
Abstract
Type I Baeyer–Villiger monooxygenases (BVMOs) strongly prefer NADPH over NADH as an electron donor. In order to elucidate the molecular basis for this coenzyme specificity, we have performed a site-directed mutagenesis study on phenylacetone monooxygenase (PAMO) from Thermobifida fusca. Using sequence alignments of type I BVMOs and crystal structures of PAMO and cyclohexanone monooxygenase in complex with NADP+, we identified four residues that could interact with the 2′-phosphate moiety of NADPH in PAMO. The mutagenesis study revealed that the conserved R217 is essential for binding the adenine moiety of the nicotinamide coenzyme while it also contributes to the recognition of the 2′-phosphate moiety of NADPH. The substitution of T218 did not have a strong effect on the coenzyme specificity. The H220N and H220Q mutants exhibited a ~3-fold improvement in the catalytic efficiency with NADH while the catalytic efficiency with NADPH was hardly affected. Mutating K336 did not increase the activity of PAMO with NADH, but it had a significant and beneficial effect on the enantioselectivity of Baeyer–Villiger oxidations and sulfoxidations. In conclusion, our results indicate that the function of NADPH in catalysis cannot be easily replaced by NADH. This finding is in line with the complex catalytic mechanism and the vital role of the coenzyme in BVMOs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Enzymes catalyzing redox reactions often use nicotinamide coenzymes as electron donors or acceptors. Even though the only structural difference between NADP(H) and NAD(H) is a phosphate group, most enzymes show strict coenzyme specificity. When considering industrial applications, coenzyme specificity is an issue due to the fact that NADPH is relatively expensive, and it is less stable when compared with NADH (Wu et al. 1986). For cost-effective applications, nicotinamide coenzyme regeneration systems are available. While various chemical (Aksu et al. 2009; de Gonzalo et al. 2005), photochemical (Lee et al. 2009; Taglieber et al. 2008), and electrochemical (Siu et al. 2007; Ruinatscha et al. 2006) regeneration strategies are being explored, the most common methodologies are enzymatic regeneration of NAD(P)H (Eckstein et al. 2004; van der Donk and Zhao 2003) and application of whole-cell biocatalysis. When using isolated enzymes, NADH-specific enzymes (or enzymes that are indifferent toward NADH or NADPH) are preferred to NADPH-specific ones due to the above-mentioned reasons.
It has been shown that relaxing or inverting coenzyme specificity of NAD(P)H-dependent enzymes can be achieved by structure-inspired mutagenesis. Xylose reductase from Pichia stipitis shows a clear preference for NADPH, but the introduction of two amino acid substitutions was sufficient to reverse its coenzyme specificity. A K21A/N272D mutant was active with NADH while it did not accept NADPH anymore (Zeng et al. 2009). Similarly, only two mutations (E68K and D69A) were needed to turn mannitol 2-dehydrogenase from Pseudomonas fluorescens, an NAD+-specific enzyme, into an NADP+-preferring dehydrogenase (Bubner et al. 2008). A significant change in the coenzyme specificity was also achieved in the case of ferredoxin-NADP+ reductase from Anabaena PCC7119. The wild-type enzyme prefers NADPH 67,500 times (when coenzyme specificity is expressed as a ratio of catalytic efficiencies with NADPH and NADH) while the engineered mutant T155G/A160T/L263P/Y303S showed only a 12-fold preference (Martínez-Júlvez et al. 2005).
Baeyer–Villiger monooxygenases (BVMOs) are particularly interesting enzymes due to their unique ability to insert an oxygen atom into the carbon–carbon bond (Kayser 2009; Mihovilovic 2006; Kamerbeek et al. 2003). Additionally, they are capable of catalyzing other types of oxidations. BVMOs belonging to the type I family contain a tightly bound flavin adenine dinucleotide (FAD) cofactor and almost exclusively accept NADPH as an electron donor. The coenzyme plays a dual role in catalysis: except for donating a hydride to the flavin cofactor, it forms part of the active site during the oxidation reaction.
The molecular basis of the strict preference toward NADPH and the high affinity for NADPH/NADP+ among BVMOs are not well understood. The only enzyme from this group that has been studied so far with respect to the coenzyme specificity is 4-hydroxyacetophenone monooxygenase (HAPMO) from P. fluorescens ACB (Kamerbeek et al. 2004). Two conserved residues were found to play an important role in the coenzyme recognition. First, R339 was shown to be crucial for the NADPH recognition since a mutation of this residue resulted in the dramatically decreased apparent affinity for NADPH. Moreover, this mutation had a negative effect on the enzyme performance with NADH. In contrast, a mutation of K439 to an alanine, a phenylalanine, or an asparagine improved to some extent the catalytic efficiency with NADH while the enzyme was less active with NADPH. It is worth noting that this study was carried out without any structural information, based solely on a comparative sequence analysis.
