored by enzymes such as ketoreductase (KR), dehydratase (DH), enoylreductase (ER) or methyltransferase (MT), etc. Type III PKSs, such as chalcone synthase (Austin and Noel 2003), are homodimeric KSs that synthesize smaller aromatic compounds in bacteria, fungi, and plants. Products synthesized by the various types of PKSs can undergo different sets of post-PKS modifications by decorative enzymes encoded in the biosynthetic pathways, such as cyclases, oxygenases, glycosyltransferases, etc., to afford the structurally diverse natural products (Olano et al. 2010).

General mechanisms involved in polyketide biosynthesis. KS ketosynthase, AT acyl transferase, ACP acyl carrier protein, KR ketoreductase, ER enoylreductase, DH dehydratase
Polyketides are synthesized mostly by soil-borne or marine actinomycetes bacteria and filamentous fungi. Type III polyketides can also be synthesized by plants. While these organisms can be prolific and impressive natural product chemists, many of them are unfortunately difficult to work with in both laboratory and industrial settings. The difficulties can be attributed to one of more of the following reasons: (1) the strain is difficult to culture (long doubling times) or domesticate; (2) the strain is genetically intractable and refractory toward common molecular biology tools; and (3) the polyketide biosynthetic pathways are weakly expressed or silent under laboratory culturing conditions, resulting in low polyketide titers. To overcome these limitations, one important goal toward engineered biosynthesis of polyketides is the establishment of robust heterologous hosts (Pfeifer and Khosla 2001).
The workhorse organism Escherichia coli has emerged to be a useful heterologous host for the reconstitution, manipulation, and optimization of polyketide biosynthesis in recent years. The choice of E. coli is an obvious one from the start due to the (1) ease of culturing and fast growth characteristics; (2) availability of superior genetic tools; (3) well-understood primary metabolism; and (4) the lack of endogenous polyketide pathways that may crosstalk or interfere with transplanted pathways. However, E. coli also has significant drawbacks as a heterologous host, including lack of compatible post-translational enzymes, such as the phosphopantetheinyl transferase that can modify foreign ACPs (Quadri et al. 1998); unavailability of polyketide building blocks; and difficulties in efficient translation and functional folding of key biosynthetic components, such as megasynthases and the P450 family of enzymes. Fortunately, many of these limitations have been addressed in recent years via metabolic engineering, protein engineering, and synthetic biology efforts. As a result, polyketides synthesized by all three types of PKSs have been produced in E. coli. While some compounds are only synthesized at milligram per liter scales, a few complex polyketides have been produced at nearly gram per liter titers after optimization (Murli et al. 2003; Leonard et al. 2008). In addition, using enzymes mined from the biosynthetic pathways, E. coli has also been engineered to be a platform for biocatalytic synthesis of polyketide-based, clinically relevant drugs (Xie et al. 2007). These successes represent important milestones in outfitting E. coli as a powerful host for polyketide biosynthesis, and a few of these examples will be highlighted in this minireview.
Biosynthesis of type I PKS in E. coli—the 6-deoxyerythronolide B synthase success story
The most successful example of using E. coli as a heterologous host for polyketide biosynthesis is that of the total biosynthesis of 6-deoxyerythronolide B (6-dEB) and erythromycin (Pfeifer et al. 2001). 6-dEB is the 14-membered macrocyclic core of the antibiotic erythromycin synthesized by Saccharopolyspora erythraea. The PKS that synthesizes 6-dEB is DEBS, which consists of three large polypeptides, each exceeding 300 kDa in molecular weight. Together, DEBS contains one loading module and six extension modules, utilizes propionyl-CoA as a starter unit and six (2S)-methylmalonyl-CoA as extender units (Fig. 2). Each module (DEBS module 1, module 2, etc.) is responsible for one chain extension cycle, as well as the reductive tailoring of resulting β-keto product. At the end of the biosynthetic assembly, a thioesterase (TE) domain fused to the C-terminus of module 6 catalyzes the macrocyclization of the linear polyketide to yield 6-dEB (Khosla et al. 2007). The cyclized 6-dEB is then modified by a series of post-PKS enzymes, including hydroxylation and glycosylation, to yield the bioactive natural products erythromycin A–D.

Biosynthesis of 6-dEB and erythromycin
Since the initial discovery of the ery genes that encode DEBSs (Cortes et al. 1990; Donadio et al. 1991), DEBS has served as the model system to study and engineer modular type I PKSs. The linear arrangement of domains and modules inspired the colinearity rule, which allows precise prediction and manipulation of the product structure from PKS protein sequence for most of the modular PKSs. Numerous 6-dEB, erythromycin, and other analogues were synthesized through domain/module deletions, insertions, and replacements (Cane 2010). These efforts were first performed in the natural producer S. erythraea (Marsden et al. 1998) and model Streptomyces hosts (Kao et al. 1994; Xue et al. 1999) with great success. However, despite these efforts, an E. coli platform for producing 6-dEB and manipulation of DEBS was still being sought after. Pfeifer et al. reported the first total biosynthesis of 6-dEB in a highly engineered E. coli strain in 2001 (Pfeifer et al. 2001). Much of the strain engineering was focused on correctly functionalize the DEBS proteins and transplanting pathways for accumulation of the methylmalonyl-CoA building blocks. First, the sfp gene from Bacillus subtilis (Lambalot et al. 1996) was integrated into the chromosome of E. coli under a T7 promoter to assure the complete phosphopantetheinylation of the seven ACP domains in recombinant DEBS. To increase the concentrations of propionyl-CoA and methylmalonyl-CoA, the propionate catabolism genes in the prpRBCD operon were deleted, while a T7 promoter was inserted before the prpE gene, of which the encoded PrpE can convert propionate to propionyl-CoA. The resulting strain was named BAP1 and will also be mentioned later in this review. In addition, the propionyl-CoA carboxylase (pcc) gene from Streptomyces coelicolor was coexpressed to produce (2S)-methylmalonyl-CoA from propionyl-CoA. Therefore, both (2S)-methylmalonyl-CoA and propionyl-CoA can be synthesized and accumulated inside BAP1 when exogenous propionate was supplied.
