
Abstract
The CD1 and MHC systems are specialized for lipid and peptide display, respectively. Here, we review evidence showing how cellular CD1a, CD1b, CD1c, and CD1d proteins capture and display many cellular lipids to T cell receptors (TCRs). Increasing evidence shows that CD1-reactive T cells operate outside two classical immunogenetic concepts derived from the MHC paradigm. First, because CD1 proteins are non-polymorphic in human populations, T cell responses are not restricted to the donor’s genetic background. Second, the simplified population genetics of CD1 antigen-presenting molecules can lead to simplified patterns of TCR usage. As contrasted with donor-restricted patterns of MHC-TCR interaction, the donor-unrestricted nature of CD1-TCR interactions raises the prospect that lipid agonists and antagonists of T cells could be developed.
Similar content being viewed by others

Donor-unrestricted T cells
The major histocompatibility complex (MHC) contains polymorphic genes that encode antigen presenting proteins (Neefjes et al. 2011). MHC I and II proteins fold in three dimensions to form antigen-binding grooves that capture processed peptides and display them to αβ T cells. Due to extremely high polymorphism in MHC genes, the full collection of peptide antigen display platforms expressed by all members of the human species is extraordinarily diverse (Suri et al. 2006; Rossjohn et al. 2015). MHC polymorphism likely represents an evolved function that confers advantages by allowing T cells to respond to nearly any microbial pathogen, including viruses that the host has never previously encountered.
T cell receptor (TCR) expression within genetically identical populations, such as inbred mouse strains, can show simplified and recognizable patterns (Nikolich-Zugich et al. 2004). However, in considering outbred human populations, the interdonor variation of MHC proteins creates extreme complexities in the patterns of peptide antigens displayed on antigen-presenting cells (APCs) (Trowsdale and Knight 2013). This interdonor complexity makes it difficult to define immunodominant antigens for any given type of infection or autoimmune disease in human populations. Also, in considering the responding TCR repertoires, the variable (V) and joining (J) genes present in the favored TCRs used by any given patient, even in response to the same pathogen, can differ substantially (Nikolich-Zugich et al. 2004; Rossjohn et al. 2015). Likewise for human diseases in which T cells are known to play a central role, stereotyped patterns of TCRs on expanded populations of T cells are difficult to identify at the population level. Because most research on αβ T cells focuses on their MHC-encoded targets, these complex patterns of TCR expression and donor-restricted antigen recognition are sometimes viewed as general properties of αβ T cells. However, αβ T cells also recognize CD1, MR1, HLA-E, and other proteins, which are encoded by non-polymorphic genes (Sullivan et al. 2006; Hansen et al. 2007; Van Rhijn et al. 2015a). As contrasted with Doherty and Zinkernagel’s groundbreaking experiments that defined donor restriction of MHC-presented viral antigens (Zinkernagel and Doherty 1974), T cell response to non-polymorphic antigen-presenting molecules is not restricted to the genetic background of the donor. These basic observations lead to the immunogenetic concept of donor-unrestricted T cells. The most widely known examples are NKT cells that respond to CD1d (Bendelac et al. 1995; Kawano et al. 1997), and mucosal-associated invariant T (MAIT) cells that recognize MR1 (Porcelli et al. 1993; Treiner et al. 2003; Huang et al. 2005).
Beyond public TCRs
TCRs are considered “public” when several donors share a certain MHC allele and are infected with the same kind of pathogen that expresses an immunodominant antigen (Venturi et al. 2008). Outside of superantigen-reactive T cells, we know of no pattern of MHC-restricted TCR conservation that is expressed among the majority of humans studied. Thus, the concept of MHC-restricted TCRs is invoked in the particular situation of shared antigen exposure and matched MHC genes, which is not the usual setting of T cell activation in disease. In contrast, the V and J rearrangements that define NKT and MAIT TCRα chains are conserved broadly across most humans or mice, based on their recognition of a universally expressed antigen-presenting molecule (Porcelli et al. 1993; Tilloy et al. 1999; Huang et al. 2005; Van Rhijn et al. 2015a; Van Rhijn and Moody 2015; Boudinot et al. 2016). These invariant TCRs are much more public than any MHC-restricted TCR and can be thought of as universal in a species. Here, we review the diversity of cellular ligands captured and presented by human CD1 proteins to human TCRs. Looking beyond CD1d and NKT cells, this review considers the question of whether human CD1a, CD1b, and CD1c also present lipid ligands to donor-unrestricted T cells and whether the responding T cells show interdonor TCR conservation that is public or universal (Cox et al. 2009; Huang et al. 2011; de Jong et al. 2014).
Human CD1 gene polymorphism
CD1 proteins are often described non-polymorphic antigen-presenting molecules. The term ‘non-polymorphic’ is not literally true, as limited single-nucleotide polymorphisms (SNPs) in the α1 domain (Aruffo and Seed 1989; Zajonc et al. 2005a) and non-coding regions (Seshadri et al. 2014) of human CD1a genes are known. Also, some donor-specific differences have also been deduced from non-synonymous mutations from the exon 2 of CD1a and CD1d genes (Han et al. 1999). Therefore, inter-individual differences in CD1 expression and function could, in the future, be found to influence immunologic diseases and host response. However, because CD1 SNPs are uncommon in human populations, human CD1 proteins can be considered functionally monomorphic for most key aspects of their function. This immunogenetic feature of the CD1 system represents a major distinction from the MHC system. MHC polymorphism controls rejection of transplanted solid organs and bone marrow and donor-specific patterns of antigen response (Ayala Garcia et al. 2012). MHC polymorphism generates highly diverse TCR usage by differing donors in response to a single pathogen or protein antigen (Nikolich-Zugich et al. 2004). Therefore, a major question for non-polymorphic proteins that mediate T cell activation, including CD1, MR1, and HLA-E, is whether the lack of polymorphism actually translates into simplified patterns of TCR usage and antigen response among genetically diverse humans. For CD1d and NKT cells, highly similar TRAV10+ TCRs are polyclonally expanded in nearly all humans (Bendelac et al. 1995; Kawano et al. 1997; Rossjohn et al. 2012; Zajonc and Girardi 2015), but the TCR patterns in the larger repertoire selected by CD1a, CD1b, and CD1c are just beginning to be explored (de Jong et al. 2010; de Lalla et al. 2011; Kasmar et al. 2011; Kasmar et al. 2013; Ly et al. 2013; Van Rhijn et al. 2013; Van Rhijn et al. 2014).
TCR co-recognition of CD1-lipid
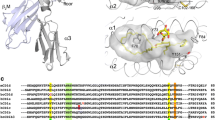
Like MHC I, CD1 heavy chains bind β2-microglobulin (β2M) and fold to form two anti-parallel α-helices, which create side walls located above a β-sheet floor (Zeng et al. 1997). The hollow groove is accessed by one or more narrow portals so that lipids are seated such that they are positioned both within and outside the outer surface of CD1 (Fig. 1). The simplest and oldest model of lipid antigen recognition can be described as TCR co-recognition of CD1-lipid complexes. Here, αβ TCRs contact one hybrid surface comprised of the outer walls of the α1- and α2-helices as well as elements of the bound lipid that protrude between the helices to lie on the outside of CD1 proteins. This mode of antigen recognition (co-recognition) was long predicted and then ruled in by a ternary crystal structure of CD1d-α-galactosyl ceramide-TCR (Borg et al. 2007) and several ternary CD1d-lipid-TCRs that followed (Wang et al. 2010; Mallevaey et al. 2011; Pellicci et al. 2011).

T cell receptor binding to MHC I-peptide, CD1a-lipid, and CD1d-lipid complexes. a A TCR binding to HLA-A2 (Garboczi et al. 1996) takes a typical footprint located near the center of the MHC I platform. b The CD1a autoreactive TCR BK6 binds on the left side of CD1a protein, where it contacts the A’ roof of CD1a but not the bound lipid ligand (Birkinshaw et al. 2015). c An invariant NKT TCR takes an extreme rightward approach to CD1d, where the TCR α chain contacts the protruding galactose and the TCR β chain extends past the CD1d platform (Borg et al. 2007)
In its most basic elements, this ternary interaction can be compared to the classical mechanism by which TCRs contact peptide-MHC (Fig. 1). In TCR co-recognition, both the antigen and the antigen-presenting molecule contact the TCR and control T cell response. While most studies of CD1 have emphasized αβ TCR response, the earliest study of human CD1 proteins in vitro demonstrated γδ T cell recognition of CD1c by an unknown mechanism (Porcelli et al. 1989). Recent reports have demonstrated that human polyclonal Vδ1+ γδ T cells can contact CD1d or CD1c using their TCRs to co-recognize CD1 in complex with α-galactosyl ceramide, sulfatide, and phosphopmycoketide antigens (Bai et al. 2012; Luoma et al. 2013; Uldrich et al. 2013; Roy et al. 2016). The following sections detail the basis of CD1-lipid and CD1-lipid TCR interactions, defining key contrasts with the peptide-MHC model and setting the stage for understanding important exceptions to the general co-recognition model that have arisen recently.
Promiscuous lipid anchoring versus precise peptide anchoring
Certain basic differences in the biochemical nature of peptide and amphipathic lipids lead to distinct modes of antigen capture and display. In MHC I, non-amer peptides usually represent the optimal fit to the width of the groove (Hansen et al. 2010; Blum et al. 2013), whereas the ends of the MHC II groove are not blocked and so bind longer peptides (>14 mers) that extend beyond the platform as “ragged ends” (Mohan and Unanue 2012; Blum et al. 2013). Peptides are anchored by charge-charge and hydrogen-bonding interactions that result from precisely positioned residues on the walls and floor of MHC ligand-binding grooves, which interact with spatially defined residues in the peptide sequence. These highly specific and position-dependent interactions have allowed the definition of anchor residues from antigenic peptides and their binding pockets from MHC-encoded antigen-presenting molecules (Madden et al. 1991; Miley et al. 2004; Jones et al. 2006). In this way, TCRs bind to and discriminate the amino acid sequences so they are directly recognizing products of genetic codes, which are subject to mutation, selection, and rapid evolution.
In contrast, the inner surface of the CD1 cavity is lined with non-polar residues that mediate relatively non-specific hydrophobic interactions with the lipid tails of bound antigens (Zeng et al. 1997; Rossjohn et al. 2012; Zajonc and Girardi 2015). The aliphatic hydrocarbons present in the alkyl and polyketide tails of antigens are comprised of repeating methylene units (Moody et al. 2005; Van Rhijn et al. 2015a). Thus, hydrophobic surfaces in CD1 can interact similar to the proximal, center, or distal ends of alkyl chains, so there is no equivalent of residue-specific motifs or anchor motifs as discovered for MHC molecules (Stern and Wiley 1994; Hansen et al. 2007).
This more promiscuous, position-independent mode of binding results in more diverse positioning of alkyl chains within CD1 proteins. For example, the same kind of antigen can take a clockwise or counterclockwise orientation within the toroidal A’ pocket of CD1d (Wu et al. 2006; Li et al. 2010; Birkholz et al. 2015). Lipid ligands show flexible lipid positioning and association of multiple lipids within one CD1 molecule, especially for CD1b, which has an unusually large cavity with four interconnected pockets that has been described as a “maze for alkyl chains” (Gadola et al. 2002) (Fig. 2). Although the aliphatic hydrocarbon chains are extremely flexible, clefts and individual pockets within CD1 proteins place an upper limit on the size of lipid antigens as reviewed previously (Ly and Moody 2014). Lipids are the products of enzymes rather than genes so are not subject to rapid change by mutation and instead change slowly over evolutionary time.

Human CD1 clefts are larger than MHC grooves. Cutaway views of human CD1a (Zajonc et al. 2003), CD1b (Gadola et al. 2002), CD1c (Mansour et al. 2016), and CD1d (Zajonc et al. 2005b) structures are shown in comparison to the transparent structure of HLA-A2 protein (Garboczi et al. 1996). The depth of ligand-binding grooves (black dashed line) is represented by a measurement of distance between atoms on roofs and floors, and the calculated volumes of ligand-binding clefts are shown in blue
Deep CD1 lipid-binding clefts
A second basic difference between MHC and CD1 antigen-presenting molecules is the depth of the groove and the extent to which antigens penetrate to the interior of the CD1 protein (Fig. 2). For example, the peptide-binding grooves in MHC proteins are typically 4–6 Å deep, as measured vertically from the top of the α-helices to the β-sheet floor. Thus, for HLA-A2-TAX and H-2Kb-dEV8 complexes, TAX and dEV8 peptides are seated in a position that is largely atop the MHC protein (Garboczi et al. 1996; Garcia et al. 1996). As first noted by Wilson and colleagues, the broken α2 helix and other features cause the mouse CD1d α-helices to ride higher above the β-sheet floor of CD1, creating a deep cleft (Zeng et al. 1997; Moody et al. 2005). The larger vertical depth of CD1 clefts was subsequently found among all four of the human CD1 antigen-presenting molecules (Gadola et al. 2002; Zajonc et al. 2005a; Scharf et al. 2010). Depending on the CD1 isoforms and positions of measurement, the depth of the groove ranges up to 12 or even 19 Å (Fig. 2). Thus, CD1 clefts are deeper and therefore more spacious (1280–2200 Å3) than MHC grooves.
CD1a, CD1c, and CD1d clefts are somewhat similar to the interior volume (1280–1780 Å3) and are defined as having two pockets, A’ and F’, which correspond in position to the A and F pockets of MHC I (Zeng et al. 1997; Moody et al. 2005; Rossjohn et al. 2012; Zajonc and Girardi 2015). In contrast, CD1b is much larger with an interior volume approximating 2200 Å3. CD1b has four defined pockets, A’, F’, C’, and the T’ pocket, which is named for its “tunneling” function at the bottom of the CD1b groove (Fig. 2). In summary, peptides sit between the α-helices of MHC I and II; lipid antigens reside more substantially within the globular head of CD1 proteins.