The only type I BVMO reported to accept NADH as an electron donor so far is the MekA monooxygenase from Pseudomonas veronii MEK700. This enzyme still prefers NADPH, but significant activity has been observed when NADH was used as a coenzyme. MekA shows the K M for NADH of 29 μM, which is comparable to the K M value for NADPH (11 μM). The authors explained the observed relaxed coenzyme specificity by the lack of conservation of residues typical in BVMOs (counterparts of K439 and R440 from HAPMO; Völker et al. 2008).
Phenylacetone monooxygenase (PAMO) from Thermobifida fusca is an attractive BVMO since it is a robust and thermostable protein, and its crystal structure has been solved (Malito et al. 2004). However, it shows a 6,000-fold preference toward NADPH over NADH (vide infra). Unfortunately, despite the fact that NADP+ stays bound to the protein throughout the catalytic cycle and it is beneficial for the stability of BVMOs (van den Heuvel et al. 2005), the coenzyme was absent in the elucidated crystal structure of PAMO. This prevented a detailed analysis of NADPH binding. Recently, however, two structures of cyclohexanone monooxygenase from Rhodococcus sp. strain HI-31 (CHMO Rhodo ) in complex with NADP+ have been elucidated. These structures have provided for the first time direct information on how NADPH is bound in BVMOs (Mirza et al. 2009).
Herein, we report on identification of residues that are involved in the coenzyme recognition in PAMO. We decided to use the structures of CHMO Rhodo (sharing 43% sequence identity with PAMO) to predict residues involved in the binding of NADP+ in PAMO. In addition, we explored the sequence information of related BVMOs. This formed the basis for the design and characterization of a series of single and double mutants of PAMO in order to investigate the molecular basis for the NADPH specificity and, if possible, to increase its catalytic efficiency with NADH. Our data confirm the role of specific residues in NADPH recognition in PAMO, and are in agreement with the proposed complex role of NADP(H) in the catalytic mechanism of BVMOs.
Materials and methods
Enzymes and reagents
Oligonucleotide primers were purchased from Sigma. dNTPs were obtained from Clontech. Pfu Turbo polymerase was from Stratagene. DpnI was from New England Biolabs. All other chemicals were obtained from Acros Organics, Sigma-Aldrich, Julich Chiral Solutions GmbH, and Roche Diagnostics GmbH. Escherichia coli TOP10 from Invitrogen was used as a host for DNA manipulations and protein expression. (±)-3-Methyl-4-phenylbutan-2-one and (±)-1-phenylpropan-2-yl acetate were prepared as described (Rodríguez et al. 2009).
Construction of PAMO mutants
The PyMol software was used to align and visualize structures, and to prepare figures. The structure of PAMO (1W4X) was superimposed with the structures of CHMO Rhodo in open and closed forms (3GWF and 3GWD, respectively). Alignment of structures was restricted to the NADP-binding domains of the two proteins.
Mutants were prepared by QuikChange® site-directed mutagenesis using the primers listed in Table 1. The mutagenesis reactions were performed according to the manufacturer’s recommendations. A modified pBAD plasmid containing pamo gene with C-terminal His-tag (Fraaije et al. 2005) was used as a template for mutagenesis reactions. The plasmid containing pamo gene with H220Q mutation was used to construct double mutants H220Q/K336H and H220Q/K336N. Introduced mutations were verified by sequencing (GATC Biotech, Konstanz, Germany).
Protein expression and purification
E. coli TOP10 transformed with plasmids containing the wild-type or mutant pamo genes were grown overnight at 37°C in 5 mL of Luria–Bertani medium supplemented with 50 μg mL−1 ampicillin. On the next day, the 5-mL cultures were used to inoculate 500 mL of Terrific Broth medium supplemented with 50 μg mL−1 ampicillin and 0.02% (w v−1) l-arabinose for induction of protein expression. After cultivation for 16 h at 37°C, cells were harvested, washed with 50 mM Tris/HCl pH 7.5, and centrifuged again. Cell pellets were stored at −20°C.