Upon transformation of the plasmids that encode all three of the DEBS megasynthases, 6-dEB was successfully produced in E. coli for the first time at a titer of 20 mg/L. 6-dEB was produced ~100 mg/L by high cell density fed-batch fermentation upon the coexpression of an accessory thioesterase TEII from S. erythraea (Pfeifer et al. 2002). Lau et al. found that excess ammonia concentration dramatically decreased the E. coli cell productivity; hence, ammonia was maintained below 40 mM throughout the fermentation process, while high phosphate concentration was maintained to support cell growth (Lau et al. 2004). When DEBS proteins were expressed in BAP1 from two plasmids (Murli et al. 2003) of different origins of replications, the titer was further improved to 1.1 g/L, which was comparable to that by the best S. coelicolor host.
Many metabolic engineering efforts that aim at improving the yield of 6-dEB soon followed the initial success by Pfeifer and Khosla. For example, the B12-dependent methylmalonyl-CoA mutase-epimerase pathway from Propionibacterium shermanii was introduced into BAP1 (Dayem et al. 2002) (Fig. 2). This pathway can first convert succinyl-CoA to (2R)-methylmalonyl-CoA, which can then be epimerized to (2S)-methylmalonyl-CoA. The advantage of this pathway is that the extender unit (2S)-methylmalonyl-CoA can be provided independently of the starter unit propionyl-CoA. Wang et al. overexpressed the S-adenosylmethionine synthetase MetK from Streptomyces spectabilis, and the specific production of 6-dEB was improved from 10.86 to 20.08 mg/L/OD600 (Wang et al. 2007). The authors attributed the improvement in 6-dEB titers to the increased synthesis of signaling molecules such as AI-2. Recently, Zhang et al. deleted E. coli ygfH gene, which encodes the propionyl-CoA:succinate CoA transferase and increased 6-dEB titer in the parent strain from 65 to 129 mg/L under shake flask conditions (Zhang et al. 2010). Wang and Pfeifer (2008) also integrated the pcc genes (pccB and accA1) and the entire genes encoding DEBS (~30 kb) into the E. coli chromosome to create a plasmid-free strain YW9. This strain produced 6-dEB at a titer of 0.52 mg/L at 30°C and remarkably can produce 0.11 mg/L 6-dEB in E. coli even at 37°C. Some other metabolic engineering attempts, including overexpressing the E. coli methylmalonyl-CoA mutase, sbm, or deleting the methylmalonyl-CoA decarboxylase ygfG, did not have an positive impact on the production of 6-dEB. Similarly, overexpressing the malonyl/methylmalonyl-CoA ligase (MatB) pathway also did not lead to any improvement, although methylmalonyl-CoA was accumulated to 90% of the intracellular acyl-CoA pool (Murli et al. 2003).
Utilizing the E. coli platform, Pfeifer et al. (2001) were able to generate an analogue of 6-dEB by replacing the loading module with that from rif PKS. Taking advantage of the broad substrate specificity of the DEBS loading didomain, Kennedy et al. (2003) overexpressed the acetoacetyl-CoA:acetyl-CoA transferase AtoD in E. coli. When butyrate was supplied, butyryl-CoA was synthesized as the starter unit and the corresponding 6-dEB analogue (15-methyl-6-dEB) was generated by E. coli. Furthermore, by inactivating the loading didomain of DEBS followed by supplementation of various acyl-thioesters starter units, additional 6-dEB analogues were produced (Murli et al. 2005).
To complete erythromycin biosynthesis in E. coli, Peiru et al. identified 16 genes from the megalomicin (meg) biosynthetic pathway that encoded the tailoring enzymes that can convert 6-dEB to erythromycin C, as well as a rRNA methyltransferase (ErmE) that was necessary to confer host self-resistance (Peiru et al. 2005). Two additional plasmids encoding the l-mycarose and d-desosamine operons were constructed. The l-mycarose operon contains genes necessary for synthesis of TDP-l-mycarose, a erythronolide B TDP-mycarose glycosyltransferase, and 6dEB 6-hydroxylase; whereas the d-desosamine operon contains genes required for the synthesis of TDP-d-desosamine, a TDP-d-desosamine glycosyltransferase, and 6dEB 12-hydroxylase. Then, an engineered E. coli strain expressing DEBS and all the necessary tailoring enzymes (23 genes in total) was constructed, which produced the antibacterial erythromycin C at a titer of 0.4 mg/L and erythromycin D at that of 0.5 mg/L. The final conversion to the most desired erythromycin A, which requires the action of an O-methyltransferase, has not been achieved to date.