The binding clefts present in the four types of human CD1 proteins differ in their volume and architecture (Fig. 2). Detailed reviews of CD1 cleft architecture have been published recently (Brennan et al. 2013; Ly and Moody 2014; Salio and Cerundolo 2015; Van Rhijn et al. 2015a), so this review highlights only key distinguishing features of each CD1 isoform. Mouse and human CD1d have similar structures. CD1d provides the archetype in which the A’ and F’ pockets sit side by side below the raised position of the bent α2 helix (Fig. 1c).
The most obviously distinct cleft is that present in CD1b, which is notable for its large size and division into four (rather than two) named pockets. The A’ pocket is connected with F’ pocket through C’ pocket and the T’ tunnel. The highly interconnected nature of the four pockets led to its description as a maze for alkyl chains (Gadola et al. 2002). The ATF superchannel, which is unique to CD1b, allows capture of the very long-chain (∼C72–C54) mycolic acids, which wind their way through the channel (Batuwangala et al. 2004). In contrast, the F’ pocket of CD1a is connected only to the A’ pocket, which unlike other CD1 proteins, abruptly terminates rather than encircling the A’ pole to form a complete toroid. Thus, the CD1a cleft is shaped like a J-shaped tube with one entrance and no secondary exit to the outer surface of the protein. Because there is only one path through the interior of the CD1a protein, it has been proposed to act as a “molecular ruler” to select alkyl chains of a defined length (Zajonc et al. 2003). CD1c is notable for its open, accessible, and flexible architecture. Whereas all CD1 proteins have an entry point over the F’ pocket, ligands can enter the CD1c cavity at any of several points, known as the D’, E’, and F’ portals (Scharf et al. 2010; Mansour et al. 2016). Upon binding to cholesteryl lipid ligands, the CD1c protein undergoes a marked overall conformation change that has been compared to a venus fly trap swallowing its prey (Mansour et al. 2016).
Narrow portals versus wide grooves
A third basic difference between MHC and CD1 cavities is the extent to which the interior of the protein communicates with the outer surface, where TCR interactions occur. MHC cavities are known as grooves because they are long, narrow, and open at the top, allowing peptides to be exposed for recognition across most of the width of the protein (Figs. 2 and 3). The openings in CD1 proteins that allow lipids to protrude are often called grooves, but considering the actual physical shape, “groove” is a misnomer. Grooves are usually understood as being are longer than they are wide, and they are open at the top. The CD1 clefts located between the α-helices are neither long nor fully open at the top. Instead, CD1 proteins are partially closed, creating small, rounded openings which are called portals.

CD1 antigen display platforms are laterally asymmetric. Portals in human CD1a (Zajonc et al. 2003), CD1b (Gadola et al. 2002), CD1c (Mansour et al. 2016), and CD1d (Zajonc et al. 2005b) are located on the right side of CD1 antigen display platform. The A’ roofs are located at the left sides of the CD1 platforms. This organization of the display platform creates a situation in which approaching TCRs encounter CD1 surfaces on the left and protruding antigen on the right. In contrast, classical MHC molecules like HLA-A2 protein (Garboczi et al. 1996) bind peptides in a laterally symmetric way so that the antigen is accessible on both sides of the midline
The A’ roof
The partially closed nature of the CD1 platform results from the presence of a structure called the A’ roof, which is so named because it protects the contents of the interior of the A’ pocket from directly contacting the outer surface of CD1 proteins (Fig. 3). This roof structure is formed by amino acids on the α1-helices, which reach across to residues on the α2 helix to form interdomain tethers. For CD1b, experiments suggest that these tethers can be interrupted through the effects of low pH, which protonates acidic residues, thereby releasing charge-charge interactions with basic residues on the contralateral helix (Relloso et al. 2008). This release of interdomain tethers is thought to reduce steric hindrance for ligands entering the groove, because tether mutations allow CD1b binding of larger lipids, increased rates of lipid loading and increased rates of lipid unloading (Relloso et al. 2008). Thus, unlike the sides of the clefts that form from rigid secondary structural elements (the α1- and α2-helices), the A’ roof is a tenuous structure with a regulatory function. Highlighting the weak structural nature of the A’ roof, binary crystal structures of CD1a-sphingomyelin complexes show that sphingomyelin can penetrate the A’ roof by disrupting a triad of residues that form interdomain interactions (Birkinshaw et al. 2015). The A’ roof is present in CD1a, CD1b, CD1c, and CD1d, and it has no equivalent in MHC I or MHC II structures. Thus, the A’ roof can be considered a distinct architectural feature that defines CD1 clefts as being distinct from other antigen-presenting molecules (Fig. 3) (Gadola et al. 2002; Zajonc et al. 2003; Zajonc et al. 2005b; Scharf et al. 2010; Mansour et al. 2016).
TCR contact with hydrophilic head groups
Most CD1 ligands are amphipathic lipids that carry polar or charged head groups comprised of one or more carbohydrate units, a peptide, a sulfate ester, or a phosphate ester. Typically, the polar and rigid head groups are not fully inserted into the CD1 cavity. Instead, amphipathic lipids are positioned with their fatty acyl, sphingosine, polyketide, or cholesteryl moieties residing within the CD1 pockets, allowing the hydrophilic head groups to protrude upwards toward the surface of CD1. More specifically, the lipid anchors typically transition to the hydrophilic head groups at the top of the F’ pocket, near a structure known as the F’ portal, which connects the interior of the groove to the outer surface of CD1 (Figs. 2 and 3). In most cases, the hydrophilic head groups extend to the outer surface of CD1 and contribute to the TCR contact surface. For example, extensive studies of CD1d-α-galactosyl ceramide-TCR, the α-anomerically linked galactose unit protrudes through the F’ portal and lies near the right edge of the CD1d platform, where it is positioned underneath the binding footprint of the NKT TCR (Borg et al. 2007).
In agreement with the co-recognition model, early work on glycolipids presented by CD1b and CD1d showed marked specificity for the protruding glucose or galactose head groups on glucose monomycolate or α-hexosyl ceramides (Kawano et al. 1997; Moody et al. 1997; Borg et al. 2007). In contrast, T cells could recognize lipid analogs with changes in the length and structure of the lipid tails. Thus, the most basic model of amphipathic lipid display emphasizes a CD1 anchoring function of lipid tails, leading to presentation of carbohydrate, peptide, phosphate, or sulfate moieties, which interact in a highly specific manner with TCRs (Rossjohn et al. 2012; Zajonc and Girardi 2015). This classical co-recognition model now dominates thinking about the mechanism of T cell activation by CD1-lipid (Fig. 1) (Cox et al. 2009; Huang et al. 2011; de Jong et al. 2014). The following sections emphasize that certain CD1-lipid complexes diverge from the typical TCR co-recognition mechanism. In particular, recent studies of T cell autoreactive response to CD1a proteins or self lipids have provided evidence for dominant negative control of T cell response by certain non-permissive lipid ligands, as well as T cell activation by mechanisms in which the lipid appears to be ignored (de Jong et al. 2014; Birkinshaw et al. 2015). Combining these unexpected structural and functional findings, alternate models of CD1 recognition by TCRs are emerging.
Lateral asymmetry of CD1 antigen display platforms
The A’ roof creates a kind of lateral asymmetry in the CD1 antigen display mechanism, which is readily apparent from inspection of the α-helical surface of CD1 (Fig. 3, red and white). By convention, CD1 proteins are shown with the A’ roof on the left and the F’ portal on the right. Since antigens emerge from the F’ portal, their origin on the platform is shifted to the right of center. Unlike the broad span of peptides across the lateral dimension of MHC proteins, the head groups of many CD1 antigens are more vertically oriented and so occupy a smaller area of the platform. Thus, the final position of head groups, which emerge from the right side of the platform, is determined by whether they lean right or left as they exit the portal. Thus, considering all binary CD1-lipid structures solved to date, the antigen footprints on the CD1 surface skew rightward (Rossjohn et al. 2015; Van Rhijn et al. 2015a). The left side of the platform is formed by the A’ roof, so the exposed surface is mostly CD1 itself, without overlying ligand. The situation in which the CD1 antigen display platform is closed on the left (Fig. 3, red) and open to antigen on the right (Fig. 3, white) raises a new hypothesis. If TCRs can bind using left- or right-sided footprints, then left-sided TCRs would more extensively contact the unliganded surface of CD1, and right-sided footprints would more extensively contact antigen (Fig. 1b, c).
Certain aspects of the left-right binding model have been experimentally ruled in. All human CD1 platforms are asymmetric (Fig. 3), and not all CD1 reactive TCRs bind near the center of the platform. For example, one invariant NKT TCR binds near the right edge of the CD1 platform, such that the TCR α chain mostly contacts the protruding galactose (Borg et al. 2007). The TCR β chain takes an extreme rightward position (Fig. 1c). The first TCR footprint on CD1a, shown using the clone BK6, demonstrates a left-shifted footprint such that the TCR sits down on the A’ roof of CD1a (Fig. 1b). In fact, the BK6 TCR fails to contact the lipid ligand or ligands in the cleft (Birkinshaw et al. 2015). Thus, despite the relatively small number of TCR footprints on CD1 solved to date, extreme right and left skewing is already demonstrated to occur. This left-right shift model emphasizes that left-skewing TCRs may be more autoreactive, but it does not seek to explain all self or non-self interactions in terms of lateral translation of TCRs. Near the middle of the platform, TCRs and antigens interact in different ways. For example, CD1d-reactive αβ and γδ TCRs bind nearer the middle of CD1d, contacting both CD1 protein and bound lipid (Mallevaey et al. 2011; Rossjohn et al. 2012; Luoma et al. 2013; Uldrich et al. 2013).
T cell autoreactivity to CD1
Attention usually focuses on the subset of foreign lipid ligands, which are considered antigens because they activate T cells through direct contact with TCRs. Regulatory functions of CD1-restricted T cells in inflammatory diseases and cancers supports the existence of self-lipid antigens (Godfrey and Kronenberg 2004; Dellabona et al. 2015). CD1 autoreactivity was identified in the first report of T cell response to CD1 proteins, as shown by the CD1a-dependent lysis of target cells by the cytotoxic αβ T cell line BK6 (Porcelli et al. 1989; Porcelli et al. 1993). The mechanisms of self lipid presentation have been clearly observed for CD1a (de Jong et al. 2010; de Lalla et al. 2011), CD1b (Van Rhijn et al. 2015b), and CD1d (Brossay et al. 1998; Mattner et al. 2005; Brigl et al. 2006; Mallevaey et al. 2011). Several studies suggest that CD1a autoreactivity is particularly common as compared with autoreactive T cells recognizing CD1b, CD1c, or CD1d (de Jong et al. 2010; de Lalla et al. 2011; de Jong et al. 2014). Although few CD1b-autoreactive T cells have been seen in patient studies, recent studies have identified CD1b autoreactivity in transgenic mice and humans, providing evidence that phosphatidylgycerol is a self antigen (Shamshiev et al. 1999; De Libero et al. 2005; Van Rhijn et al. 2015b). A potentially important CD1c-presented self lipid antigen is methyl-lysophosphatidic acid, which is presented by CD1c to activate T cells that kill acute leukemia cells (Lepore et al. 2014). Exogenous α-galactosylceramide was described from marine sponge and so was considered to be a foreign antigen (Kawano et al. 1997). However, α-glycosylceramides were also recently identified from mammalian cells (Kain et al. 2014).
In vitro, CD1 autoreactivity can be demonstrated when T cell activation is dependent on the presence of cellular or plate bound CD1 protein but occurs in the absence of an added foreign antigen (de Jong et al. 2014). Autoreactivity is often considered to be explained by TCR recognition of chemically defined self antigens. That is, the TCR is thought to contact bound lipid antigen and specifically discriminate its structure. However, in theory, autoreactivity could also be explained by TCR contact with the antigen-presenting molecule itself, using a mechanism in which no bound ligand is contacted. Such a model is infrequently invoked for peptide-MHC in autologous systems, perhaps because bound peptides broadly occupy the antigen display platform and there is little room on the surface for TCRs to bind in a manner that does not contact peptide. However, the A’ roof structure, which is present among all four CD1 antigen-presenting molecules and absent in MHC I and II, could create a large antigen-free landing pad for CD1-reactive TCRs (Fig. 3).
An alternate model: absence of interference
Direct TCR recognition of the CD1 surface received indirect support from functional data showing that CD1a autoreactive T cell clones were activated by squalene, wax esters, and free fatty acids (de Jong et al. 2014). These T cell agonist lipids lack any hydrophilic head groups that usually comprise TCR epitopes. Instead, these lipids are relatively small and hydrophobic, and they were recognized in a cross-reactive fashion by T cell clones. These observations raised the hypothesis that such lipids might not directly contact the TCR but instead nest deeply within CD1a and do not interfere with direct TCR-CD1a contact. In this absence of interference model, the activating property of the lipid comes not from contacting the TCR but instead from not contacting the TCR. The lipid takes on the role of a permissive ligand, which displaces other non-permissive ligands that might block TCR contact with CD1a.
Structural proof for the absence of interference mechanism was obtained through the CD1a-lipid-TCR studies published last year (Birkinshaw et al. 2015). The BK6 TCR binds on the left side of the CD1a platform, over the A’ roof (Fig. 1b). In this mode of binding, the TCR exclusively contacts CD1a and fails to bind lysophosphatidylcholine or fatty acids sitting within in the cleft. Further, the purification of TCR-CD1a complexes demonstrated that many dozens of self lipids with varying mass and polarity can be bound in CD1-TCR complexes. This suggests that the TCR ignores the bound self lipid. However, the lipids might contribute in an indirect way to TCR activation even in the absence of TCR contact. Lipids can alter the surface of the CD1 protein by pushing outward on key residues or closing up loose ternary structures (Garcia-Alles et al. 2006; McCarthy et al. 2007; Garcia-Alles et al. 2011; Mansour et al. 2016).