The proteins were purified as follows: Each cell pellet was resuspended in 20 mL of 50 mM Tris/HCl pH 7.5 containing 100 μM FAD. Cells were disrupted by sonication and subsequently subjected to centrifugation (15,000×g for 45 min at 4°C, JA-17 rotor, Beckman Coulter). Clarified cell extract was loaded on 2 mL of Ni2+ Sepharose HP (GE Healthcare) pre-equilibrated with the Tris buffer and incubated for 30 min at 4°C with rotating. Then, the column was washed with two to three column volumes of 50 mM Tris/HCl pH 7.5, followed by two to three column volumes of 50 mM Tris/HCl pH 7.5 containing 5 mM imidazole. The protein was eluted using 50 mM Tris/HCl pH 7.5 with 500 mM imidazole. Fractions containing yellow protein were applied on a pre-equilibrated Econo-Pac 10DG desalting column (Bio-Rad). The desalted protein was stored at −80°C.
The purity of protein samples was assessed with SDS-polyacrylamide gel electrophoresis. Extinction coefficients of FAD bound in PAMO mutants were determined as described previously (Fraaije et al. 2005). UV–Vis absorption spectra were collected on a Perkin-Elmer Lambda Bio40 spectrophotometer.
Steady-state kinetic analysis
Protein samples used for kinetic analyses were first incubated for 20 min at 50°C to ensure full activity (Fraaije et al. 2005). Enzyme activity was measured in 50 mM Tris/HCl pH 7.5 at 25°C by following the decrease in coenzyme absorption at 340 nm (ε NAD(P)H, 340 = 6.22 mM−1 cm−1). In the case of some mutants, concentrations of NAD(P)H required to reach the maximal reaction rates resulted in absorption values exceeding 1 U. In these experiments, the decrease in coenzyme absorption was measured at 370 or 390 nm (ε NAD(P)H, 370 = 2.7 mM−1 cm−1, ε NAD(P)H, 390 = 0.43 mM−1 cm−1). Activity measurements were typically performed using 0.001–2.5 μM enzyme, 100 μM NADPH, 1 mM phenylacetone, and 1% (v v −1) DMSO. Kinetic parameters of the mutants with NADH and NADPH were determined using coenzyme concentration varying in the range of 1–3,000 μM. The obtained observed rate constants were fitted to the Michaelis–Menten equation using SigmaPlot for Windows version 10.0.
Analysis of enzymatic selectivity
In order to study the enantioselectivity of the wild-type and mutant PAMO, enzyme-mediated oxidations were performed with (±)-3-methyl-4-phenylbutan-2-one and thioanisole in the presence of NADPH or NADH. Glucose dehydrogenase (5 U) and glucose (20 mM) were employed to regenerate the reduced coenzyme. Reactions with (±)-3-methyl-4-phenylbutan-2-one (10 mM) were conducted in 50 mM Tris/HCl pH 8.0, at 30°C and 250 rpm in the presence of NADPH (20 or 500 μM) or NADH (5 mM). For analysis, reaction mixtures were extracted with ethyl acetate and analyzed on a Hewlett-Packard 6890 Series II chromatograph to determine the conversion and the enantiomeric excesses of (R)-3-methyl-4-phenylbutan-2-one and (S)-1-phenylpropan-2-yl acetate using a Restek RtβDEXse column (Rodríguez et al. 2009). The E value was calculated using the formula described by Straathof and Jongejan (1997).
Thioanisole (10 mM) was oxidized in 50 mM Tris/HCl pH 9.0, at 30°C and 250 rpm, in the presence of NADPH (200 μM) or NADH (5 mM) and the glucose dehydrogenase regeneration system. The reaction mixtures were extracted with ethyl acetate and analyzed by GC using a HP-1 column to determine the progress in conversion. HPLC on a Hewlett-Packard 1100 LC liquid chromatograph equipped with a Chiralcel OD column was employed to determine the optical purity of the formed (R)-methyl phenyl sulfoxide (Rioz-Martínez et al. 2010).
Results
Design and biochemical characterization of PAMO mutants
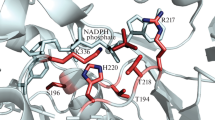
A model of PAMO with NADP+ was prepared by structural alignment of the NADP-binding domains of PAMO and CHMO Rhodo (Fig. 1a). The superposition showed highly similar architecture of the NADP domains of the two proteins as indicated by the root mean square deviation values of 1.4 Å. Structural alignments of PAMO-CHMOopen or PAMO-CHMOclosed did not differ significantly. As the NADP-binding domains of PAMO and CHMO Rhodo are highly similar in structure and both enzymes are efficient in utilizing NADPH as coenzyme, it is expected that the binding mode of NADPH is analogous in both enzymes.