Biosynthesis of other complete and partial type I polyketides in E. coli
Epothilones are potent anticancer agents (tubulin inhibitors) that were originally isolated from Sorangium cellulosum (Bollag et al. 1995). Epothilones are synthesized by a hybrid PKS-nonribosomal peptide synthetases (NRPS) pathway. The NRPS module synthesizes the thiazole portion of the molecule, while the PKS portion assembles the macrolide core. The 56-kb epo gene cluster consists of one loading module (EpoA), one NRPS module (EpoB), eight PKS modules in three polypeptides (EpoD, EpoE, and EpoF) and a P450 epoxidase (EpoK) (Julien et al. 2000; Tang et al. 2000). The size of the entire cluster therefore presents a challenge to reconstitute in E. coli. Boddy et al. (2004) used a precursor directed method to first demonstrate epothilone production from the BAP1 strain. This was accomplished using a three-plasmid system to express EpoD module 6, EpoE, and EpoF in soluble forms. To mimic the upstream substrate of EpoD module 6, an advanced N-acetyl cysteamine (SNAC) thioester precursor was synthesized to be recognized by the KS domain of module 6. When the precursor was supplemented to the BAP1 strain, epothilone C was produced at a titer of 0.7 mg/L, which is comparable to that of the wild-type native host.
Subsequently, Mutka et al. (2006) expressed the full-length epo PKS-NRPS in E. coli and observed production of epothilone C and D. To accomplish this milestone, difficulties in molecular biology were first overcome through custom gene synthesis and strategic restriction site placement. At the same time, the GC-rich epo genes were codon optimized to ensure efficient translation. To further improve the protein expression and folding, the T7 promoter was changed to the arabinose-inducible PBAD promoter, and chaperone proteins were coexpressed. Soluble expression of larger portions of the epo PKS modules allowed them to perform the precursor-directed biosynthesis using much simpler SNAC-based substrates. Feeding of (E)-2-methyl-3-(2-methylthiazol-4-yl) acrylic acid to BAP1 encoding the last seven modules (EpoD, EpoE, and EpoF) of the epo biosynthetic pathway resulted in the expected synthesis of epothilone C. With this platform, novel epothilone analogues can be generated in a more straightforward fashion through the feeding of custom-synthesized precursors. The epothilone example therefore shows that synthetic biology approaches can play key roles in overcoming difficulties associated with reconstitution of large pathway into E. coli.
The epothilone PKS currently represents the largest modular type I PKS reconstituted in E. coli. Other large macrolides require more PKS modules and therefore are even more daunting obstacles. One such example is the important antibacterial agent rifamycin, of which the biosynthetic gene cluster is ~90 kb (Schupp et al. 1998; August et al. 1998). The biosynthesis pathways of rifamycin and other ansamycins are primed by 3-amino-5-hydroxybenzoic acid (AHBA), which is synthesized by a dedicated metabolic pathway consists of seven enzymes. AHBA then serves as the starter unit for the first megasynthases in the rif pathway, RifA, which is a ~530 kDa NRPS/PKS megasynthases and cannot be solubly expressed in E. coli. Toward heterologously reconstituting the rif pathway in BAP1, Khosla and coworkers first successfully expressed a cassette of seven genes to produce AHBA. Among them, two of the genes were cloned from the ansamycin pathway because the corresponding RifG and RifJ cannot be expressed solubly (Watanabe et al. 2003). To overcome the enormous size of RifA, the same research group first dissected RifA into two smaller, bimodular enzymes (~240 and ~290 kDa). To subsequently reestablish communication and substrate transfer between the separated proteins, intermodular peptide linkers from the DEBS pathway (those between DEBS modules 2 and 3) were appended to the C- and N-termini of the upstream and downstream RifA modules, respectively (Watanabe et al. 2003). This linker approach is an important strategy that can be used to split otherwise unmanageable megasynthases into considerably smaller pieces while maintaining the crucial crosstalk and specificity. Finally, by combining the AHBA pathway and the reengineered RifA parts, the key intermediate P8_1-OG was produced in BAP1 at a titer of 2.5 mg/L.
Biosynthesis of type II polyketides in E. coli
In contrast to the success of reconstituting type I PKSs, E. coli has been a complete disappointment as a host for type II PKSs from actinomycete despite intense efforts by many groups. The main obstacle is the insoluble expression of the \( {\hbox{K}}{{\hbox{S}}_\alpha } - {\hbox{K}}{{\hbox{S}}_\beta } \) heterodimer (combined size of ~90 kDa), which is part of the minimal PKS and synthesizes the full poly-β-ketone backbone (Hertweck et al. 2007). \( {\hbox{K}}{{\hbox{S}}_\alpha } \) is the active subunit and catalyzes the repeated Claisen-like condensations, while \( {\hbox{K}}{{\hbox{S}}_\beta } \) has been associated with chain length determination (Tang et al. 2003). Different attempts to express \( {\hbox{K}}{{\hbox{S}}_\alpha } - {\hbox{K}}{{\hbox{S}}_\beta } \) solubly in E. coli, including translational fusion to other proteins, to each other, or to dimerizing peptides, have all resulted in 100% of the highly expressed complex residing in the insoluble fraction of E. coli total protein. The exact reason for the insoluble expression is not known, but we hypothesize that it could be due to incompatibilities between the rates of protein synthesis, subunit folding, and heterodimerization, the latter of which is likely the most important factor.