The absence of interference model also relies on the existence of non-permissive lipids that block CD1-TCR contact. Mass spectrometry studies have identified lipids with large head groups, including globosides, gangliosides, and sphingomyelin, which bind to CD1a and CD1d, and block T cell activation (Cox et al. 2009; Rossjohn et al. 2012; de Jong et al. 2014; Birkinshaw et al. 2015). The large head groups could sterically inhibit the approach of TCRs to CD1. A second mechanism of T cell inhibition is suggested by the structure of CD1a sphingomyelin (Birkinshaw et al. 2015). Here, the choline head group pushes through the A’ roof of CD1a to alter a triad of tethering residues, directly demonstrating roof disruption by a non-permissive ligand. Overall, these alternative models emphasize direct contact of TCRs with CD1 itself, where stimulatory or inhibitory lipids play indirect roles in activation by taking positions below or to the side of the TCR-CD1 contact zone. As outlined below, the chemical identification of non-permissive ligands might offer an approach to selective inhibition of T cell responses in vivo.
Cellular control of CD1-lipid complex formation
CD1-presented antigens were discovered in bacteria, and most studies of T cell response emphasize a role of lipid antigen recognition in host defense (Brennan et al. 2013; Van Rhijn et al. 2015a). However, CD1 proteins also capture self lipid ligands as they transit the secretory and endosomal pathways of cells (Cox et al. 2009; Yuan et al. 2009; Huang et al. 2011; de Jong et al. 2014). A self lipid ligand eluted from mouse CD1d1 was identified as a glycosylphosphatidylinositol (GPI) and described as the “the” CD1d ligand (Joyce et al. 1998). Thus, early studies considered the possibility that membrane phospholipids related in structure to phosphatidylinositol dominate other lipid types in their occupancy of CD1 clefts. However, the CD1 system has no known lipid equivalent of the class II invariant chain-derived peptide (CLIP) that binds in the groove of MHC II proteins (Denzin and Cresswell 1995). Instead, most evidence now argues against the idea that one specialized lipid uniformly occupies the cleft of most CD1 proteins until exogenous antigens are encountered. For example, several mass spectrometry studies of natural ligands eluted from cellular CD1 proteins have demonstrated many classes of phospholipids, di- and triacylglycerols, glycolipids, glycophospholipids, sphingolipids, and terpene lipids (Cox et al. 2009; Yuan et al. 2009; Huang et al. 2011; Facciotti et al. 2012; Zeissig et al. 2012; de Jong et al. 2014). Further, recently developed quantitative lipidomics methods can reliably count the number of both named and unnamed lipids that are detected at equivalent mass and retention time in replicate elution experiments (Huang et al. 2011; de Jong et al. 2014). Quantitative methods indicate that at a minimum, many hundreds of distinct molecular species of self lipid are captured by CD1a, CD1b, CD1c, and CD1d proteins when expressed in human cells. Thus, the CD1 system is capable of broad survey and display of the cellular lipids.
Dynamic lipid ligand capture
Increasing evidence suggests that while CD1 proteins can promiscuously bind most types of cellular lipids, including those with straight chains or ringed structures (Van Rhijn et al. 2015b; Mansour et al. 2016), APCs have mechanisms that edit the spectrum of lipids display. For example, proteins that influence lipid entry into cells or access to the hydrophobic clefts of CD1 molecules control the efficiency of cellular antigen display. Extracellular lipid-binding proteins such as apolipoprotein B augment lipoprotein particle uptake by myeloid dendritic cells (van den Elzen et al. 2005; Moore and Tabas 2011). Cell surface lectins, including the mannose receptor (Prigozy et al. 1997), langerin (Hunger et al. 2004), and SigLecs (Kawasaki et al. 2014), influence the uptake of glycolipids and their delivery to the endosomal network. Intracellular lipid transfer proteins, including saposins and microsomal triglyceride transfer protein (Brozovic et al. 2004; Kang and Cresswell 2004; Winau et al. 2004; Zhou et al. 2004b; Dougan et al. 2005; Dougan et al. 2007b), promote the transfer of lipids from a bilayer or other aggregated states to lipid-protein complexes, which allow insertion of a single lipid into the cleft of CD1 proteins (Zeissig et al. 2010; Salio et al. 2013).
The loading and unloading of lipids into the cleft is also controlled by the effects of pH on CD1 structure. The pH of loading and unloading interactions is in turn controlled by the extent to which each CD1 protein traffics through the secretory pathway at neutral pH versus acidic compartments of the endosomal network. This complex topic has been separately reviewed (Sugita et al. 1999; Jayawardena-Wolf et al. 2001; Moody and Porcelli 2003; Brigl and Brenner 2004). In summary, CD1b, CD1c, and CD1d proteins have tyrosine-based motifs in their cytoplasmic tails, which bind adaptor protein (AP) complexes and promote trafficking of CD1 proteins to late endosomes and lysosomes (Jackman et al. 1998; Chiu et al. 2002; Sugita et al. 2002; Lawton et al. 2005). Human CD1a lacks the tyrosine motif and so resides predominantly at the cell surface and in early endosomal compartments, where it can directly capture many lipids at neutral pH (Sugita et al. 1999; Manolova et al. 2006). At the other extreme, human CD1b binds two AP complexes (AP-2, AP-3) and most prominently traffics to lysosomes (Jackman et al. 1998; Briken et al. 2002). CD1b cannot accept large lipids when present at the surface, but its trafficking to lysosomes promotes loading and unloading of small lipids, as well as the selective capture of lipids with very long alkyl chains (C80) (Moody et al. 2002). To a lesser extent, the low pH of lysosomes promotes lipid exchange and exogenous antigen capture by human CD1c and CD1d, which bind to AP-2 and localize somewhat specifically to LAMP1+ compartments (Briken et al. 2000; Sugita et al. 2000).
Low pH influences CD1 structure directly through several mechanisms, including anionic residues at positions 80 and 86 in CD1b that form interdomain tethers that connect across from α1- to α2-helices to enclose lipids within CD1b (Relloso et al. 2008). Acidic pH promotes protonation of the tethering residues, which likely releases the interdomain tethers, which is thought to dilate the entry portal to allow increased lipid access and release. A second mechanism favoring lipid turnover in lysosomes is the acid-mediated cleavage and activation of saposins, which bind and transfer membrane lipids into the groove of CD1 proteins (Kang and Cresswell 2004; Winau et al. 2004; Zhou et al. 2004b; Leon et al. 2012; Salio et al. 2013). Increased lipid binding and release from CD1d has also been observed in cells (Bai et al. 2009; Im et al. 2009). These molecular mechanisms suggest a model for CD1b, CD1c, and CD1d proteins in which newly transcribed CD1 proteins normally capture endogenous self lipids. After trafficking to the acidic compartments in the endocytic network, they become particularly receptive to exogenous lipids acquired by phagocytosis, fluid phase endocytosis, or receptor mediate capture. In contrast, CD1a resides mainly at the cell surface or early endosomes, where it captures lipids without a strong influence of acid-driven mechanisms (Moody and Porcelli 2003; Manolova et al. 2006; Cernadas et al. 2010).
CD1 and the problem of size
The MHC system uses a global and easily understood sizing mechanism to match peptide antigens to the volume of MHC grooves. For MHC I, the proteasome and other peptidases trim full-length proteins into 8–10 mer peptides that match the volume of the groove (Hansen et al. 2010; Blum et al. 2013). MHC II uses endosomal peptidases that generate longer and more varied peptides that can sit in several different registers within the open-ended groove of MHC II (Suri et al. 2006; Blum et al. 2013).
Experimental data regarding the optimal sizing of self lipid ligands in CD1 clefts derive from two sources: crystallographic studies of CD1-lipid complexes generated in vitro and lipids loaded onto CD1 proteins in cells. Crystal studies provide detailed information regarding the seating of lipids within CD1 grooves, but the complexes are necessarily produced from homogenous lipid preparations favored by the experimentalist. For crystallography, lipids are loaded onto CD1 proteins under artificial, acellular conditions that involve catalyzed protein folding or treatment with detergents and other loading cofactors. In contrast, cellular studies involve naturally occurring and heterogenous lipids within cells but do not directly assess lipid positioning in the cleft. In recent years, these two techniques, which have complementary strengths and weaknesses, are converging to produce a coherent picture of lipid display by all four CD1 proteins and a specialized mode of lipid display that is unique to the large cleft present in human CD1b proteins.
As highlighted above, lipid ligands sit more completely within the globular head of CD1 proteins with narrow portals that control their entry and exit (Figs. 2 and 3) (Zeng et al. 1997; Gadola et al. 2002; Zajonc et al. 2005a; Scharf et al. 2010). Ligands of CD1 proteins can range from 12 carbons (C12) with GMM to an intermediate length of C56 or to long-chain mycolyl lipids that exceed C80 (Beckman et al. 1994; Moody et al. 1997; Moody et al. 2002; Moody et al. 2005; Cheng et al. 2006; Layre et al. 2009). If lipids are not trimmed to fit the CD1 cleft volume, then the general question arises as to how diverse, biologically occurring lipids of differing chain length are sized to fit the cleft. Interestingly, the main answer seems to be that the cleft volumes of CD1a, CD1c, and CD1d are optimized to capture the common membrane lipids without chemical modification of lipid anchors prior to loading. Particularly large lipids up to C80 can be captured by the groove of CD1b, and small lipids can bind together with chaperone lipids such that two or more ligands fill the groove (Fig. 4). The following sections highlight cellular lipid capture and new insights into two types of chaperone lipids: spacers and scaffolds.

CD1b can bind large or small antigens. The transparent surface of CD1b proteins (Gadola et al. 2002; Batuwangala et al. 2004) shows the different orientation of associated ligands, a C80 glucose monomycolate (green, left panel) or a smaller ganglioside (green, right panel) bound with two detergent molecules (pink and blue). In the CD1b-glucose monomoycolate structure, one lipid fills the groove. In the ganglioside structure, the antigenic lipid protrudes through the F’ portal, but the scaffold and spacer lipids are confined to the interior of the groove
Spacers and scaffold lipids
Carbohydrate head groups of some glycolipids do undergo enzymatic trimming to reveal epitopes (Prigozy et al. 2001; Zhou et al. 2004a; de la Salle et al. 2005). However, aliphatic hydrocarbon chains are chemically inert, and even enzymatic digestion of lipid chains would require specialized enzymatic interactions, such as cycles of β-oxidation in dendritic cells (Pearce and Everts 2015). Accordingly, to date, there are no clear examples of lipid anchors that undergo trimming as part of the antigen-loading process. In fact, one particularly large ∼C80 GMM antigen was proven not to undergo lipid shortening prior to loading onto CD1b (Cheng et al. 2006). Instead, it appears that lipids bind with unmodified lipid anchors. In fact, the volumes of CD1a, CD1c, and CD1d grooves (1280–1780 Å3) are a good match to the size of the lipid anchors present in common diacylglyerides and sphingolipids present in cells (∼C32–C46) (Lazzarini et al. 2015).
However, the volume of CD1b (2200 Å3) is much larger, raising the question of which kinds of endogenous self lipids might bind. Few self lipids exist, which posses the combined lipid length (∼C76) that is needed to fill the CD1b groove. Increasing functional and crystallographic evidence suggests that CD1b can bind two or more endogenous lipids so that their combined length occupies the full groove (Gadola et al. 2002; Garcia-Alles et al. 2006; Garcia-Alles et al. 2011; Huang et al. 2011). In particular, crystal structures indicate that amphipathic antigens bind in the “upper chamber” of CD1b comprised by the A’ and F’ pockets. Highly hydrophobic lipids with no hydrophilic head groups, such as diacylglycerides and dideoxyceramides (Huang et al. 2011), sit deeply within the CD1b groove, such as the T’ tunnel. Based on their location at the bottom of the groove and their apparent positioning to provide “upward support” to the antigenic lipid, they are known as so scaffold lipids (Fig. 4).
An early observation in the CD1b antigen processing system was that short-chain antigens can load into CD1b, when it is present at the surface of APCs. However, specialized reactions occurring in lysosomes are needed to load lipids with long alkyl chains (Moody et al. 2002). Combining these older observations on ligand size with the newer discovery of scaffold lipids, a new and integrated hypothesis can explain both findings. That is, two functionally distinct chambers may be present in CD1b. Loading of small (∼C32) lipids might require ejection of the amphipathic lipid from the upper chamber, comprised of the A’ and F’ pockets, whereas loading of long-chain lipids (∼C80) might require pH-mediated ejection of this lipid and the spacer lipid to clear both the upper and lower chambers (Ly and Moody 2014). Experimental data using chain length analogs and diacylglyerides provide proof of principle support for this model (Huang et al. 2011), but it has not yet been tested on the broader range of self and foreign lipids for the CD1b system.
In addition to the specific idea that scaffold lipids push upwards on antigenic lipids (Fig. 4), a more general “spacer” function of lipid chaperones is inferred from the study of in vitro generated CD1-lipid complexes. Crystal structures involving CD1a, CD1b, CD1c, and CD1d show examples of single chain or other small lipids that do not fill the entire cleft, as well as electron densities that sit beside the antigen (Gadola et al. 2002; Zajonc et al. 2005a; Zajonc et al. 2005b; Garcia-Alles et al. 2011; Birkinshaw et al. 2015). Such spacer lipids include free fatty acids and may have a more general role in allowing lipids of differing length to bind CD1 proteins (Birkinshaw et al. 2015). Moving from consideration of cellular mechanisms of antigen capture, the following discussion highlights the known types of donor-unrestricted T cells, starting with the well known NKT cells and moving to more recently discovered T cell types.
Invariant NKT cells
Invariant NKT cells (also known as type I NKT cells) were identified in mice based on the conserved use of TCR β chains (Fowlkes et al. 1987). Subsequent studies identified the other two key aspects that now comprise the modern definition of NKT cells: strict TCR α sequence conservation and CD1d reactivity (Imai et al. 1986; Bendelac et al. 1995; Kawano et al. 1997). In vivo, NKT cells expand as populations of similar but non-clonal TCRs with nearly identical TCR α sequences. In humans the TCR α chain is typically encoded by TRAV10 joined to TRAJ18, and a biased usage of TCR β genes, often TRBV25 (Brennan et al. 2013). NKT cells are proven to be positively selected by CD1d proteins in mice and presumably a similar mechanism occurs in humans (Chiu et al. 2002).