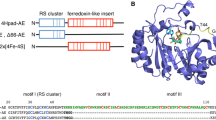
a Superposition of CHMO Rhodo (3GWF), complexed with NADP+, on the NADPH-binding domain of PAMO (1W4X). CHMO is shown in cyan, PAMO in green, and NADP+ in magenta. Residues within 8 Å from the 2′-phosphate group are shown in a stick representation; residues targeted in the mutagenesis study are labeled. b Partial sequence alignment of PAMO, CHMO Acineto , CHMO Rhodo , HAPMO, and MekA. Residues subjected to mutagenesis are indicated with a triangle; conserved residues are with gray background. Coenzyme preference in favor of NADPH is included
Inspection of the structural superpositions revealed several residues located within 8 Å around the 2′-phosphate group of NADP+ in PAMO. Some of these residues, possessing charged or polar side chains and pointing toward the 2′-phosphate group, could be involved in interaction with this phosphate moiety and therefore contribute to the discrimination between NADPH and NADH: R217, T218, H220, and K336. Using this information as well as sequence data and previous studies on the coenzyme specificity of HAPMO, we designed 13 single mutants: R217A, R217L, T218A, H220A, H220D, H220E, H220F, H220N, H220Q, H220T, H220W, K336H, and K336N. Some of these mutations were combined during the next round of mutagenesis: H220Q/K336H and H220Q/K336N.
All the mutant proteins could be overexpressed in soluble form in E. coli TOP10 at levels similar to wild-type PAMO. The His-tagged mutant enzymes were purified using the Ni2+ Sepharose. All the mutants displayed the typical UV/Vis flavin absorption spectrum, which indicates that the mutations did not affect correct folding and tight binding of the FAD cofactor. The mutant proteins also exhibited similar thermostability when compared with the wild-type enzyme.
Steady-state kinetic analysis of PAMO mutants
In order to analyze the effect of the mutations on the coenzyme specificity, steady-state kinetic parameters with NADH and NADPH were determined for all the mutants. The kinetic data are summarized in Table 2. In the previous study developed for HAPMO, it was found that replacement of R339 (which corresponds to R217 in PAMO) with an alanine caused a dramatic drop in activity (Kamerbeek et al. 2004). This residue is highly conserved among BVMOs, and inspection of the model of PAMO with NADP+ bound (Fig. 1a) reveals that this arginine can interact with the adenine moiety and possibly also with the 2′-phosphate group. We decided to mutate this residue to an alanine to investigate its role in PAMO. A mutation to a leucine was included as well to check whether this amino acid could position the adenine part while the lack of a positive charge would specifically disrupt the binding of 2′-phosphate group of NADPH. For both R217 mutants, the K M, NADPH increased by three orders of magnitude while the k cat decreased only slightly. As a result, the catalytic efficiency with NADPH (k cat/K M) decreased 570-fold in the case of R217A and 4,000-fold in the case of R217L. The catalytic efficiencies with NADH also decreased, again mainly due to the high K M, NADH values. However, the decrease was less dramatic (4- to 6-fold). This disproportion in effects caused a shift in the coenzyme specificity, rendering mutants that are active with both coenzymes and displaying comparable albeit poor efficiencies.
From the model of PAMO with NADP+ (Fig. 1a), it was concluded that T218 could be involved in interactions with the 2′-phosphate group. We mutated this residue to an alanine to probe whether it is indeed important for the coenzyme recognition. The T218A mutant showed only a 2-fold increase in both the K M and the k cat with NADH. For NADPH, the K M increased 2-fold while the k cat was slightly higher. Consequently, the coenzyme specificity was not significantly changed, which suggests that this residue plays only a marginal role in coenzyme recognition.
Sequence alignment of PAMO, CHMO, MekA, and HAPMO showed that only PAMO contains a histidine at position 220 (Fig. 1b). Thus, we first mutated H220 to a glutamine (as in CHMO and MekA) and to an asparagine (as in HAPMO). The kinetic parameters of these mutants with NADPH were hardly affected; only the K M, NADPH of H220Q was slightly increased. Both mutants showed improved affinity toward NADH (with the K M, NADH decreased ~2 times in the case of H220N and ~3.5 for H220Q), and the k cat, NADH of H220N was slightly increased. Hence, the catalytic efficiencies of these mutants with NADH were improved ~3-fold when compared to the wild-type enzyme. Encouraged by these initial results, which indicated that H220 is indeed involved in the coenzyme recognition, we prepared more mutants.
Analysis of the binding mode of NADH and NADPH in proteins has shown that NADH-specific enzymes exhibit a few common features. One common property of the NADH binding pockets is the presence of an aspartate or a glutamate that is able to interact with two hydroxyl groups of the adenine ribose moiety (Carugo and Argos 1997). Therefore, we attempted to create such a specific interaction in order to improve the NADH recognition in PAMO. The position 220 appeared as a good candidate as the side chain of H220 points toward the ribose ring, and the distance between ribose and H220 is around 4.5 Å. Moreover, the H220N and H220Q mutants were more efficient with NADH, which indicates that mutations to similar amino acids may also be beneficial.