Recently, our group engineered a fungal nonreducing PKS capable of synthesizing the poly-β-ketone backbone in E. coli, thereby indirectly bypassed the need for soluble \( {\hbox{K}}{{\hbox{S}}_\alpha } - {\hbox{K}}{{\hbox{S}}_\beta } \) expression (Zhang et al. 2008). Using the bikaverin synthase PKS4 from Gibberella fujikuroi (Linnemannstons et al. 2002), which is a megasynthase that can be expressed solubly in E. coli (Ma et al. 2007), Ma and coworkers first showed that ketoreductase (act KR) and cyclases associated with bacterial type II PKS can modify the polyketide backbone synthesized by PKS4. However, the built-in product template (PT) domains of PKS4 cyclized most of the products in a fungal-specific regioselectivity, which is considerably different than those of type II PKS products (Thomas 2001). To inactivate the built-in, fungal-specific cyclization activities of PKS4, Zhang et al. extracted the minimal PKS components (including the KS-AT didomain and ACP) from PKS4 (Zhang et al. 2008). These components were efficient in synthesizing a complete nonaketide backbone in E. coli. Combining the PKS4 minimal PKS with an assortment of type II PKS tailoring enzymes led to the synthesis of compounds in BAP1 that were previously only observed from Streptomyces, including PK8 (Kramer et al. 1997) and SEK26 (McDaniel et al. 1995). The E. coli platform can be easily scaled up under fermentation conditions and afforded milligram quantities of aromatic polyketides. Other fungal nonreducing PKSs of different chain-length specificities may be similarly engineered toward the synthesis of aromatic polyketides of different sizes in E. coli.
Biosynthesis of plant type III polyketides in E. coli
The type III PKSs provide the key structural scaffolds of a variety of plant secondary metabolites by catalyzing decarboxylative condensation between the starter unit, such as p-coumaroyl-CoA or cinnamoyl-CoA, and the extended unit malonyl-CoA. Depending on the specific activities of the type III PKSs, the product can be cyclized into different structures. The chalcone synthases (CHS) cyclize the products into flavanones, which can be further modified into to a variety of flavonoids (Weisshaar and Jenkins 1998). Stilbene synthase (STS) can cyclize the polyketide via a different regioselectivity to produce the stilbene backbone, which is the key intermediate in the biosynthesis of stilbenoids (Austin et al. 2004). Curcuminoid synthase (CUS) represents the non-cyclization type of type III PKSs that only catalyzes condensation reactions to generate curcuminoids (Abe and Morita 2010).
Horinouchi and coworkers used E. coli as a heterologous host for different plant type III PKSs and generated libraries of plant-specific polyketides from both simple amino acid and carboxylic acid precursors using different type III PKSs. First, an E. coli strain was engineered to overexpress phenylalanine ammonia lyase (PAL) from Rhodotorula rubra and 4-coumarate:coenzyme A ligase (4CL) from S. coelicolor A3(2). When supplemented with phenylalanine or tyrosine, the required starter units cinnamoyl-CoA or p-coumaroyl-CoA can be produced, respectively. Using this host, coexpression of CHS from the licorice plant Glycyrrhiza echinata resulted in the productions of pinocembrin chalcone and naringenin chalcone (Fig. 3; Hwang et al. 2003; Miyahisa et al. 2005). Similarly, resveratrol and pinosylvin were synthesized by replacing CHS with Arachis hypogaea STS and overexpressing Corynebacterium glutamicum acetyl-CoA carboxylase (ACC) (Fig. 3; Katsuyama et al. 2007b). Finally, the coexpression of PAL, 4CL, CUS, and ACC in E. coli produced bisdemethoxycurcumin and dicinnamoylmethane (Fig. 3; Katsuyama et al. 2008). Hence, once a pathway for the biosynthesis of precursors is established, the E. coli host can be directed toward the synthesis of different type III polyketides by varying the choice of the key PKS.

Biosynthesis of flavonoids, stilbenoids, and curcuminoids
To produce structurally more diverse compounds, precursor-directed biosynthesis strategy was performed based on the above system. Various aromatic carboxylic acids were supplied and led to the synthesis of corresponding unnatural compounds (Katsuyama et al. 2007a, 2010). Additional coexpression of post-PKS modification enzymes introduced by a multi-plasmid approach can further tailor the natural or unnatural flavonols, flavones, methylated resveratrols, etc. (Miyahisa et al. 2006; Leonard et al. 2006; Yan et al. 2007).