NKT cells respond to synthetic α-linked hexosyl ceramides identified from synthetic compound libraries (Kawano et al. 1997). Most glycosyl sphingosines in mammals show a β-anomeric linkage, so the α-linkage emerged as a key chemical feature of NKT cell antigens, which determines their recognition and denotes them as artificial or foreign lipids. However, recent studies have identified small amounts of α-linked hexosyl ceramides present in the gut flora or in human tissues, suggesting that α-linked sphingolipids are present at low levels in mammals. Also, natural glycolipid antigens have been identified in non-pathogenic sphingomonas species (Kinjo et al. 2005; Mattner et al. 2005; Rossjohn et al. 2015) and bacteroides species that live in the gastrointestinal tract and self mammalian tissues (Wieland Brown et al. 2013; An et al. 2014; Olszak et al. 2014; Telesford et al. 2015). However, the chemical identification of any single positively selecting lipid antigen or the sources of the antigen or antigens that control in vivo response remains controversial.
NKT cells have a number of characteristic secondary features, including expansion of cell numbers in the gastrointestinal tract, high rates of cytokine production, expression of promyelocytic leukemia zinc finger (PLZF), and transcriptional programs that overlap with those seen in NK cells (Savage et al. 2008; Cohen et al. 2013; Kim et al. 2015). The deletion of NKT cells in mice by knockout of CD1d- or TCR-joining regions results in many altered immunological outcomes in vivo in mice, a topic that has been reviewed in detail (Barral and Brenner 2007) and considered in another chapter of this issue (Crosby and Kronenberg 2016). Invariant NKT cells are less abundant in humans as compared to mice, but their activation and function has been examined by the use of α-galactosyl ceramide analogs in pre-clinical cancer studies. For example, α-galactosylceramide was broadly used to design various therapeutic methods, including lipid injection, transfer of pulsed antigen-presenting cells, and in vitro activated NKT cells, which have achieved limited success (Salio et al. 2014). NKT cells have been implicated in human asthma (Akbari et al. 2006) and mouse models (Akbari et al. 2003) of asthma, although the number of NKT cells in humans with asthma has been questioned (Thomas et al. 2006; Thomas et al. 2010).
Diverse NKT cells
Diverse NKT cells (also known as type II NKT cells) express a more variable αβ TCR repertoire and do not recognize α-galactosylceramide. For example, the existence of CD1d reactive T cells that lack the stereotyped TCR present in invariant NKT cells has been long known (Chiu et al. 1999; Tatituri et al. 2013). The oligoclonality and antigen-specific nature of diverse NKT cells has also been shown with mouse TRAV7 and TRAV9 encoded TCRs recognizing CD1d and the self-glycolipid known as sulfatide (Chiu et al. 1999; Arrenberg et al. 2010). Staining of human diverse NKT cells with CD1d-β-glycosylceramide or glycosylsphingosine tetramers has been observed (Nair et al. 2015). Although showing a relatively diverse TCR repertoire, type II NKT cells share similar phenotypic and functional features with iNKT cells, including high autoreactivity (Gumperz et al. 2000; Brigl et al. 2006), PLZF-dependent development (Zhao et al. 2014), and rapid secretion of a wide range of cytokines after stimulation (Zhao et al. 2014). Many diverse NKT cells show an activated or memory phenotype, consistent to the rapid activation and secretion of cytokines (Zhang et al. 2011). Diverse NKT cells appear to be more abundant in humans than invariant NKT cells, as shown with CD1d tetramer or dimer staining (Chang et al. 2008; Nair et al. 2015). In addition to sulfatide and glycosylceramide species, other self and exogenous lipids, including mammalian, bacterial, and pollen-derived phospholipids, have also been identified as antigens for diverse NKT cells as reviewed recently (Macho-Fernandez and Brigl 2015; Bandyopadhyay et al. 2016).
NKT cells in vivo
In addition to the functions of NKT cells in cancer, infection and autoimmunity models, which have been previously reviewed (Viale et al. 2012; Taniguchi et al. 2015), recently published data implicate the role for NKT cells in metabolism through their actions in adipose tissue and the liver. Although invariant NKT cells are present in relatively low numbers in human blood, they are present in very large numbers in adipose tissues (Ji et al. 2012; Lynch et al. 2012; Schipper et al. 2012). Further, the number of NKT cells in adipose tissues of lean individuals is higher than that of fat individuals (Lynch et al. 2012). Invariant NKT cells appear to be protective against obesity and insulin resistance, which occurs through secreting anti-inflammatory cytokines (Ji et al. 2012; Lynch et al. 2012; Schipper et al. 2012). Cytokines have downstream influence on the functions of macrophages and regulatory T cells (Ji et al. 2012; Lynch et al. 2014).
Notably, NKT cells are also enriched in microvascular compartments of the liver and play important roles in antitumor defenses (Seino et al. 2006), and antiviral responses and pathogenesis of chronic liver diseases as detailed below. Invariant NKTs were protective in viral hepatitis by secreting IFN-γ to inhibit the proliferation of hepatitis B and C virus in liver cells (Sprengers et al. 2008; Ye et al. 2009). This protective function of iNKT cells is induced by the stimulation of CD1d-presented host lysophospholipids (Zeissig et al. 2012). In liver inflammation, invariant NKT cells were described as pathogenic for chronic injury but potential protective to acute injury (Gao et al. 2009; Bandyopadhyay et al. 2016). For example, the iNKT-deficient mice develop less alcohol-induced steatosis with reduced neutrophil infiltration (Cui et al. 2015). The administration of an anti-CD1d-blocking antibody significantly suppresses this pro-inflammatory cascade and ameliorates alcohol-induced steatosis (Mathews et al. 2016). Interestingly, sulfatide-activated diverse NKT cells can dampen liver inflammation and suppress the pro-inflammatory responses induced by invariant NKT cells in mouse alcoholic liver disease (Maricic et al. 2015). Similarly, autoimmune hepatitis is associated with IL-17 producing iNKT cells, which can also be antagonized by diverse NKT cells (Takeda et al. 2000; Halder et al. 2007). The opposing roles and interplay between invariant and diverse NKTs in liver inflammation suggests that the specificity of lipid antigens and sequences of TCR can be important for functional outcomes (Bandyopadhyay et al. 2016). Therefore, future diagnostic or therapeutic applications based on NKT cells in liver diseases likely require further determination of CD1d-presented lipid antigens and better understanding of the apparently distinct functions of invariant or diverse NKT cells (Zeissig et al. 2012; Maricic et al. 2015).
TCR-defined T cell types
The two most widely known human T cell subtypes that are defined by stereotyped TCRs are NKT cells, which are discussed above, and mucosa-associated invariant T (MAIT) cells, which recognize MR1. MR1-reactive T cells that express nearly invariant TCR α chains, typically with TRAV1-2-TRAJ33 junction (Tilloy et al. 1999; Martin et al. 2009; Dusseaux et al. 2011). NKT cells and MAIT cells share three key features: recognition of non-polymorphic antigen-presenting molecules, expansion of similar but non-clonal TCRs within one individual (intradonor TCR conservation), and expansion of similar but non-clonal TCR in most or all humans (interdonor TCR conservation). Until recently, NKT cells and MAIT cells were considered to be the only two specialized carve out subtypes that are distinguished from the larger αβ T cell repertoire based on TCR expression.
However, this immunogenetic concept of TCR-defined T cell compartments of the human αβ T cell repertoire is now expanding in two ways (Van Rhijn et al. 2015a). First, for T cells that recognize the CD1d-α-galactosyl ceramide (Kawano et al. 1997; Borg et al. 2007) or MR1-5-(2-oxopropylideneamino)-6-d-ribitylaminouracil complexes (Kjer-Nielsen et al. 2012), new V and J gene combinations are being described that are similar to but do not formally meet the usual definitions of invariant NKT cell and MAIT cell TCRs. For example, the TCR β chain repertoire of MAIT cells was defined as typically using TRAV1-2 rearranged to TRAJ33. However, MAIT cells with TRAJ20 or TRAJ12 are now described (Patel et al. 2013; Reantragoon et al. 2013). A recent study shows that MR1-reactive T cells can express TCRs not encoded by TRAV1-2, which is an immunogenetic feature previously thought to be essential for MR1 recognition (Gherardin et al. 2016). Also, NKT cells expressing the atypical V gene, TRAV13, can bind to CD1d-α-galactosyl ceramide (Uldrich et al. 2011). These discoveries were enabled by bypassing the conventional TCR-based experimental methods and instead using CD1d and MR1 tetramers to more broadly ask which kinds of TCRs bind antigen complexes. Such atypical TCRs recognizing typical antigens are incrementally expanding the definitions of so-called invariant TCRs. The extent to which these represent physiologically distinct responses, which are different from the classical TCR definitions, is not yet known.
A second expansion of the concept of TCR-defined T cell types is based on the fact that non-polymorphic antigen-presenting molecules other than CD1d and MR1 are widely expressed in humans, including HLA-E, CD1a, CD1b, or CD1c, yet the extent to which they might activate T cells with intradonor and interdonor stereotyped TCRs has not been widely investigated (Van Rhijn et al. 2015a). T cells showing both of these properties would become candidates for new TCR-defined components of the human immune system. Several recent studies set the stage for work in this area, including work by Van Rhijn showing that TCR β chains can be broadly conserved among half or more of donors studied (Van Rhijn et al. 2015b). Also, human CD1a, CD1b, and CD1c tetramers bound to Mycobacterium tuberculosis-derived lipids have been recently validated for use in tuberculosis patients (Kasmar et al. 2009; Kasmar et al. 2011; Kasmar et al. 2013; Ly et al. 2013; Van Rhijn et al. 2015b). Tetramer-based studies of polyclonal T cells in the ex vivo state have allowed for discovery of new TCR-defined T cell types in the CD1b-reactive human T cell repertoire.
GEM T cells express TRAV1-2
Early, small-scale surveys of TCRs expressed on T cells recognizing group 1 CD1 proteins bound to various lipid antigens failed to identify stereotyped patterns of V or J gene usage (Grant et al. 1999), leading to the idea that CD1a, CD1b, and CD1c proteins might stimulate T cells that are fundamentally more diverse in their TCR usage, as contrasted with invariant NKT cells. However, when CD1b tetramers were loaded with one kind of mycobacterial GMM antigen, two patterns of intradonor and interdonor TCR conservation were readily observed among genetically unrelated latent tuberculosis patients. TCRs comprised of nearly invariant sequences derived from TRAV1-2 joined to TRAJ9, with some apparent bias for expression of TRBV6-2-encoded TCR β chains, were identified in genetically unrelated donors. Based on their reliance on germline-encoded TCRs, relative lack of N-region additions, and specificity for mycobacterial lipids, these T cells were named germline-encoded mycolyl reactive (GEM) T cells (Van Rhijn et al. 2013).
GEM TCRs show high affinity (∼1 μM) binding to CD1b-GMM and secrete interferon-γ and TNF, when stimulated (Van Rhijn et al. 2013). Their possible role in human host response to M. tuberculosis infection is now being investigated. GEM TCRs share the properties of intradonor conservation, interdonor conservation, and recognition of non-polymorphic antigen-presenting molecules, so they can be considered an invariant TCR type in the human repertoire. However, the earliest studies of their functions suggest an immunological role that is distinct from the immunoregulatory properties of MAIT cells and NKT cells. The latter two T cell types are expanded to large numbers in peripheral blood, and they express activation markers at baseline and respond rapidly in large numbers to purified antigen or pathogens that express key antigens (Brennan et al. 2013; Van Rhijn et al. 2013; Gold et al. 2014). In contrast, GEM T cells recognize a foreign antigen and appear to be less abundant in the blood of healthy blood donors in the absence of infection (Van Rhijn et al. 2013). Small-scale human studies suggest GEM T cells might be a kind of extremely public donor unrestricted T cell involved in human host response to a defined foreign antigen from mycobacteria, rather than an innate T cell type recognizing self lipids.
LDN5-like T cells
Shortly after the discovery of GEM T cells, CD1b-GMM tetramers also isolated T cells from latent tuberculosis expressing lower affinity TCRs with TCR β chains encoded by the TRBV4-1 V gene (Van Rhijn et al. 2014). These polyclonal T cells from genetically unrelated donors show TCR patterns that were also seen two decades prior in a T cell clone known as LDN5, which was isolated from an Mycobacterium leprae-infected skin lesions. Thus, what was previously viewed as a unique TRAV17-TRAJ9-encoded TCR α chain paired with a TRABV4-1-encoded β chain might be more typical of a polyclonal response of relatively low affinity TCRs binding CD1b-GMM from two related mycobacterial pathogens. Therefore, these newly discovered polyclonal T cells were designated LDN5-like T cells. Taken together, identification of GEM T cells and LDN5-like T cells document the existence of two interdonor-conserved TCRs recognizing one foreign glycolipid. The atypical NKT cell and MAIT cell TCRs discussed above show some variations in V and J gene usage but still possess key aspects of the dominant TCR motif. In contrast, these two TCR types are entirely unrelated in their V and J gene usage yet recognize the same kind of antigen complex. Given the speed and ease with which stereotyped TCRs could be identified in the CD1b-reactive repertoire, the study of the relatively uncharted field of CD1a and CD1c-reactive TCRs might also reveal interdonor TCR conservation in the future.
Role of CD1a, CD1b, and CD1c in disease
In addition to the proposed role of NKT cells and CD1d in human disease discussed above, the widespread expression of CD1a, CD1b, and CD1c in human tissues (Dougan et al. 2007a), as well as increasing evidence for the activation of polyclonal T cells recognizing these CD1 isoforms (de Jong et al. 2010; de Lalla et al. 2011; Van Rhijn et al. 2015b), now raise the possibility that CD1a-, CD1b-, or CD1c-restricted T cells might be involved in human disease. Although mice lack CD1a, CD1b, and CD1c, the development of human CD1 transgenic mice (Felio et al. 2009; Kobayashi et al. 2012; Zhao et al. 2015) as well as guinea pigs (Hiromatsu et al. 2002) represent viable models for disease-focused investigation. Considering humans as a model for study of disease, the most intensive investigation of the disease-relevant CD1-restricted T cells has focused on the infections of M. tuberculosis and other bacteria.