To our disappointment, the H220E mutant performed worse than wild-type PAMO with both coenzymes. The most pronounced effect was the dramatic increase in the K M, NADPH (240-fold) while the substitution did not improve the performance with NADH. The H220D mutant showed a slightly improved catalytic efficiency with NADH, mostly due to a 2-fold higher k cat when compared with wild-type PAMO. Again, the K M, NADH was slightly higher whereas the K M, NADPH increased drastically (50-fold). The substantial decrease in the catalytic efficiency with NADPH and the retention of the activity with NADH make the H220D mutant 180-fold more specific toward NADH than the wild-type enzyme. For exploring effects, more substitutions of H220 were examined: H220A, H220F, H220T, and H220W. However, none of these mutants performed significantly better with NADH when compared with the wild-type protein. Only the H220A mutation caused a slight increase in the k cat for both coenzymes.
The amino acid K336 in PAMO corresponds to K439 in HAPMO, which has been shown to be important for the coenzyme specificity of the latter enzyme (Kamerbeek et al. 2004). All characterized BVMOs possess a lysine or an arginine at this position, with the exception of MekA, which contains a histidine (Fig. 1b). The lack of the conserved lysine could be an explanation for the increased preference of MekA toward NADH. Therefore, we decided to mutate K336 in PAMO to an asparagine and a histidine. However, the mutations did not result in any improvement in the NADH recognition. The k cat values for these mutants decreased with the exception of k cat, NADH of K336N, which was unaffected. The K M values of the mutants for both coenzymes increased. Similarly, merging one of the most beneficial mutations H220Q with K336H or K336N did not bring any positive effect on the NADH recognition, even though the combination of a glutamine and a histidine at analogous positions is present in MekA.
Enantioselectivity of PAMO mutants
We decided to investigate whether the mutations introduced in PAMO have an influence on the enantioselective behavior of the enzyme. Several studies have shown that apparently gentle mutations (Torres Pazmiño et al. 2007) or subtle changes in the medium (de Gonzalo et al. 2006; Rodríguez et al. 2008) can result in a different enantioselective behavior of PAMO. Wild-type PAMO and a selection of mutants (R217A, T218A, H220E, H220N, K336N) were tested in (a) the asymmetric sulfoxidation of thioanisole in the presence of NADPH or NADH (Table 3) and (b) the kinetic resolution of racemic 3-methyl-4-phenylbutan-2-one by enantioselective Baeyer–Villiger oxidation (Table 4).
Wild-type PAMO converted thioanisole preferentially into (R)-sulfoxide with ee p = 44% and 84% conversion when NADPH was used as coenzyme while 42% optical purity and 14% conversion were obtained with NADH. Most of the mutants tested showed a similar enantioselectivity (44 ± 5% ee p) with the exception of K336N, which gave an ee p of 58% and 64% with NADPH and NADH, respectively. By comparing the degree of conversion, the mutants showed much lower activities with respect to the wild type enzyme in the presence of NADPH or NADH, with the exception of the H220N. This biocatalyst led to similar conversions when employing NADPH while a 3-fold increase in conversion with respect to the wild-type enzyme was achieved when using NADH (37% after 30 h). This is in agreement with the improved performance of this mutant with NADH.
The enzymatic oxidation of racemic 3-methyl-4-phenylbutan-2-one catalyzed by wild-type PAMO and its mutants was also analyzed. When using the wild-type enzyme, this ketone was oxidized with 51% conversion after 1 h using NADPH while with NADH only 32% conversion was obtained after 3 h. For most mutants, lower conversions were observed with both coenzymes. With respect to enantioselectivity, wild-type PAMO and most of the mutants exhibited E values of 55 ± 8. Again, the K336N mutant showed a superior enantioselectivity as indicated by the E values of 92 and 96 when using NADPH and NADH, respectively.