To improve titer of flavanone biosynthesis in E. coli, Koffas and coworkers designed a series of experiments to optimize the availability of the key building block malonyl-CoA. Malonyl-CoA can be synthesized from acetyl-CoA by the enzyme ACC using biotin as a cofactor. First, ACC from Photorhabdus luminescens was coexpressed with Petroselinum crispum 4CL, Petunia hybrida CHS, and Medicago sativa CHI, which resulted in the production of pinocembrin and naringenin with titers of 196 and 67 mg/L, respectively. Second, the biotin ligase (BPL) from P. luminescens was coexpressed and improved the titer of pinocembrin to 367 mg/L. Third, overexpression of E. coli endogenous acetyl-CoA synthetase resulted in further improvement of titer of pinocembrin up to 429 mg/L when supplemented with acetate at low level (Leonard et al. 2007). Alternatively, malonyl-CoA can also be synthesized from malonyl-CoA synthase MatB from malonate. When Rhizobium trifolii MatB and dicarboxylate carrier protein (MatC) were coexpressed with 4CL, CHS, and CHI in E. coli, which was supplemented with malonate, the maximal titer of pinocembrin reached 710 mg/L (Leonard et al. 2008). It is worth noting that the high titer was achieved by also adding 0.2 mM of cerulenin, which suppresses the activities of FabB/F that competes for the malonyl-CoA pool for fatty acid biosynthesis.
Biocatalytic synthesis of simvastatin in E. coli
In addition to serving as a heterologous host for the various PKSs discussed above, E. coli has also been engineered to catalyze the semisynthesis of important, polyketide-derived drug molecules. One example is the biocatalytic synthesis of simvastatin, an important cholesterol-lowing drug, which is the active ingredient of the blockbuster drug Zocor. Simvastatin is a statin, which is a family of compounds that that can inhibit the 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, the enzyme that catalyzes the rate-limiting step in the biosynthesis of cholesterol (Freeman 2006). The first statin approved by the FDA, lovastatin (Mevacor), is a fungal polyketide produced by Aspergillus terreus (Hendrickson et al. 1999; Kennedy et al. 1999). Simvastatin is a semi-synthesized derivative of lovastatin and is a more potent inhibitor of HMG-CoA reductase while exhibiting considerable less side effects in treating hypercholesterolemia (Manzoni and Rollini 2002). The only difference between lovastatin and simvastatin is the substitution of α-S-methylbutyrate with α-dimethylbutyrate side chain at C-8 position of the decaline core (Fig. 4). Currently, simvastatin is produced by a multistep chemical synthesis starting from lovastatin.

Synthesis of simvastatin using E. coli as the whole-cell biocatalyst
LovD is an acyltransferase found in the lovastatin biosynthetic pathway and is responsible for catalyzing the last step of lovastatin biosynthesis by acylating monacolin J with the α-S-methylbutyryl side chain synthesized by an iterative type I PKS LovF (Xie et al. 2009). Therefore, LovD was considered as a potential enzyme that can transfer a dimethylbutyryl side chain to monacolin J and afford simvastatin via the biocatalytic approach. To confirm the activities of LovD, Xie et al. (2006) cloned and overexpressed LovD in E. coli. LovD was shown to regioselectively transfer α-S-methylbutyryl acyl group to monacolin J to produce lovastatin via a ping-pong mechanism. The authors demonstrated that LovD had broad substrate specificities toward the acyl carrier, the acyl substrate, and the decalin acyl acceptor. For example, it was able to accept membrane permeable SNAC thioesters, such as α-dimethyl-butyryl-SNAC (DMB-SNAC) as the acyl donor. Using E. coli as a whole-cell biocatalytic host, monacolin J was converted to simvastatin using a LovD expression strain and feeding of DMB-SNAC.
To improve the efficiency of the acyltransfer reaction and decrease the cost of the α-dimethylbutyryl acyl donor, further optimization of the substrate was performed (Xie and Tang 2007). Among the various thioesters assayed in this effort, the authors determined that α-dimethylbutyryl-S-methyl-3-mercaptopropionate (DMB-SMMP) is a superior substrate considering both the high initial turnover rate and the low raw material cost (Fig. 4). When DMB-SMMP was supplemented to the E. coli host, nearly quantitative conversion (>99%) of monacolin J to simvastatin was achieved at 6 g/L. Subsequently, an engineered E. coli strain YT2, which contains a bioH knockout, was selected as the whole-cell host (Xie et al. 2007). BioH was previously reported as a carboxylesterase involved in the biotin biosynthetic pathway (Sanishvili et al. 2003). It was found that BioH rapidly hydrolyzed DMB-SMMP into dimethylbutyryl mercaptopropionic acid (DMB-SMPA; Fig. 4), which significantly lowered the overall turnover rate of the transformation. Using the YT2 strain as the host, the rate of simvastatin synthesis was doubled.
Directed evolution was then used to improve the LovD catalytic activity (Gao et al. 2009). The authors employed a combination of error prone polymerase chain reaction (ep-PCR), saturation mutagenesis, and site mutagenesis to generate LovD variants. E. coli libraries expressing mutant LovD were obtained, and the whole-cell activities of converting monacolin J to simvastatin were assayed by utilizing an agar-based diffusion screening assay. This method relied on the property that simvastatin inhibited the growth of the embedded Neurospora crassa. The best mutant G7 displayed ~11-fold higher increase in whole-cell activity compared to the wild-type LovD. Using this mutant, ~30 g/L monacolin J can be quantitatively converted to simvastatin within 1 day. The authors also attempted to provide a plausible explanation for the basis of enhanced catalytic efficiency of LovD by solving the crystal structures of parent LovD, an improved mutant G5, and G5 co-crystallized with lovastatin and simvastatin. Comparisons between these structures revealed that beneficial mutations promoted a more compact conformation that was favorable for catalysis. Using the combined strategies of protein engineering, metabolic engineering, and substrate optimization, the E. coli based platform is now more efficient, more cost-effective and environmentally friendlier than the synthetic approaches previously used in the semisynthesis of simvastatin.