T cells from asymptomatic latent tuberculosis patients significantly respond to mycobacterial lipid antigen preparations, in comparison to minimally detectable or absent responses from active tuberculosis prior to chemotherapy (Moody et al. 2000; Gilleron et al. 2004; Layre et al. 2009). Development of tetramer or dextramer reagents have recently detected the existence of polyclonal mycobacterial lipid-reactive T cells in patients with latent tuberculosis (Kasmar et al. 2011; Kasmar et al. 2013; Ly et al. 2013; Kasprowicz et al. 2016).
Future question: a network of invariant T cell types?
Among the many known ligands and antigens for the group 1 CD1 system, GMM is the only antigen that has been systematically investigated for conserved human TCR patterns (Van Rhijn et al. 2015a). However, the discovery of two TCR motifs with one antigen supports future efforts to characterize TCR usage in the repertoires of T cells responding to CD1a, CD1b, and CD1c. A large number of lipids bind to cellular human CD1 proteins (Cox et al. 2009; Huang et al. 2011; de Jong et al. 2014). The combinatorial nature of CD1-lipid complex formation suggests that currently unknown antigen complexes exist and the possibility of identifying further invariant TCRs is real. CD1a and CD1c tetramers have been recently validated (Kasmar et al. 2013; Ly et al. 2013; Roy et al. 2014; Roy et al. 2016). Early reports of CD1c-mediated T cell response document possible overexpression of TRBV7-8 and TRBV7-9 among responding T cells, which might represent an incipient motif for TCRs responding to CD1c (Roy et al. 2014). Thinking beyond MAIT cells, NKT cells, and GEM T cells, it is possible to consider a model in which the combinatorial interactions of CD1 and lipids to form structurally diverse complexes on the surface of DCs and B cells could support a network of interdonor-conserved TCRs in humans.
Future applications
Turning to practical applications, the simplified population genetics of CD1 proteins and the responding TCRs raises a new possibility. Is the CD1 system “druggable” such that lipid ligands might be designed that optimally bind to CD1 proteins and thereby block or activate interdonor-conserved T cells? In concept, antigen-specific therapy is highly desirable because it could bypass the many side effects common to general blockers or activators of T cell response, such as cyclosporine or anti-CD3 monoclonal antibodies. However, peptide antigen therapy has not advanced significantly due in part to a key immunogenetic barrier: design of peptides that bind equivalently to allelic variants is complex, and with some exceptions, is not feasible (Lalvani and Pareek 2010). This barrier is lacking in the CD1 system due to the nearly uniform expression of CD1 proteins in humans. Second, whereas NKT cells and MAIT cells are activated in many situations, the stereotyped TCRs present on GEM and LDN5-like T cells recognize a pathogen-specific antigen, raising the possibility that detection of the TCR expansions could be used for diagnosis of mycobacterial disease (Van Rhijn et al. 2013).
The known phenotypic characteristics and activating ligands of invariant NKT cells and MAIT cell might allow design of lipid or small molecule therapies. NKT cells have a pre-activated phenotype that might allow bypassing of negative regulatory pathways found in MHC-restricted T cells, so therapeutic design of lipid activators might take advantage of the observed quick response phenotype. For example, α-galactosylceramide-stimulated invariant NKT cell provides adjuvant effect, while co-administered with peptide antigens for better stimulating conventional T cell responses (Fujii et al. 2003; Silk et al. 2004; Salio et al. 2014). Considering the more recently described lipid antigens, the CD1a-presented skin lipid, squalene (de Jong et al. 2014), is a widely used adjuvant known as MF59, which enhances the efficacy of vaccines (Fox and Haensler 2013). Last, whereas design of T cell activators requires detailed knowledge of the responding TCRs, design of T cell blocking ligands could be accomplished through CD1-lipid binding assays using lipids with large head groups. For example, sphingomyelin has been observed to block CD1-mediated T cells in several contexts and so might be developed as a T cell inhibitor (de Jong et al. 2014; Birkinshaw et al. 2015). In these ways, the simplified immunogenetic structure of the CD1 system supports new approaches to T cell-focused therapies.
References
Akbari O, Stock P, Meyer E, Kronenberg M, Sidobre S, Nakayama T, Taniguchi M, Grusby MJ, DeKruyff RH, Umetsu DT (2003) Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyperreactivity. Nat Med 9:582–588
Akbari O, Faul JL, Hoyte EG, Berry GJ, Wahlstrom J, Kronenberg M, DeKruyff RH, Umetsu DT (2006) CD4+ invariant T-cell-receptor+ natural killer T cells in bronchial asthma. N Engl J Med 354:1117–1129
An D, Oh SF, Olszak T, Neves JF, Avci FY, Erturk-Hasdemir D, Lu X, Zeissig S, Blumberg RS, Kasper DL (2014) Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell 156:123–133
Arrenberg P, Halder R, Dai Y, Maricic I, Kumar V (2010) Oligoclonality and innate-like features in the TCR repertoire of type II NKT cells reactive to a beta-linked self-glycolipid. Proc Natl Acad Sci U S A 107:10984–10989
Aruffo A, Seed B (1989) Expression of cDNA clones encoding the thymocyte antigens CD1a, b, c demonstrates a hierarchy of exclusion in fibroblasts. J Immunol 143:1723–1730
Ayala Garcia MA, Gonzalez Yebra B, Lopez Flores AL, Guani Guerra E (2012) The major histocompatibility complex in transplantation. J Transplant 2012:842141
Bai L, Sagiv Y, Liu Y, Freigang S, Yu KO, Teyton L, Porcelli SA, Savage PB, Bendelac A (2009) Lysosomal recycling terminates CD1d-mediated presentation of short and polyunsaturated variants of the NKT cell lipid antigen alphaGalCer. Proc Natl Acad Sci U S A 106:10254–10259
Bai L, Picard D, Anderson B, Chaudhary V, Luoma A, Jabri B, Adams EJ, Savage PB, Bendelac A (2012) The majority of CD1d-sulfatide-specific T cells in human blood use a semiinvariant Vdelta1 TCR. Eur J Immunol 42:2505–2510
Bandyopadhyay K, Marrero I, Kumar V (2016) NKT cell subsets as key participants in liver physiology and pathology. Cell Mol Immunol 13:337–346
Barral DC, Brenner MB (2007) CD1 antigen presentation: how it works. Nat Rev Immunol 7:929–941
Batuwangala T, Shepherd D, Gadola SD, Gibson KJ, Zaccai NR, Fersht AR, Besra GS, Cerundolo V, Jones EY (2004) The crystal structure of human CD1b with a bound bacterial glycolipid. J Immunol 172:2382–2388
Beckman EM, Porcelli SA, Morita CT, Behar SM, Furlong ST, Brenner MB (1994) Recognition of a lipid antigen by CD1-restricted alpha beta+ T cells. Nature 372:691–694
Bendelac A, Lantz O, Quimby ME, Yewdell JW, Bennink JR, Brutkiewicz RR (1995) CD1 recognition by mouse NK1+ T lymphocytes. Science 268:863–865
Birkholz A, Nemcovic M, Yu ED, Girardi E, Wang J, Khurana A, Pauwels N, Farber E, Chitale S, Franck RW, Tsuji M, Howell A, Van Calenbergh S, Kronenberg M, Zajonc DM (2015) Lipid and carbohydrate modifications of alpha-galactosylceramide differently influence mouse and human type I natural killer T cell activation. J Biol Chem 290:17206–17217
Birkinshaw RW, Pellicci DG, Cheng TY, Keller AN, Sandoval-Romero M, Gras S, de Jong A, Uldrich AP, Moody DB, Godfrey DI, Rossjohn J (2015) Alphabeta T cell antigen receptor recognition of CD1a presenting self lipid ligands. Nat Immunol 16:258–266
Blum JS, Wearsch PA, Cresswell P (2013) Pathways of antigen processing. Annu Rev Immunol 31:443–473
Borg NA, Wun KS, Kjer-Nielsen L, Wilce MC, Pellicci DG, Koh R, Besra GS, Bharadwaj M, Godfrey DI, McCluskey J, Rossjohn J (2007) CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor. Nature 448:44–49
Boudinot P, Mondot S, Jouneau L, Teyton L, Lefranc MP, Lantz O (2016) Restricting nonclassical MHC genes coevolve with TRAV genes used by innate-like T cells in mammals. Proc Natl Acad Sci U S A 113:E2983–E2992
Brennan PJ, Brigl M, Brenner MB (2013) Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol 13:101–117
Brigl M, Brenner MB (2004) CD1: antigen presentation and T cell function. Annu Rev Immunol 22:817–890
Brigl M, van den Elzen P, Chen X, Meyers JH, Wu D, Wong CH, Reddington F, Illarianov PA, Besra GS, Brenner MB, Gumperz JE (2006) Conserved and heterogeneous lipid antigen specificities of CD1d-restricted NKT cell receptors. J Immunol 176:3625–3634
Briken V, Jackman RM, Watts GF, Rogers RA, Porcelli SA (2000) Human CD1b and CD1c isoforms survey different intracellular compartments for the presentation of microbial lipid antigens. J Exp Med 192:281–288
Briken V, Jackman RM, Dasgupta S, Hoening S, Porcelli SA (2002) Intracellular trafficking pathway of newly synthesized CD1b molecules. EMBO J 21:825–834
Brossay L, Tangri S, Bix M, Cardell S, Locksley R, Kronenberg M (1998) Mouse CD1-autoreactive T cells have diverse patterns of reactivity to CD1+ targets. J Immunol 160:3681–3688
Brozovic S, Nagaishi T, Yoshida M, Betz S, Salas A, Chen D, Kaser A, Glickman J, Kuo T, Little A, Morrison J, Corazza N, Kim JY, Colgan SP, Young SG, Exley M, Blumberg RS (2004) CD1d function is regulated by microsomal triglyceride transfer protein. Nat Med 10:535–539
Cernadas M, Cavallari M, Watts G, Mori L, De Libero G, Brenner MB (2010) Early recycling compartment trafficking of CD1a is essential for its intersection and presentation of lipid antigens. J Immunol 184:1235–1241
Chang DH, Deng H, Matthews P, Krasovsky J, Ragupathi G, Spisek R, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV (2008) Inflammation-associated lysophospholipids as ligands for CD1d-restricted T cells in human cancer. Blood 112:1308–1316
Cheng TY, Relloso M, Van Rhijn I, Young DC, Besra GS, Briken V, Zajonc DM, Wilson IA, Porcelli S, Moody DB (2006) Role of lipid trimming and CD1 groove size in cellular antigen presentation. EMBO J 25:2989–2999
Chiu YH, Jayawardena J, Weiss A, Lee D, Park SH, Dautry-Varsat A, Bendelac A (1999) Distinct subsets of CD1d-restricted T cells recognize self-antigens loaded in different cellular compartments. J Exp Med 189:103–110
Chiu YH, Park SH, Benlagha K, Forestier C, Jayawardena-Wolf J, Savage PB, Teyton L, Bendelac A (2002) Multiple defects in antigen presentation and T cell development by mice expressing cytoplasmic tail-truncated CD1d. Nat Immunol 3:55–60
Cohen NR, Brennan PJ, Shay T, Watts GF, Brigl M, Kang J, Brenner MB (2013) Shared and distinct transcriptional programs underlie the hybrid nature of iNKT cells. Nat Immunol 14:90–99
Cox D, Fox L, Tian R, Bardet W, Skaley M, Mojsilovic D, Gumperz J, Hildebrand W (2009) Determination of cellular lipids bound to human CD1d molecules. PLoS One 4:e5325
Crosby CM, Kronenberg M (2016) Invariant natural killer T cells: front line fighters in the war against pathogenic microbes. Immunogenetics. doi:10.1007/s00251-016-0933-y.