Discussion
The number of BVMOs cloned and characterized is growing rapidly. These versatile oxidative biocatalysts can be utilized in a wide range of chemo-, regio-, and/or enantioselective oxidation reactions. However, for synthetic applications, one has to keep in mind that for each conversion, a reduced nicotinamide coenzyme is required. Therefore, it is desirable to engineer an NADH-specific or NADH/NADPH-indifferent BVMO. Despite the fact that almost all type I BVMOs strongly prefer NADPH as an electron donor, there is little information on what determines this strict coenzyme preference. The preference for NADPH over NADH is typically quite strong when expressed as ratio of the k cat/K M values for both coenzymes. For CHMO Acineto , HAPMO, and PAMO, the preference for NADPH is >500 (Fig. 1b). Unfortunately, the coenzyme preference of CHMO Rhodo has not been determined yet. Only for one BVMO a relatively relaxed preference has been reported: MekA shows only a 6-fold preference for NADPH. Inspired by these observations and the recent elucidation of the crystal structures of PAMO and CHMO Rhodo , we have performed a systematic mutagenesis study on PAMO in order to pinpoint residues that determine the coenzyme specificity. Alanine scanning of R217, T218, and H220 suggests that T218 and H220 do not significantly contribute to the recognition of NADPH or NADH. Contrarily, R217 was found to play a pivotal role in binding both coenzymes. Replacing this arginine by an alanine or a leucine resulted in a nearly fully abolished recognition of both coenzymes (K M ≫ 100 μM). This finding is in line with the observed positioning of the analogous arginine (R209) in CHMO Rhodo (Fig. 2). In both NADP+-complexed CHMO Rhodo crystal structures, the arginine lies on top of the adenine ring of NADP+ serving as a lock to keep the adenine bound. Except for interacting with the adenine, it also can form favorable electrostatic interactions with the 2′-phosphate of NADP+. This is confirmed by the observation that the K M, NADPH increased by three orders of magnitude while the K M, NADH only increased ~3-fold upon replacing R217. The dual role of R217 in locking the adenine part of the nicotinamide coenzymes while interacting with the 2′-phosphate of NADPH for a large part dictates that structurally related BVMOs prefer NADPH binding.

Binding of NADP+ in CHMOclosed (a) and CHMOopen (b) structures. NADP+ is shown in purple
Inspired by sequence alignment of PAMO with MekA and CHMO, we have analyzed several H220 mutants. Of all the created mutants, the only mutant that showed an improved affinity for NADH (K M, NADH) while at the same time it displayed a decreased affinity for NADPH (K M, NADPH) was H220Q. This mutation resulted in a 3-fold increase in the catalytic efficiency with NADH. This effect is in line with the observation that MekA, exhibiting a relaxed coenzyme specificity, also contains a glutamine at this position. Other mutations, e.g., H220A and H220E, did not bring any significant positive effect on the NADH recognition. In fact, the H220E and H220D mutants had a significantly impaired recognition of NADPH, which may be caused by electrostatic repulsion of the 2′-phosphate (see Fig. 1a). The glutamine and asparagine mutations appear to create a more favorable interaction between the residue at the position 220 and the adenine ribose moiety.
Another candidate that could play a role in the coenzyme recognition is K336. This lysine, preceding the catalytically crucial R337, is highly conserved among BVMOs. Based on the inspection of the model of PAMO with NADPH, it was foreseen that this lysine may interact with the 2′-phosphate group. Replacing this residue only decreased the catalytic efficiency of the respective mutants for both coenzymes. The substitutions had a relatively large effect on the K M, NADPH, which indicates that K336 indeed is required for the effective recognition of NADPH. Combining the H220Q and K336H mutations, as present in MekA, did not improve the performance with NADH. Despite its conservation, K336 is not essential for catalysis since mutations at this position did not destroy the activity. Interestingly, this lysine appears to be important for shaping the active site as the enantioselective behavior of PAMO was found to be improved in the K336 mutant. This was found for a sulfoxidation reaction (wt-PAMO, 44% ee; K336N PAMO, 64% ee) and a Baeyer–Villiger oxidation (wt-PAMO, E = 55; K336N PAMO, E = 96). This finding can be explained by the fact that the residue next to K336 is R337, which was proposed to play a role in substrate positioning in the active site (Torres Pazmiño et al. 2008). Shortening a loop that is located next to K336-R337 has previously been also shown to affect the substrate specificity (Bocola et al. 2005). It can be rationalized that mutation at position 336 also influences the positioning of substrate. Thereby, this mutation promotes reaction/production of one enantiomer.