Conclusions
E. coli is a highly versatile microorganism for reconstituting and engineering natural product biosynthetic pathways. Despite some limitations in protein expression, building block availability, and intracellular capacity for lipophilic compounds, combined efforts from different disciplines of biotechnology have rendered E. coli a suitable host for many PKSs. With the identification of an exploding number of biosynthetic pathways from recently sequenced organisms, engineered E. coli strains discussed in this review will play more important roles in the identification, dissection, and manipulation of new PKS machineries in the future.
References
Abe I, Morita H (2010) Structure and function of the chalcone synthase superfamily of plant type III polyketide synthases. Nat Prod Rep 27(6):809–838
August PR, Tang L, Yoon YJ, Ning S, Muller R, Yu TW, Taylor M, Hoffmann D, Kim CG, Zhang XH, Hutchinson CR, Floss HG (1998) Biosynthesis of the ansamycin antibiotic rifamycin: deductions from the molecular analysis of the rif biosynthetic gene cluster of amycolatopsis mediterranei s699. Chem Biol 5(2):69–79
Austin MB, Noel AJP (2003) The chalcone synthase superfamily of type III polyketide synthases. Nat Prod Rep 20(1):79–110
Austin MB, Bowman ME, Ferrer JL, Schroder J, Noel JP (2004) An aldol switch discovered in stilbene synthases mediates cyclization specificity of type III polyketide synthases. Chem Biol 11(9):1179–1194
Baltz RH (2006) Molecular engineering approaches to peptide, polyketide and other antibiotics. Nat Biotechnol 24(12):1533–1540
Boddy CN, Hotta K, Tse ML, Watts RE, Khosla C (2004) Precursor-directed biosynthesis of epothilone in Escherichia coli. J Am Chem Soc 126(24):7436–7437
Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM (1995) Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res 55(11):2325–2333
Cane DE (2010) Programming of erythromycin biosynthesis by a modular polyketide synthase. J Biol Chem 285:27517–27523
Cane DE, Walsh CT, Khosla C (1998) Harnessing the biosynthetic code: combinations, permutations, and mutations. Science 282(5386):63–68
Cortes J, Haydock SF, Roberts GA, Bevitt DJ, Leadlay PF (1990) An unusually large multifunctional polypeptide in the erythromycin-producing polyketide synthase of Saccharopolyspora erythraea. Nature 348(6297):176–178
Dayem LC, Carney JR, Santi DV, Pfeifer BA, Khosla C, Kealey JT (2002) Metabolic engineering of a methylmalonyl-coa mutase-epimerase pathway for complex polyketide biosynthesis in Escherichia coli. Biochemistry 41(16):5193–5201
Donadio S, Katz L (1992) Organization of the enzymatic domains in the multifunctional polyketide synthase involved in erythromycin formation in Saccharopolyspora erythraea. Gene 111(1):51–60
Donadio S, Staver MJ, McAlpine JB, Swanson SJ, Katz L (1991) Modular organization of genes required for complex polyketide biosynthesis. Science 252(5006):675–679
Freeman MW (2006) Statins, cholesterol, and the prevention of coronary heart disease. FASEB J 20(2):200–201
Gao X, Xie XK, Pashkov I, Sawaya MR, Laidman J, Zhang WJ, Cacho R, Yeates TO, Tang Y (2009) Directed evolution and structural characterization of a simvastatin synthase. Chem Biol 16(10):1064–1074
Hendrickson L, Davis CR, Roach C, Nguyen DK, Aldrich T, McAda PC, Reeves CD (1999) Lavastatin biosynthesis in Aspergillus terreus: characterization of blocked mutants, enzyme activities and a multifunctional polyketide synthase gene. Chem Biol 6(7):429–439
Hertweck C, Luzhetskyy A, Rebets Y, Bechthold A (2007) Type II polyketide synthases: gaining a deeper insight into enzymatic teamwork. Nat Prod Rep 24(1):162–190
Hutchinson CR (1997) Biosynthetic studies of daunorubicin and tetracenomycin c. Chem Rev 97(7):2525–2535
Hwang EI, Kaneko M, Ohnishi Y, Horinouchi S (2003) Production of plant-specific flavanones by Escherichia coli containing an artificial gene cluster. Appl Environ Microbiol 69(5):2699–2706
Julien B, Shah S, Ziermann R, Goldman R, Katz L, Khosla C (2000) Isolation and characterization of the epothilone biosynthetic gene cluster from Sorangium cellulosum. Gene 249(1–2):153–160
Kao CM, Katz L, Khosla C (1994) Engineered biosynthesis of a complete macrolactone in a heterologous host. Science 265(5171):509–512
Katsuyama Y, Funa N, Horinouchi S (2007a) Precursor-directed biosynthesis of stilbene methyl ethers in Escherichia coli. Biotechnol J 2(10):1286–1293
Katsuyama Y, Funa N, Miyahisa I, Horinouchi S (2007b) Synthesis of unnatural flavonoids and stilbenes by exploiting the plant biosynthetic pathway in Escherichia coli. Chem Biol 14(6):613–621