Cui K, Yan G, Xu C, Chen Y, Wang J, Zhou R, Bai L, Lian Z, Wei H, Sun R, Tian Z (2015) Invariant NKT cells promote alcohol-induced steatohepatitis through interleukin-1beta in mice. J Hepatol 62:1311–1318
de Jong A, Pena-Cruz V, Cheng TY, Clark RA, Van Rhijn I, Moody DB (2010) CD1a-autoreactive T cells are a normal component of the human alphabeta T cell repertoire. Nat Immunol 11:1102–1109
de Jong A, Cheng TY, Huang S, Gras S, Birkinshaw RW, Kasmar AG, Van Rhijn I, Pena-Cruz V, Ruan DT, Altman JD, Rossjohn J, Moody DB (2014) CD1a-autoreactive T cells recognize natural skin oils that function as headless antigens. Nat Immunol 15:177–185
de la Salle H, Mariotti S, Angenieux C, Gilleron M, Garcia-Alles LF, Malm D, Berg T, Paoletti S, Maitre B, Mourey L, Salamero J, Cazenave JP, Hanau D, Mori L, Puzo G, De Libero G (2005) Assistance of microbial glycolipid antigen processing by CD1e. Science 310:1321–1324
de Lalla C, Lepore M, Piccolo FM, Rinaldi A, Scelfo A, Garavaglia C, Mori L, De Libero G, Dellabona P, Casorati G (2011) High-frequency and adaptive-like dynamics of human CD1 self-reactive T cells. Eur J Immunol 41:602–610
De Libero G, Moran AP, Gober HJ, Rossy E, Shamshiev A, Chelnokova O, Mazorra Z, Vendetti S, Sacchi A, Prendergast MM, Sansano S, Tonevitsky A, Landmann R, Mori L (2005) Bacterial infections promote T cell recognition of self-glycolipids. Immunity 22:763–772
Dellabona P, Consonni M, de Lalla C, Casorati G (2015) Group 1 CD1-restricted T cells and the pathophysiological implications of self-lipid antigen recognition. Tissue Antigens 86:393–405
Denzin LK, Cresswell P (1995) HLA-DM induces CLIP dissociation from MHC class II alpha beta dimers and facilitates peptide loading. Cell 82:155–165
Dougan SK, Salas A, Rava P, Agyemang A, Kaser A, Morrison J, Khurana A, Kronenberg M, Johnson C, Exley M, Hussain MM, Blumberg RS (2005) Microsomal triglyceride transfer protein lipidation and control of CD1d on antigen-presenting cells. J Exp Med 202:529–539
Dougan SK, Kaser A, Blumberg RS (2007a) CD1 expression on antigen-presenting cells. Curr Top Microbiol Immunol 314:113–141
Dougan SK, Rava P, Hussain MM, Blumberg RS (2007b) MTP regulated by an alternate promoter is essential for NKT cell development. J Exp Med 204:533–545
Dusseaux M, Martin E, Serriari N, Peguillet I, Premel V, Louis D, Milder M, Le Bourhis L, Soudais C, Treiner E, Lantz O (2011) Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood 117:1250–1259
Facciotti F, Ramanjaneyulu GS, Lepore M, Sansano S, Cavallari M, Kistowska M, Forss-Petter S, Ni G, Colone A, Singhal A, Berger J, Xia C, Mori L, De Libero G (2012) Peroxisome-derived lipids are self antigens that stimulate invariant natural killer T cells in the thymus. Nat Immunol 13:474–480
Felio K, Nguyen H, Dascher CC, Choi HJ, Li S, Zimmer MI, Colmone A, Moody DB, Brenner MB, Wang CR (2009) CD1-restricted adaptive immune responses to Mycobacteria in human group 1 CD1 transgenic mice. J Exp Med 206:2497–2509
Fowlkes BJ, Kruisbeek AM, Ton-That H, Weston MA, Coligan JE, Schwartz RH, Pardoll DM (1987) A novel population of T-cell receptor alpha beta-bearing thymocytes which predominantly expresses a single V beta gene family. Nature 329:251–254
Fox CB, Haensler J (2013) An update on safety and immunogenicity of vaccines containing emulsion-based adjuvants. Expert Rev Vaccines 12:747–758
Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM (2003) Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med 198:267–279
Gadola SD, Zaccai NR, Harlos K, Shepherd D, Castro-Palomino JC, Ritter G, Schmidt RR, Jones EY, Cerundolo V (2002) Structure of human CD1b with bound ligands at 2.3 A, a maze for alkyl chains. Nat Immunol 3:721–726
Gao B, Radaeva S, Park O (2009) Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol 86:513–528
Garboczi DN, Ghosh P, Utz U, Fan QR, Biddison WE, Wiley DC (1996) Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature 384:134–141
Garcia KC, Degano M, Stanfield RL, Brunmark A, Jackson MR, Peterson PA, Teyton L, Wilson IA (1996) An alphabeta T cell receptor structure at 2.5 A and its orientation in the TCR-MHC complex. Science 274:209–219
Garcia-Alles LF, Versluis K, Maveyraud L, Vallina AT, Sansano S, Bello NF, Gober HJ, Guillet V, de la Salle H, Puzo G, Mori L, Heck AJ, De Libero G, Mourey L (2006) Endogenous phosphatidylcholine and a long spacer ligand stabilize the lipid-binding groove of CD1b. EMBO J 25:3684–3692
Garcia-Alles LF, Collmann A, Versluis C, Lindner B, Guiard J, Maveyraud L, Huc E, Im JS, Sansano S, Brando T, Julien S, Prandi J, Gilleron M, Porcelli SA, de la Salle H, Heck AJ, Mori L, Puzo G, Mourey L, De Libero G (2011) Structural reorganization of the antigen-binding groove of human CD1b for presentation of mycobacterial sulfoglycolipids. Proc Natl Acad Sci U S A 108:17755–17760
Gherardin NA, Keller AN, Woolley RE, Le Nours J, Ritchie DS, Neeson PJ, Birkinshaw RW, Eckle SB, Waddington JN, Liu L, Fairlie DP, Uldrich AP, Pellicci DG, McCluskey J, Godfrey DI, Rossjohn J (2016) Diversity of T cells restricted by the MHC class I-related molecule MR1 facilitates differential antigen recognition. Immunity 44:32–45
Gilleron M, Stenger S, Mazorra Z, Wittke F, Mariotti S, Bohmer G, Prandi J, Mori L, Puzo G, De Libero G (2004) Diacylated sulfoglycolipids are novel mycobacterial antigens stimulating CD1-restricted T cells during infection with Mycobacterium tuberculosis. J Exp Med 199:649–659
Godfrey DI, Kronenberg M (2004) Going both ways: immune regulation via CD1d-dependent NKT cells. J Clin Invest 114:1379–1388
Gold MC, McLaren JE, Reistetter JA, Smyk-Pearson S, Ladell K, Swarbrick GM, Yu YY, Hansen TH, Lund O, Nielsen M, Gerritsen B, Kesmir C, Miles JJ, Lewinsohn DA, Price DA, Lewinsohn DM (2014) MR1-restricted MAIT cells display ligand discrimination and pathogen selectivity through distinct T cell receptor usage. J Exp Med 211:1601–1610
Grant EP, Degano M, Rosat JP, Stenger S, Modlin RL, Wilson IA, Porcelli SA, Brenner MB (1999) Molecular recognition of lipid antigens by T cell receptors. J Exp Med 189:195–205
Gumperz JE, Roy C, Makowska A, Lum D, Sugita M, Podrebarac T, Koezuka Y, Porcelli SA, Cardell S, Brenner MB, Behar SM (2000) Murine CD1d-restricted T cell recognition of cellular lipids. Immunity 12:211–221
Halder RC, Aguilera C, Maricic I, Kumar V (2007) Type II NKT cell-mediated energy induction in type I NKT cells prevents inflammatory liver disease. J Clin Invest 117:2302–2312
Han M, Hannick LI, DiBrino M, Robinson MA (1999) Polymorphism of human CD1 genes. Tissue Antigens 54:122–127
Hansen TH, Huang S, Arnold PL, Fremont DH (2007) Patterns of nonclassical MHC antigen presentation. Nat Immunol 8:563–568
Hansen TH, Connolly JM, Gould KG, Fremont DH (2010) Basic and translational applications of engineered MHC class I proteins. Trends Immunol 31:363–369
Hiromatsu K, Dascher CC, LeClair KP, Sugita M, Furlong ST, Brenner MB, Porcelli SA (2002) Induction of CD1-restricted immune responses in guinea pigs by immunization with mycobacterial lipid antigens. J Immunol 169:330–339
Huang S, Gilfillan S, Cella M, Miley MJ, Lantz O, Lybarger L, Fremont DH, Hansen TH (2005) Evidence for MR1 antigen presentation to mucosal-associated invariant T cells. J Biol Chem 280:21183–21193
Huang S, Cheng TY, Young DC, Layre E, Madigan CA, Shires J, Cerundolo V, Altman JD, Moody DB (2011) Discovery of deoxyceramides and diacylglycerols as CD1b scaffold lipids among diverse groove-blocking lipids of the human CD1 system. Proc Natl Acad Sci U S A 108:19335–19340
Hunger RE, Sieling PA, Ochoa MT, Sugaya M, Burdick AE, Rea TH, Brennan PJ, Belisle JT, Blauvelt A, Porcelli SA, Modlin RL (2004) Langerhans cells utilize CD1a and langerin to efficiently present nonpeptide antigens to T cells. J Clin Invest 113:701–708
Im JS, Arora P, Bricard G, Molano A, Venkataswamy MM, Baine I, Jerud ES, Goldberg MF, Baena A, Yu KO, Ndonye RM, Howell AR, Yuan W, Cresswell P, Chang YT, Illarionov PA, Besra GS, Porcelli SA (2009) Kinetics and cellular site of glycolipid loading control the outcome of natural killer T cell activation. Immunity 30:888–898
Imai K, Kanno M, Kimoto H, Shigemoto K, Yamamoto S, Taniguchi M (1986) Sequence and expression of transcripts of the T-cell antigen receptor alpha-chain gene in a functional, antigen-specific suppressor-T-cell hybridoma. Proc Natl Acad Sci U S A 83:8708–8712
Jackman RM, Stenger S, Lee A, Moody DB, Rogers RA, Niazi KR, Sugita M, Modlin RL, Peters PJ, Porcelli SA (1998) The tyrosine-containing cytoplasmic tail of CD1b is essential for its efficient presentation of bacterial lipid antigens. Immunity 8:341–351
Jayawardena-Wolf J, Benlagha K, Chiu YH, Mehr R, Bendelac A (2001) CD1d endosomal trafficking is independently regulated by an intrinsic CD1d-encoded tyrosine motif and by the invariant chain. Immunity 15:897–908
Ji Y, Sun S, Xu A, Bhargava P, Yang L, Lam KS, Gao B, Lee CH, Kersten S, Qi L (2012) Activation of natural killer T cells promotes M2 macrophage polarization in adipose tissue and improves systemic glucose tolerance via interleukin-4 (IL-4)/STAT6 protein signaling axis in obesity. J Biol Chem 287:13561–13571
Jones EY, Fugger L, Strominger JL, Siebold C (2006) MHC class II proteins and disease: a structural perspective. Nat Rev Immunol 6:271–282
Joyce S, Woods AS, Yewdell JW, Bennink JR, De Silva AD, Boesteanu A, Balk SP, Cotter RJ, Brutkiewicz RR (1998) Natural ligand of mouse CD1d1: cellular glycosylphosphatidylinositol. Science 279:1541–1544
Kain L, Webb B, Anderson BL, Deng S, Holt M, Costanzo A, Zhao M, Self K, Teyton A, Everett C, Kronenberg M, Zajonc DM, Bendelac A, Savage PB, Teyton L (2014) The identification of the endogenous ligands of natural killer T cells reveals the presence of mammalian alpha-linked glycosylceramides. Immunity 41:543–554
Kang SJ, Cresswell P (2004) Saposins facilitate CD1d-restricted presentation of an exogenous lipid antigen to T cells. Nat Immunol 5:175–181
Kasmar A, Van Rhijn I, Moody DB (2009) The evolved functions of CD1 during infection. Curr Opin Immunol 21:397–403
Kasmar AG, van Rhijn I, Cheng TY, Turner M, Seshadri C, Schiefner A, Kalathur RC, Annand JW, de Jong A, Shires J, Leon L, Brenner M, Wilson IA, Altman JD, Moody DB (2011) CD1b tetramers bind {alpha}{beta} T cell receptors to identify a mycobacterial glycolipid-reactive T cell repertoire in humans. J Exp Med 208:1741–1747
Kasmar AG, Van Rhijn I, Magalhaes KG, Young DC, Cheng TY, Turner MT, Schiefner A, Kalathur RC, Wilson IA, Bhati M, Gras S, Birkinshaw RW, Tan LL, Rossjohn J, Shires J, Jakobsen S, Altman JD, Moody DB (2013) Cutting edge: CD1a tetramers and dextramers identify human lipopeptide-specific T cells ex vivo. J Immunol 191:4499–4503
Kasprowicz VO, Cheng TY, Ndung’u T, Sunpath H, Moody DB, Kasmar AG (2016) HIV disrupts human T cells that target mycobacterial glycolipids. J Infect Dis 213:628–633
Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, Ueno H, Nakagawa R, Sato H, Kondo E, Koseki H, Taniguchi M (1997) CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science 278:1626–1629
Kawasaki N, Rillahan CD, Cheng TY, Van Rhijn I, Macauley MS, Moody DB, Paulson JC (2014) Targeted delivery of mycobacterial antigens to human dendritic cells via Siglec-7 induces robust T cell activation. J Immunol 193:1560–1566
Kim EY, Lynch L, Brennan PJ, Cohen NR, Brenner MB (2015) The transcriptional programs of iNKT cells. Semin Immunol 27:26–32
Kinjo Y, Wu D, Kim G, Xing GW, Poles MA, Ho DD, Tsuji M, Kawahara K, Wong CH, Kronenberg M (2005) Recognition of bacterial glycosphingolipids by natural killer T cells. Nature 434:520–525
Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L, Bhati M, Chen Z, Kostenko L, Reantragoon R, Williamson NA, Purcell AW, Dudek NL, McConville MJ, O’Hair RA, Khairallah GN, Godfrey DI, Fairlie DP, Rossjohn J, McCluskey J (2012) MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 491(7426):717–723