PAMO seems to have evolved into a truly NADPH-specific BVMO. Some improvement in the NADH recognition can be achieved by introducing single mutations, but the catalytic performance of the engineered mutants with NADH is still far from that of wild-type PAMO with NADPH. The resilience of PAMO and other BVMOs to be engineered into NADH-dependent enzymes may relate to the complex catalytic mechanism and the structural role of the coenzyme in the active site. It has been shown that the nicotinamide coenzyme in BVMOs and related monooxygenases has a moonlighting role in catalysis (Mirza et al. 2009; Alfieri et al. 2008). Except for providing the necessary reducing equivalents, the coenzyme is bound throughout the catalytic cycle to assist, as a surrogate active site residue, in catalysis. This is reflected by the fact that, only in the presence of NADP+ in the active site, PAMO catalyzes highly enantioselective oxidations. PAMO-mediated catalysis using an artificial reductant merely resulted in poor conversions with no significant enantioselectivity (de Gonzalo et al. 2005). The complex role of NADP+ is also confirmed by the structural data obtained with CHMO Rhodo , which has revealed a delicate sliding of the coenzyme in the binding pocket during catalysis (Mirza et al. 2009). This implies that the NADPH-binding pocket should accommodate alternate binding modes of the coenzyme. The rearrangement of the NADP+ binding pocket is nicely exemplified by the alternate positioning of the lysine in CHMO Rhodo that is analogous to K336 in PAMO. This lysine moves by 7 Å upon a subtle movement of the coenzyme (Fig. 2). Altogether, the available data hint to a role of NADPH in BVMO catalysis that cannot easily be replaced by NADH. It is doubtful whether a more comprehensive enzyme redesign approach, e.g., targeting more residues, will easily yield a BVMO that is efficient with NADH. Nevertheless, some of the described mutations may provide a good starting point for such effort, and the identification of the role of K336 in tuning enantioselectivity may be valuable in generating mutants with altered (enantio)selective properties.
References
Aksu S, Arends IWCE, Hollman F (2009) A new regeneration system for oxidized nicotinamide cofactors. Adv Synth Catal 351:1211–1216
Alfieri A, Malito E, Orru R, Fraaije MW, Mattevi A (2008) Revealing the moonlighting role of NADP in the structure of a flavin-containing monooxygenase. Proc Natl Acad Sci USA 105:6572–6577
Bocola M, Schulz F, Leca F, Vogel A, Fraaije MW, Reetz MT (2005) Converting phenylacetone monooxygenase into phenylcyclohexanone monooxygenase by rational design: towards practical Baeyer–Villiger monooxygenases. Adv Synth Catal 347:979–986
Bubner P, Klimacek M, Nidetzky B (2008) Structure-guided engineering of the coenzyme specificity of Pseudomonas fluorescens mannitol 2-dehydrogenase to enable efficient utilization of NAD(H) and NADP(H). FEBS Lett 582:233–237
Carugo O, Argos P (1997) NADP-dependent enzymes. I: conserved stereochemistry of cofactor binding. Proteins 28:10–28
de Gonzalo G, Ottolina G, Carrea G, Fraaije MW (2005) [Cp*Rh(bpy)(H2O)]2+ as a coenzyme substitute in enzymatic oxidations catalyzed by Baeyer–Villiger monooxygenases. Chem Commun 29:3724–3726
de Gonzalo G, Ottolina G, Zambianchi F, Fraaije MW, Carrea G (2006) Biocatalytic properties of Baeyer–Villiger monooxygenases in aqueous–organic media. J Mol Catal B Enzym 39:91–97
Eckstein M, Dauβman T, Kragl U (2004) Recent developments in the NAD(P)H regeneration for enzymatic reductions in one and two phase systems. Biocat Biotrans 22:89–96
Fraaije MW, Wu J, Heuts DP, van Hellemond EW, Spelberg JH, Janssen DB (2005) Discovery of a thermostable Baeyer–Villiger monooxygenase by genome mining. Appl Microbiol Biotechnol 66:393–400
Kamerbeek NM, Janssen DB, van Berkel WJH, Fraaije MW (2003) Baeyer–Villiger monooxygenases, an emerging family of flavin-dependent monooxygenases. Adv Synth Catal 345:667–678
Kamerbeek NM, Fraaije MW, Janssen DB (2004) Identifying determinants of NADPH specificity in Baeyer–Villiger monooxygenases. Eur J Biochem 271:2107–2116
Kayser MM (2009) “Designer reagents” recombinant microorganisms: a new powerful tool for organic synthesis. Tetrahedron 65:947–974