Katsuyama Y, Matsuzawa M, Funa N, Horinouch S (2008) Production of curcuminoids by Escherichia coli carrying an artificial biosynthesis pathway. Microbiology 154:2620–2628
Katsuyama Y, Hirose Y, Funa N, Ohnishi Y, Horinouchi S (2010) Precursor-directed biosynthesis of curcumin analogs in Escherichia coli. Biosci Biotechnol Biochem 74(3):641–645
Kennedy J, Auclair K, Kendrew SG, Park C, Vederas JC, Hutchinson CR (1999) Modulation of polyketide synthase activity by accessory proteins during lovastatin biosynthesis. Science 284(5418):1368–1372
Kennedy J, Murli S, Kealey JT (2003) 6-deoxyerythronolide b analogue production in Escherichia coli through metabolic pathway engineering. Biochemistry 42(48):14342–14348
Khosla C, Tang Y, Chen AY, Schnarr NA, Cane DE (2007) Structure and mechanism of the 6-deoxyerythronolide b synthase. Annu Rev Biochem 76:195–221
Kramer PJ, Zawada RJX, McDaniel R, Hutchinson CR, Hopwood DA, Khosla C (1997) Rational design and engineered biosynthesis of a novel 18-carbon aromatic polyketide. J Am Chem Soc 119(4):635–639
Lambalot RH, Gehring AM, Flugel RS, Zuber P, LaCelle M, Marahiel MA, Reid R, Khosla C, Walsh CT (1996) A new enzyme superfamily—the phosphopantetheinyl transferases. Chem Biol 3(11):923–936
Lau J, Tran C, Licari P, Galazzo J (2004) Development of a high cell-density fed-batch bioprocess for the heterologous production of 6-deoxyerythronolide b in Escherichia coli. J Biotechnol 110(1):95–103
Leonard E, Yan Y, Koffas MA (2006) Functional expression of a p450 flavonoid hydroxylase for the biosynthesis of plant-specific hydroxylated flavonols in Escherichia coli. Metab Eng 8(2):172–181
Leonard E, Lim KH, Saw PN, Koffas MAG (2007) Engineering central metabolic pathways for high-level flavonoid production in Escherichia coli. Appl Environ Microbiol 73(12):3877–3886
Leonard E, Yan Y, Fowler ZL, Li Z, Lim CG, Lim KH, Koffas MAG (2008) Strain improvement of recombinant Escherichia coli for efficient production of plant flavonoids. Mol Pharm 5(2):257–265
Linnemannstons P, Schulte J, del Mar PM, Proctor RH, Avalos J, Tudzynski B (2002) The polyketide synthase gene pks4 from Gibberella fujikuroi encodes a key enzyme in the biosynthesis of the red pigment bikaverin. Fungal Genet Biol 37(2):134–148
Ma SM, Zhan J, Watanabe K, Xie X, Zhang W, Wang CC, Tang Y (2007) Enzymatic synthesis of aromatic polyketides using pks4 from Gibberella fujikuroi. J Am Chem Soc 129(35):10642–10643
Manzoni M, Rollini N (2002) Biosynthesis and biotechnological production of statins by filamentous fungi and application of these cholesterol-lowering drugs. Appl Environ Microbiol 58(5):555–564
Marsden AFA, Wilkinson B, Cortes J, Dunster NJ, Staunton J, Leadlay PF (1998) Engineering broader specificity into an antibiotic-producing polyketide synthase. Science 279(5348):199–202
McDaniel R, Ebertkhosla S, Hopwood DA, Khosla C (1993) Engineered biosynthesis of novel polyketides. Science 262(5139):1546–1550
McDaniel R, Ebertkhosla S, Hopwood DA, Khosla C (1995) Rational design of aromatic polyketide natural-products by recombinant assembly of enzymatic subunits. Nature 375(6532):549–554
Miyahisa I, Kaneko M, Funa N, Kawasaki H, Kojima H, Ohnishi Y, Horinouchi S (2005) Efficient production of (2s)-flavanones by Escherichia coli containing an artificial biosynthetic gene cluster. Appl Environ Microbiol 68(4):498–504
Miyahisa I, Funa N, Ohnishi Y, Martens S, Moriguchi T, Horinouchi S (2006) Combinatorial biosynthesis of flavones and flavonols in Escherichia coli. Appl Environ Microbiol 71(1):53–58
Murli S, Kennedy J, Dayem LC, Carney JR, Kealey JT (2003) Metabolic engineering of Escherichia coli for improved 6-deoxyerythronolide b production. J Ind Microbiol Biotechnol 30(8):500–509
Murli S, MacMillan KS, Hu ZH, Ashley GW, Dong SD, Kealey JT, Reeves CD, Kennedy J (2005) Chemobiosynthesis of novel 6-deoxyerythronolide b analogues by mutation of the loading module of 6-deoxyerythronolide b synthase 1. Appl Environ Microbiol 71(8):4503–4509
Mutka SC, Carney JR, Liu YQ, Kennedy J (2006) Heterologous production of epothilone c and d in Escherichia coli. Biochemistry 45(4):1321–1330
Olano C, Mendez C, Salas JA (2009) Antitumor compounds from actinomycetes: from gene clusters to new derivatives by combinatorial biosynthesis. Nat Prod Rep 26(5):628–660
Olano C, Mendez C, Salas JA (2010) Post-pks tailoring steps in natural product-producing actinomycetes from the perspective of combinatorial biosynthesis. Nat Prod Rep 27(4):571–616