Kobayashi C, Shiina T, Tokioka A, Hattori Y, Komori T, Kobayashi-Miura M, Takizawa T, Takahara K, Inaba K, Inoko H, Takeya M, Dranoff G, Sugita M (2012) GM-CSF-independent CD1a expression in epidermal Langerhans cells: evidence from human CD1A genome-transgenic mice. J investigative dermatol 132:241–244
Lalvani A, Pareek M (2010) A 100 year update on diagnosis of tuberculosis infection. Br Med Bull 93:69–84
Lawton AP, Prigozy TI, Brossay L, Pei B, Khurana A, Martin D, Zhu T, Spate K, Ozga M, Honing S, Bakke O, Kronenberg M (2005) The mouse CD1d cytoplasmic tail mediates CD1d trafficking and antigen presentation by adaptor protein 3-dependent and -independent mechanisms. J Immunol 174:3179–3186
Layre E, Collmann A, Bastian M, Mariotti S, Czaplicki J, Prandi J, Mori L, Stenger S, De Libero G, Puzo G, Gilleron M (2009) Mycolic acids constitute a scaffold for mycobacterial lipid antigens stimulating CD1-restricted T cells. Chemist biol 16:82–92
Lazzarini A, Macchiarulo A, Floridi A, Coletti A, Cataldi S, Codini M, Lazzarini R, Bartoccini E, Cascianelli G, Ambesi-Impiombato FS, Beccari T, Curcio F, Albi E (2015) Very-long-chain fatty acid sphingomyelin in nuclear lipid microdomains of hepatocytes and hepatoma cells: can the exchange from C24:0 to C16:0 affect signal proteins and vitamin D receptor? Mol Biol Cell 26:2418–2425
Leon L, Tatituri RV, Grenha R, Sun Y, Barral DC, Minnaard AJ, Bhowruth V, Veerapen N, Besra GS, Kasmar A, Peng W, Moody DB, Grabowski GA, Brenner MB (2012) Saposins utilize two strategies for lipid transfer and CD1 antigen presentation. Proc Natl Acad Sci U S A 109:4357–4364
Lepore M, de Lalla C, Gundimeda SR, Gsellinger H, Consonni M, Garavaglia C, Sansano S, Piccolo F, Scelfo A, Haussinger D, Montagna D, Locatelli F, Bonini C, Bondanza A, Forcina A, Li Z, Ni G, Ciceri F, Jeno P, Xia C, Mori L, Dellabona P, Casorati G, De Libero G (2014) A novel self-lipid antigen targets human T cells against CD1c(+) leukemias. J Exp Med 211:1363–1377
Li Y, Girardi E, Wang J, Yu ED, Painter GF, Kronenberg M, Zajonc DM (2010) The Valpha14 invariant natural killer T cell TCR forces microbial glycolipids and CD1d into a conserved binding mode. J Exp Med 207:2383–2393
Luoma AM, Castro CD, Mayassi T, Bembinster LA, Bai L, Picard D, Anderson B, Scharf L, Kung JE, Sibener LV, Savage PB, Jabri B, Bendelac A, Adams EJ (2013) Crystal structure of Vdelta1 T cell receptor in complex with CD1d-sulfatide shows MHC-like recognition of a self-lipid by human gammadelta T cells. Immunity 39:1032–1042
Ly D, Moody DB (2014) The CD1 size problem: lipid antigens, ligands, and scaffolds. Cell Mol Life Sci 71:3069–3079
Ly D, Kasmar AG, Cheng TY, de Jong A, Huang S, Roy S, Bhatt A, van Summeren RP, Altman JD, Jacobs WR Jr, Adams EJ, Minnaard AJ, Porcelli SA, Moody DB (2013) CD1c tetramers detect ex vivo T cell responses to processed phosphomycoketide antigens. J Exp Med 210:729–741
Lynch L, Nowak M, Varghese B, Clark J, Hogan AE, Toxavidis V, Balk SP, O’Shea D, O’Farrelly C, Exley MA (2012) Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity 37:574–587
Lynch L, Michelet X, Zhang S, Brennan PJ, Moseman A, Lester C, Besra G, Vomhof-Dekrey EE, Tighe M, Koay HF, Godfrey DI, Leadbetter EA, Sant’Angelo DB, von Andrian U, Brenner MB (2014) Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T cells and macrophages in adipose tissue. Nat Immunol 16(1):85–95
Macho-Fernandez E, Brigl M (2015) The extended family of CD1d-restricted NKT cells: sifting through a mixed bag of TCRs, antigens, and functions. Front Immunol 6:362
Madden DR, Gorga JC, Strominger JL, Wiley DC (1991) The structure of HLA-B27 reveals nonamer self-peptides bound in an extended conformation. Nature 353:321–325
Mallevaey T, Clarke AJ, Scott-Browne JP, Young MH, Roisman LC, Pellicci DG, Patel O, Vivian JP, Matsuda JL, McCluskey J, Godfrey DI, Marrack P, Rossjohn J, Gapin L (2011) A molecular basis for NKT cell recognition of CD1d-self-antigen. Immunity 34:315–326
Manolova V, Kistowska M, Paoletti S, Baltariu GM, Bausinger H, Hanau D, Mori L, De Libero G (2006) Functional CD1a is stabilized by exogenous lipids. Eur J Immunol 36:1083–1092
Mansour S, Tocheva AS, Cave-Ayland C, Machelett MM, Sander B, Lissin NM, Molloy PE, Baird MS, Stubs G, Schroder NW, Schumann RR, Rademann J, Postle AD, Jakobsen BK, Marshall BG, Gosain R, Elkington PT, Elliott T, Skylaris CK, Essex JW, Tews I, Gadola SD (2016) Cholesteryl esters stabilize human CD1c conformations for recognition by self-reactive T cells. Proc Natl Acad Sci U S A 113:E1266–E1275
Maricic I, Sheng H, Marrero I, Seki E, Kisseleva T, Chaturvedi S, Molle N, Mathews SA, Gao B, Kumar V (2015) Inhibition of type I natural killer T cells by retinoids or following sulfatide-mediated activation of type II natural killer T cells attenuates alcoholic liver disease in mice. Hepatology 61:1357–1369
Martin E, Treiner E, Duban L, Guerri L, Laude H, Toly C, Premel V, Devys A, Moura IC, Tilloy F, Cherif S, Vera G, Latour S, Soudais C, Lantz O (2009) Stepwise development of MAIT cells in mouse and human. PLoS Biol 7:e54
Mathews S, Feng D, Maricic I, Ju C, Kumar V, Gao B (2016) Invariant natural killer T cells contribute to chronic-plus-binge ethanol-mediated liver injury by promoting hepatic neutrophil infiltration. Cell Mol Immunol 13:206–216
Mattner J, Debord KL, Ismail N, Goff RD, Cantu C 3rd, Zhou D, Saint-Mezard P, Wang V, Gao Y, Yin N, Hoebe K, Schneewind O, Walker D, Beutler B, Teyton L, Savage PB, Bendelac A (2005) Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature 434:525–529
McCarthy C, Shepherd D, Fleire S, Stronge VS, Koch M, Illarionov PA, Bossi G, Salio M, Denkberg G, Reddington F, Tarlton A, Reddy BG, Schmidt RR, Reiter Y, Griffiths GM, van der Merwe PA, Besra GS, Jones EY, Batista FD, Cerundolo V (2007) The length of lipids bound to human CD1d molecules modulates the affinity of NKT cell TCR and the threshold of NKT cell activation. J Exp Med 204:1131–1144
Miley MJ, Messaoudi I, Metzner BM, Wu Y, Nikolich-Zugich J, Fremont DH (2004) Structural basis for the restoration of TCR recognition of an MHC allelic variant by peptide secondary anchor substitution. J Exp Med 200:1445–1454
Mohan JF, Unanue ER (2012) Unconventional recognition of peptides by T cells and the implications for autoimmunity. Nat Rev Immunol 12:721–728
Moody DB, Porcelli SA (2003) Intracellular pathways of CD1 antigen presentation. Nat Rev Immunol 3:11–22
Moody DB, Reinhold BB, Guy MR, Beckman EM, Frederique DE, Furlong ST, Ye S, Reinhold VN, Sieling PA, Modlin RL, Besra GS, Porcelli SA (1997) Structural requirements for glycolipid antigen recognition by CD1b-restricted T cells. Science 278:283–286
Moody DB, Ulrichs T, Muhlecker W, Young DC, Gurcha SS, Grant E, Rosat JP, Brenner MB, Costello CE, Besra GS, Porcelli SA (2000) CD1c-mediated T-cell recognition of isoprenoid glycolipids in Mycobacterium tuberculosis infection. Nature 404:884–888
Moody DB, Briken V, Cheng TY, Roura-Mir C, Guy MR, Geho DH, Tykocinski ML, Besra GS, Porcelli SA (2002) Lipid length controls antigen entry into endosomal and nonendosomal pathways for CD1b presentation. Nat Immunol 3:435–442
Moody DB, Zajonc DM, Wilson IA (2005) Anatomy of CD1-lipid antigen complexes. Nat Rev Immunol 5:387–399
Moore KJ, Tabas I (2011) Macrophages in the pathogenesis of atherosclerosis. Cell 145:341–355
Nair S, Boddupalli CS, Verma R, Liu J, Yang R, Pastores GM, Mistry PK, Dhodapkar MV (2015) Type II NKT-TFH cells against Gaucher lipids regulate B-cell immunity and inflammation. Blood 125:1256–1271
Neefjes J, Jongsma ML, Paul P, Bakke O (2011) Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 11:823–836
Nikolich-Zugich J, Slifka MK, Messaoudi I (2004) The many important facets of T-cell repertoire diversity. Nat Rev Immunol 4:123–132
Olszak T, Neves JF, Dowds CM, Baker K, Glickman J, Davidson NO, Lin CS, Jobin C, Brand S, Sotlar K, Wada K, Katayama K, Nakajima A, Mizuguchi H, Kawasaki K, Nagata K, Muller W, Snapper SB, Schreiber S, Kaser A, Zeissig S, Blumberg RS (2014) Protective mucosal immunity mediated by epithelial CD1d and IL-10. Nature 509:497–502
Patel O, Kjer-Nielsen L, Le Nours J, Eckle SB, Birkinshaw R, Beddoe T, Corbett AJ, Liu L, Miles JJ, Meehan B, Reantragoon R, Sandoval-Romero ML, Sullivan LC, Brooks AG, Chen Z, Fairlie DP, McCluskey J, Rossjohn J (2013) Recognition of vitamin B metabolites by mucosal-associated invariant T cells. Nat Commun 4:2142
Pearce EJ, Everts B (2015) Dendritic cell metabolism. Nat Rev Immunol 15:18–29
Pellicci DG, Clarke AJ, Patel O, Mallevaey T, Beddoe T, Le Nours J, Uldrich AP, McCluskey J, Besra GS, Porcelli SA, Gapin L, Godfrey DI, Rossjohn J (2011) Recognition of beta-linked self glycolipids mediated by natural killer T cell antigen receptors. Nat Immunol 12:827–833
Porcelli S, Brenner MB, Greenstein JL, Balk SP, Terhorst C, Bleicher PA (1989) Recognition of cluster of differentiation 1 antigens by human CD4-CD8-cytolytic T lymphocytes. Nature 341:447–450
Porcelli S, Yockey CE, Brenner MB, Balk SP (1993) Analysis of T cell antigen receptor (TCR) expression by human peripheral blood CD4-8- alpha/beta T cells demonstrates preferential use of several V beta genes and an invariant TCR alpha chain. J Exp Med 178:1–16
Prigozy TI, Sieling PA, Clemens D, Stewart PL, Behar SM, Porcelli SA, Brenner MB, Modlin RL, Kronenberg M (1997) The mannose receptor delivers lipoglycan antigens to endosomes for presentation to T cells by CD1b molecules. Immunity 6:187–197
Prigozy TI, Naidenko O, Qasba P, Elewaut D, Brossay L, Khurana A, Natori T, Koezuka Y, Kulkarni A, Kronenberg M (2001) Glycolipid antigen processing for presentation by CD1d molecules. Science 291:664–667
Reantragoon R, Corbett AJ, Sakala IG, Gherardin NA, Furness JB, Chen Z, Eckle SB, Uldrich AP, Birkinshaw RW, Patel O, Kostenko L, Meehan B, Kedzierska K, Liu L, Fairlie DP, Hansen TH, Godfrey DI, Rossjohn J, McCluskey J, Kjer-Nielsen L (2013) Antigen-loaded MR1 tetramers define T cell receptor heterogeneity in mucosal-associated invariant T cells. J Exp Med 210:2305–2320
Relloso M, Cheng TY, Im JS, Parisini E, Roura-Mir C, DeBono C, Zajonc DM, Murga LF, Ondrechen MJ, Wilson IA, Porcelli SA, Moody DB (2008) pH-dependent interdomain tethers of CD1b regulate its antigen capture. Immunity 28:774–786
Rossjohn J, Pellicci DG, Patel O, Gapin L, Godfrey DI (2012) Recognition of CD1d-restricted antigens by natural killer T cells. Nat Rev Immunol 12:845–857
Rossjohn J, Gras S, Miles JJ, Turner SJ, Godfrey DI, McCluskey J (2015) T cell antigen receptor recognition of antigen-presenting molecules. Annu Rev Immunol 33:169–200
Roy S, Ly D, Li NS, Altman JD, Piccirilli JA, Moody DB, Adams EJ (2014) Molecular basis of mycobacterial lipid antigen presentation by CD1c and its recognition by alphabeta T cells. Proc Natl Acad Sci U S A 111:E4648–E4657
Roy S, Ly D, Castro CD, Li NS, Hawk AJ, Altman JD, Meredith SC, Piccirilli JA, Moody DB, Adams EJ (2016) Molecular analysis of lipid-reactive Vdelta1 gammadelta T cells identified by CD1c tetramers. J Immunol 196:1933–1942
Salio M, Cerundolo V (2015) Regulation of lipid specific and vitamin specific non-MHC restricted T cells by antigen presenting cells and their therapeutic potentials. Front Immunol 6:388
Salio M, Ghadbane H, Dushek O, Shepherd D, Cypen J, Gileadi U, Aichinger MC, Napolitani G, Qi X, van der Merwe PA, Wojno J, Veerapen N, Cox LR, Besra GS, Yuan W, Cresswell P, Cerundolo V (2013) Saposins modulate human invariant natural killer T cells self-reactivity and facilitate lipid exchange with CD1d molecules during antigen presentation. Proc Natl Acad Sci U S A 110:E4753–E4761
Salio M, Silk JD, Jones EY, Cerundolo V (2014) Biology of CD1- and MR1-restricted T cells. Annu Rev Immunol 32:323–366
Savage AK, Constantinides MG, Han J, Picard D, Martin E, Li B, Lantz O, Bendelac A (2008) The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity 29:391–403