Lee SH, Nam DH, Park CB (2009) Screening xanthene dyes for visible light-driven nicotinamide adenine dinucleotide regeneration and photoenzymatic synthesis. Adv Synth Catal 351:2589–2594
Malito E, Alfieri A, Fraaije MW, Mattevi A (2004) Crystal structure of a Baeyer–Villiger monooxygenase. Proc Natl Acad Sci USA 101:13157–13162
Martínez-Júlvez M, Tejero J, Peregrina JR, Nogués I, Frago S, Gómez-Moreno C, Medina M (2005) Towards a new interaction enzyme:coenzyme. Biophys Chem 115:219–224
Mihovilovic MD (2006) Enzyme mediated Baeyer–Villiger oxidations. Curr Org Chem 10:1265–1284
Mirza IA, Yachnin BJ, Wang S, Grosse S, Bergeron H, Imura A, Iwaki H, Hasegawa Y, Lau PC, Berghuis AM (2009) Crystal structures of cyclohexanone monooxygenase reveal complex domain movements and a sliding cofactor. J Am Chem Soc 131:8848–8854
Rioz-Martínez A, Bisogno FR, Rodríguez C, de Gonzalo G, Lavandera I, Torres Pazmiño DE, Fraaije MW, Gotor V (2010) Biocatalysed concurrent production of enantioenriched compounds through parallel interconnected kinetic asymmetric transformations. Org Biomol Chem 8:1431–1437
Rodríguez C, de Gonzalo G, Torres Pazmiño DE, Fraaije MW, Gotor V (2008) Selective Baeyer–Villiger oxidation of racemic ketones in aqueous–organic media catalyzed by phenylacetone monooxygenase. Tetrahedron: Asymmetry 19:197–203
Rodríguez C, de Gonzalo G, Torres Pazmiño DE, Fraaije MW, Gotor V (2009) Baeyer–Villiger monooxygenase-catalyzed kinetic resolution of racemic alpha-alkyl benzyl ketones: enzymatic synthesis of alpha-alkyl benzylketones and alpha-alkyl benzylesters. Tetrahedron: Asymmetry 20:1168–1173
Ruinatscha R, Hoellrigl V, Otto K, Schmid A (2006) Productivity of selective electroenzymatic reduction and oxidation reactions: theoretical and practical considerations. Adv Synth Catal 348:2015–2026
Siu E, Won K, Park CB (2007) Electrochemical regeneration of NADH using conductive vanadia-silica xerogels. Biotechnol Prog 23:293–296
Straathof AJJ, Jongejan JA (1997) The enantiomeric ratio: origin, determination and prediction. Enzyme Microb Technol 21:559–571
Taglieber A, Schulz F, Hollmann F, Rusek M, Reetz MT (2008) Light-driven biocatalytic oxidation and reduction reactions: scope and limitations. ChemBioChem 9:565–572
Torres Pazmiño DE, Snajdrova R, Rial DV, Mihovilovic MD, Fraaije MW (2007) Altering the substrate specificity and enantioselectivity of phenylacetone monooxygenase by structure-inspired enzyme redesign. Adv Synth Catal 349:1361–1368
Torres Pazmiño DE, Baas BJ, Janssen DB, Fraaije MW (2008) Kinetic mechanism of phenylacetone monooxygenase from Thermobifida fusca. Biochemistry 47:4082–4093
van den Heuvel RH, Tahallah N, Kamerbeek NM, Fraaije MW, van Berkel WJ, Janssen DB, Heck AJ (2005) Coenzyme binding during catalysis is beneficial for the stability of 4-hydroxyacetophenone monooxygenase. J Biol Chem 280:32115–32121
van der Donk WA, Zhao H (2003) Recent developments in pyridine nucleotide regeneration. Curr Opin Biotechnol 14:421–426
Völker A, Kirschner A, Bornscheuer UT, Altenbuchner J (2008) Functional expression, purification, and characterization of the recombinant Baeyer–Villiger monooxygenase MekA from Pseudomonas veronii MEK700. Appl Microbiol Biotechnol 77:1251–1260
Wu JT, Wu LH, Knight JA (1986) Stability of NADPH: effect of various factors on the kinetics of degradation. Clin Chem 32:314–319
Zeng QK, Du HL, Wang JF, Wei DQ, Wang XN, Li YX, Lin Y (2009) Reversal of coenzyme specificity and improvement of catalytic efficiency of Pichia stipitis xylose reductase by rational site-directed mutagenesis. Biotechnol Lett 31:1025–1029
Acknowledgements
H.M. Dudek, D.E. Torres Pazmiño, G. de Gonzalo, and M.W. Fraaije received financial support from the EU-FP7 OXYGREEN project. Ana Rioz-Martínez (FPU Program) thanks the Spanish Ministerio de Ciencia e Innovación (MICINN) for her predoctoral fellowship which is financed by the European Social Fond.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Dudek, H.M., Torres Pazmiño, D.E., Rodríguez, C. et al. Investigating the coenzyme specificity of phenylacetone monooxygenase from Thermobifida fusca . Appl Microbiol Biotechnol 88, 1135–1143 (2010). https://doi.org/10.1007/s00253-010-2769-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2769-y