Peiru S, Menzella HG, Rodriguez E, Carney J, Gramajo H (2005) Production of the potent antibacterial polyketide erythromycin c in Escherichia coli. Appl Environ Microbiol 71(5):2539–2547
Pfeifer BA, Khosla C (2001) Biosynthesis of polyketides in heterologous hosts. Microbiol Mol Biol Rev 65(1):106–118
Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C (2001) Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science 291(5509):1790–1792
Pfeifer B, Hu ZH, Licari P, Khosla C (2002) Process and metabolic strategies for improved production of escherichia coli-derived 6-deoxyetythronolide b. Appl Environ Microbiol 68(7):3287–3292
Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P, Walsh CT (1998) Characterization of sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry 37(6):1585–1595
Sanishvili R, Yakunin AF, Laskowski RA, Skarina T, Evdokimova E, Doherty-Kirby A, Lajoie GA, Thornton JM, Arrowsmith CH, Savchenko A, Joachimiak A, Edwards AM (2003) Integrating structure, bioinformatics, and enzymology to discover function: BioH, a new carboxylesterase from Escherichia coli. J Biol Chem 278(28):26039–26045
Schupp T, Toupet C, Engel N, Goff S (1998) Cloning and sequence analysis of the putative rifamycin polyketide synthase gene cluster from Amycolatopsis mediterranei. FEMS Microbiol Lett 159(2):201–207
Staunton J, Weissman KJ (2001) Polyketide biosynthesis: a millennium review. Nat Prod Rep 18(4):380–416
Tang L, Shah S, Chung L, Carney J, Katz L, Khosla C, Julien B (2000) Cloning and heterologous expression of the epothilone gene cluster. Science 287(5453):640–642
Tang Y, Tsai SC, Khosla C (2003) Polyketide chain length control by chain length factor. J Am Chem Soc 125(42):12708–12709
Thomas R (2001) A biosynthetic classification of fungal and streptomycete fused-ring aromatic polyketides. ChemBioChem 2(9):612–627
Wang Y, Pfeifer BA (2008) 6-Deoxyerythronolide b production through chromosomal localization of the deoxyerythronolide b synthase genes in E. coli. Metab Eng 10(1):33–38
Wang Y, Boghigian BA, Pfeifer BA (2007) Improving heterologous polyketide production in Escherichia coli by overexpression of an s-adenosylmethionine synthetase gene. Appl Environ Microbiol 77(2):367–373
Watanabe K, Rude MA, Walsh CT, Khosla C (2003) Engineered biosynthesis of an ansamycin polyketide precursor in Escherichia coli. Proc Natl Acad Sci USA 100(17):9774–9778
Weisshaar B, Jenkins GI (1998) Phenylpropanoid biosynthesis and its regulation. Curr Opin Plant Biol 1(3):251–257
Weissman KJ, Leadlay PF (2005) Combinatorial biosynthesis of reduced polyketides. Nat Rev Microbiol 3(12):925–936
Xie XK, Tang Y (2007) Efficient synthesis of simvastatin by use of whole-cell biocatalysis. Appl Environ Microbiol 73(7):2054–2060
Xie XK, Watanabe K, Wojcicki WA, Wang CCC, Tang Y (2006) Biosynthesis of lovastatin analogs with a broadly specific acyltransferase. Chem Biol 13:1161–1169
Xie XK, Wong WW, Tang Y (2007) Improving simvastatin bioconversion in Escherichia coli by deletion of BioH. Metab Eng 9(4):379–386
Xie XK, Meehan MJ, Xu W, Dorrestein PC, Tang Y (2009) Acyltransferase mediated polyketide release from a fungal megasynthase. J Am Chem Soc 131(24):8388–8389
Xue Q, Ashley G, Hutchinson CR, Santi DV (1999) A multiplasmid approach to preparing large libraries of polyketides. Proc Natl Acad Sci USA 96(21):11740–11745
Yan Y, Huang L, Koffas MAG (2007) Biosynthesis of 5-deoxyflavanones in microorganisms. Biotechnol J 2(10):1250–1262
Zhang W, Li Y, Tang Y (2008) Engineered biosynthesis of bacterial aromatic polyketides in Escherichia coli. Proc Natl Acad Sci USA 105(52):20683–20688
Zhang H, Boghigian BA, Pfeifer BA (2010) Investigating the role of native propionyl-coa and methylmalonyl-coa metabolism on heterologous polyketide production in Escherichia coli. Biotechnol Bioeng 105(3):567–573
Acknowledgements
We thank Dr. Yit Heng Chooi for critical proof reading of the manuscript and insightful discussion. Research activities from our laboratory in this area are supported from funds from National Institute of Health, National Science Foundation, the Camille & Henry Dreyfus Foundation, and the David and Lucile Packard Foundation.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
X. Gao and P. Wang contributed equally to this work.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Gao, X., Wang, P. & Tang, Y. Engineered polyketide biosynthesis and biocatalysis in Escherichia coli . Appl Microbiol Biotechnol 88, 1233–1242 (2010). https://doi.org/10.1007/s00253-010-2860-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2860-4