Scharf L, Li NS, Hawk AJ, Garzon D, Zhang T, Fox LM, Kazen AR, Shah S, Haddadian EJ, Gumperz JE, Saghatelian A, Faraldo-Gomez JD, Meredith SC, Piccirilli JA, Adams EJ (2010) The 2.5 a structure of CD1c in complex with a mycobacterial lipid reveals an open groove ideally suited for diverse antigen presentation. Immunity 33:853–862
Schipper HS, Rakhshandehroo M, van de Graaf SF, Venken K, Koppen A, Stienstra R, Prop S, Meerding J, Hamers N, Besra G, Boon L, Nieuwenhuis EE, Elewaut D, Prakken B, Kersten S, Boes M, Kalkhoven E (2012) Natural killer T cells in adipose tissue prevent insulin resistance. J Clin Invest 122:3343–3354
Seino K, Motohashi S, Fujisawa T, Nakayama T, Taniguchi M (2006) Natural killer T cell-mediated antitumor immune responses and their clinical applications. Cancer Sci 97:807–812
Seshadri C, Thuong NT, Yen NT, Bang ND, Chau TT, Thwaites GE, Dunstan SJ, Hawn TR (2014) A polymorphism in human CD1A is associated with susceptibility to tuberculosis. Genes Immun 15:195–198
Shamshiev A, Donda A, Carena I, Mori L, Kappos L, De Libero G (1999) Self glycolipids as T-cell autoantigens. Eur J Immunol 29:1667–1675
Silk JD, Hermans IF, Gileadi U, Chong TW, Shepherd D, Salio M, Mathew B, Schmidt RR, Lunt SJ, Williams KJ, Stratford IJ, Harris AL, Cerundolo V (2004) Utilizing the adjuvant properties of CD1d-dependent NK T cells in T cell-mediated immunotherapy. J Clin Invest 114:1800–1811
Sprengers D, Sille FC, Derkow K, Besra GS, Janssen HL, Schott E, Boes M (2008) Critical role for CD1d-restricted invariant NKT cells in stimulating intrahepatic CD8 T-cell responses to liver antigen. Gastroenterology 134:2132–2143
Stern LJ, Wiley DC (1994) Antigenic peptide binding by class I and class II histocompatibility proteins. Structure 2:245–251
Sugita M, Grant EP, van Donselaar E, Hsu VW, Rogers RA, Peters PJ, Brenner MB (1999) Separate pathways for antigen presentation by CD1 molecules. Immunity 11:743–752
Sugita M, van Der Wel N, Rogers RA, Peters PJ, Brenner MB (2000) CD1c molecules broadly survey the endocytic system. Proc Natl Acad Sci U S A 97:8445–8450
Sugita M, Cao X, Watts GF, Rogers RA, Bonifacino JS, Brenner MB (2002) Failure of trafficking and antigen presentation by CD1 in AP-3-deficient cells. Immunity 16:697–706
Sullivan LC, Hoare HL, McCluskey J, Rossjohn J, Brooks AG (2006) A structural perspective on MHC class Ib molecules in adaptive immunity. Trends Immunol 27:413–420
Suri A, Lovitch SB, Unanue ER (2006) The wide diversity and complexity of peptides bound to class II MHC molecules. Curr Opin Immunol 18:70–77
Takeda K, Hayakawa Y, Van Kaer L, Matsuda H, Yagita H, Okumura K (2000) Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc Natl Acad Sci U S A 97:5498–5503
Taniguchi M, Harada M, Dashtsoodol N, Kojo S (2015) Discovery of NKT cells and development of NKT cell-targeted anti-tumor immunotherapy. Proc Jpn Acad Ser B Phys Biol Sci 91:292–304
Tatituri RV, Watts GF, Bhowruth V, Barton N, Rothchild A, Hsu FF, Almeida CF, Cox LR, Eggeling L, Cardell S, Rossjohn J, Godfrey DI, Behar SM, Besra GS, Brenner MB, Brigl M (2013) Recognition of microbial and mammalian phospholipid antigens by NKT cells with diverse TCRs. Proc Natl Acad Sci U S A 110:1827–1832
Telesford KM, Yan W, Ochoa-Reparaz J, Pant A, Kircher C, Christy MA, Begum-Haque S, Kasper DL, Kasper LH (2015) A commensal symbiotic factor derived from Bacteroides fragilis promotes human CD39(+)Foxp3(+) T cells and Treg function. Gut Microbes 6:234–242
Thomas SY, Lilly CM, Luster AD (2006) Invariant natural killer T cells in bronchial asthma. N Engl J Med 354:2613–2616, author reply 2613–6
Thomas SY, Chyung YH, Luster AD (2010) Natural killer T cells are not the predominant T cell in asthma and likely modulate, not cause, asthma. J Allergy Clin Immunol 125:980–984
Tilloy F, Treiner E, Park SH, Garcia C, Lemonnier F, de la Salle H, Bendelac A, Bonneville M, Lantz O (1999) An invariant T cell receptor alpha chain defines a novel TAP-independent major histocompatibility complex class Ib-restricted alpha/beta T cell subpopulation in mammals. J Exp Med 189:1907–1921
Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, Affaticati P, Gilfillan S, Lantz O (2003) Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature 422:164–169
Trowsdale J, Knight JC (2013) Major histocompatibility complex genomics and human disease. Annu Rev Genomics Hum Genet 14:301–323
Uldrich AP, Patel O, Cameron G, Pellicci DG, Day EB, Sullivan LC, Kyparissoudis K, Kjer-Nielsen L, Vivian JP, Cao B, Brooks AG, Williams SJ, Illarionov P, Besra GS, Turner SJ, Porcelli SA, McCluskey J, Smyth MJ, Rossjohn J, Godfrey DI (2011) A semi-invariant V(alpha)10(+) T cell antigen receptor defines a population of natural killer T cells with distinct glycolipid antigen-recognition properties. Nat Immunol 12:616–623
Uldrich AP, Le Nours J, Pellicci DG, Gherardin NA, McPherson KG, Lim RT, Patel O, Beddoe T, Gras S, Rossjohn J, Godfrey DI (2013) CD1d-lipid antigen recognition by the gammadelta TCR. Nat Immunol 14:1137–1145
van den Elzen P, Garg S, Leon L, Brigl M, Leadbetter EA, Gumperz JE, Dascher CC, Cheng TY, Sacks FM, Illarionov PA, Besra GS, Kent SC, Moody DB, Brenner MB (2005) Apolipoprotein-mediated pathways of lipid antigen presentation. Nature 437:906–910
Van Rhijn I, Moody DB (2015) Donor unrestricted T cells: a shared human T cell response. J Immunol 195:1927–1932
Van Rhijn I, Kasmar A, de Jong A, Gras S, Bhati M, Doorenspleet ME, de Vries N, Godfrey DI, Altman JD, de Jager W, Rossjohn J, Moody DB (2013) A conserved human T cell population targets mycobacterial antigens presented by CD1b. Nat Immunol 14:706–713
Van Rhijn I, Gherardin NA, Kasmar A, de Jager W, Pellicci DG, Kostenko L, Tan LL, Bhati M, Gras S, Godfrey DI, Rossjohn J, Moody DB (2014) TCR bias and affinity define two compartments of the CD1b-glycolipid-specific T Cell repertoire. J Immunol 192:4054–4060
Van Rhijn I, Godfrey DI, Rossjohn J, Moody DB (2015a) Lipid and small-molecule display by CD1 and MR1. Nat Rev Immunol 15:643–654
Van Rhijn I, van Berlo T, Hilmenyuk T, Cheng TY, Wolf BJ, Tatituri RV, Uldrich AP, Napolitani G, Cerundolo V, Altman JD, Willemsen P, Huang S, Rossjohn J, Besra GS, Brenner MB, Godfrey DI, Moody DB (2015b) Human autoreactive T cells recognize CD1b and phospholipids. Proc Natl Acad Sci U S A 113(2):380–385
Venturi V, Price DA, Douek DC, Davenport MP (2008) The molecular basis for public T-cell responses? Nat Rev Immunol 8:231–238
Viale R, Ware R, Maricic I, Chaturvedi V, Kumar V (2012) NKT cell subsets can exert opposing effects in autoimmunity, tumor surveillance and inflammation. Curr Immunol Rev 8:287–296
Wang J, Li Y, Kinjo Y, Mac TT, Gibson D, Painter GF, Kronenberg M, Zajonc DM (2010) Lipid binding orientation within CD1d affects recognition of Borrelia burgorferi antigens by NKT cells. Proc Natl Acad Sci U S A 107:1535–1540
Wieland Brown LC, Penaranda C, Kashyap PC, Williams BB, Clardy J, Kronenberg M, Sonnenburg JL, Comstock LE, Bluestone JA, Fischbach MA (2013) Production of alpha-galactosylceramide by a prominent member of the human gut microbiota. PLoS Biol 11:e1001610
Winau F, Schwierzeck V, Hurwitz R, Remmel N, Sieling PA, Modlin RL, Porcelli SA, Brinkmann V, Sugita M, Sandhoff K, Kaufmann SH, Schaible UE (2004) Saposin C is required for lipid presentation by human CD1b. Nat Immunol 5:169–174
Wu D, Zajonc DM, Fujio M, Sullivan BA, Kinjo Y, Kronenberg M, Wilson IA, Wong CH (2006) Design of natural killer T cell activators: structure and function of a microbial glycosphingolipid bound to mouse CD1d. Proc Natl Acad Sci U S A 103:3972–3977
Ye L, Wang X, Wang S, Wang Y, Song L, Hou W, Zhou L, Li H, Ho W (2009) CD56+ T cells inhibit hepatitis C virus replication in human hepatocytes. Hepatology 49:753–762
Yuan W, Kang SJ, Evans JE, Cresswell P (2009) Natural lipid ligands associated with human CD1d targeted to different subcellular compartments. J Immunol 182:4784–4791
Zajonc DM, Girardi E (2015) Recognition of microbial glycolipids by natural killer T cells. Front Immunol 6:400
Zajonc DM, Elsliger MA, Teyton L, Wilson IA (2003) Crystal structure of CD1a in complex with a sulfatide self antigen at a resolution of 2.15 A. Nat Immunol 4:808–815
Zajonc DM, Crispin MD, Bowden TA, Young DC, Cheng TY, Hu J, Costello CE, Rudd PM, Dwek RA, Miller MJ, Brenner MB, Moody DB, Wilson IA (2005a) Molecular mechanism of lipopeptide presentation by CD1a. Immunity 22:209–219
Zajonc DM, Maricic I, Wu D, Halder R, Roy K, Wong CH, Kumar V, Wilson IA (2005b) Structural basis for CD1d presentation of a sulfatide derived from myelin and its implications for autoimmunity. J Exp Med 202:1517–1526
Zeissig S, Dougan SK, Barral DC, Junker Y, Chen Z, Kaser A, Ho M, Mandel H, McIntyre A, Kennedy SM, Painter GF, Veerapen N, Besra GS, Cerundolo V, Yue S, Beladi S, Behar SM, Chen X, Gumperz JE, Breckpot K, Raper A, Baer A, Exley MA, Hegele RA, Cuchel M, Rader DJ, Davidson NO, Blumberg RS (2010) Primary deficiency of microsomal triglyceride transfer protein in human abetalipoproteinemia is associated with loss of CD1 function. J Clin Invest 120:2889–2899
Zeissig S, Murata K, Sweet L, Publicover J, Hu Z, Kaser A, Bosse E, Iqbal J, Hussain MM, Balschun K, Rocken C, Arlt A, Gunther R, Hampe J, Schreiber S, Baron JL, Moody DB, Liang TJ, Blumberg RS (2012) Hepatitis B virus-induced lipid alterations contribute to natural killer T cell-dependent protective immunity. Nat Med 18:1060–1068
Zeng Z, Castano AR, Segelke BW, Stura EA, Peterson PA, Wilson IA (1997) Crystal structure of mouse CD1: an MHC-like fold with a large hydrophobic binding groove. Science 277:339–345
Zhang G, Nie H, Yang J, Ding X, Huang Y, Yu H, Li R, Yuan Z, Hu S (2011) Sulfatide-activated type II NKT cells prevent allergic airway inflammation by inhibiting type I NKT cell function in a mouse model of asthma. Am J Physiol Lung Cell Mol Physiol 301:L975–L984
Zhao J, Weng X, Bagchi S, Wang CR (2014) Polyclonal type II natural killer T cells require PLZF and SAP for their development and contribute to CpG-mediated antitumor response. Proc Natl Acad Sci U S A 111:2674–2679
Zhao J, Siddiqui S, Shang S, Bian Y, Bagchi S, He Y, Wang CR (2015) Mycolic acid-specific T cells protect against Mycobacterium tuberculosis infection in a humanized transgenic mouse model. Elife 4:e08525
Zhou D, Cantu C 3rd, Sagiv Y, Schrantz N, Kulkarni AB, Qi X, Mahuran DJ, Morales CR, Grabowski GA, Benlagha K, Savage P, Bendelac A, Teyton L (2004a) Editing of CD1d-bound lipid antigens by endosomal lipid transfer proteins. Science 303:523–527
Zhou D, Mattner J, Cantu C 3rd, Schrantz N, Yin N, Gao Y, Sagiv Y, Hudspeth K, Wu YP, Yamashita T, Teneberg S, Wang D, Proia RL, Levery SB, Savage PB, Teyton L, Bendelac A (2004b) Lysosomal glycosphingolipid recognition by NKT cells. Science 306:1786–1789
Zinkernagel RM, Doherty PC (1974) Immunological surveillance against altered self components by sensitised T lymphocytes in lymphocytic choriomeningitis. Nature 251:547–548
Acknowledgments
This work is supported by the Bill and Melinda Gates Foundation, the Tuberculosis Research Unit Network (TBRU-N) (Grants AI049313, AI111224, AR048632 and AI115358). The authors are grateful for the discussions of the donor-unrestricted T cell working group of the Consortium for Tuberculosis Vaccine Development.
Author information
Authors and Affiliations
Corresponding authors
Additional information
This article is published in the Special Issue CD1, MR1, NKT, and MAIT: Evolution and Origins of Non-peptidic Antigen Recognition by T lymphocytes with Guest Editor Dr. Dirk Zajonc.
Rights and permissions
About this article

Cite this article
Huang, S., Moody, D.B. Donor-unrestricted T cells in the human CD1 system. Immunogenetics 68, 577–596 (2016). https://doi.org/10.1007/s00251-016-0942-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-016-0942-x