
Abstract
To further refine and improve biomedical research in rhesus macaques, it is necessary to increase our knowledge concerning both the degree of allelic variation (polymorphism) and diversity (gene copy number variation) in the killer cell immunoglobulin-like receptor (KIR) gene cluster. Pedigreed animals in particular should be studied, as segregation data will provide clues to the linkage of particular KIR genes/alleles segregating on a haplotype and to its gene content as well. A dual strategy allowed us to screen the presence and absence of genes and the corresponding transcripts, as well as to track differences in transcription levels. On the basis of this approach, 14 diverse KIR haplotypes have been described. These haplotypes consist of multiple inhibitory and activating Mamu-KIR genes, and any gene present on one haplotype may be absent on another. This suggests that the cost of accelerated evolution by recombination may be the loss of certain framework genes on a haplotype.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rhesus macaques (Macaca mulatta) are often used as animal models for human disease or biology: for instance, with regard to infectious diseases, autoimmunity and transplantation (Knechtle and Burlingham 2004; Bontrop and Watkins 2005; Vierboom et al. 2005). Therefore, it is necessary to refine the existing knowledge with regard to the immunogenetic factors that can influence either disease or pathology. Natural killer (NK) cells are sentinels of the immune system, influencing both innate and adaptive immune responses, and thus may play a key role in the susceptibility to and outcome of disease (Moretta et al. 2002). A group of leucocyte receptors that can determine the activation state of NK cells is the killer cell immunoglobulin-like receptors (KIR; Vilches and Parham 2002). KIR are cell surface molecules of the immunoglobulin superfamily, and their protein name is based on the number of extracellular domains, which can vary between one and three (1D–3D), and the length of their intracellular tail, which can be either long (L) or short (S) (Marsh et al. 2003; Kelley et al. 2005). Generally, the interaction between KIR and their respective ligands can have an activating or inhibitory influence on the cell, depending on the balance of these signals (Lanier 1998).
The ligands for KIR are the cell surface proteins encoded by the major histocompatibility complex (MHC) class I genes. Since these latter molecules are important for the presentation of degraded pathogen particles (intracellular peptides) to the immune system, there is an ongoing struggle involving pathogens that are trying to escape recognition by the MHC class I presentation pathway (Vossen et al. 2002). This persistent selective pressure on the part of pathogens may be one of the reasons why the MHC gene family is highly polymorphic. Since MHC and KIR interactions are entwined, these two gene systems must have had a considerable impact upon each other during evolution (Parham 2005; Single et al. 2007). Similar levels of polymorphism for the KIR region mirror the degree of polymorphism observed at the human leucocyte antigen (HLA)-A, -B and -C genes. However, whereas extensive diversity (gene copy number variation) is a hallmark of the KIR system in humans, the number of HLA class I genes is fixed in the population. In contrast, the classical MHC class I genes in rhesus macaques, Mamu-A and -B, have experienced extensive duplications (Otting et al. 2007; Karl et al. 2008; Otting et al. 2008). This aspect of gene copy number variation in concert with polymorphism gives rise to an unparalleled complexity of the MHC class I system in macaques. At present, it is not known whether this has any impact on the KIR gene complex in macaques. Moreover, some Mamu-A and -B genes are characterised by high (majors) or low (minors) expression levels, respectively (Otting et al. 2005). The extent to which this quantitative feature will influence their capacity as immune response genes is not yet understood.
Since a KIR haplotype can contain multiple genes encoding for activating or inhibitory proteins and these can be gained or lost by recombination events, there seems to be an enormous amount of plasticity in the overall KIR repertoire. Despite this plasticity, some loci are present on most haplotypes and are considered to be framework genes. Many studies have reported on the haplotype composition of KIR genes encountered within different human populations (Martin et al. 2008). This has increased substantially the number of known region configurations but has also shown that haplotype repertoires may vary between populations (Witt et al. 1999; Rajalingam et al. 2008). Since particular alleles may be linked to specific haplotypes, which are undoubtedly subject to different modes of natural selection, these alleles may be enriched in a certain population yet be absent in another.
In non-human primates, KIR genes have been described for great apes, as well as for Old and New World monkeys (Khakoo et al. 2000; Hershberger et al. 2001; Rajalingam et al. 2004; Blokhuis et al. 2009b; Cadavid and Lun 2009). Comparative genetic analysis has revealed that the mechanisms responsible for expansion and contraction of the KIR region are most likely recombination events. These events can lead to the birth of new genes with a hybrid character or to the shuffling of particular exons. Further allelic variation can be added by occurrence of point mutations. Point mutations can lead to single amino acid changes or may have more drastic effects if the polymorphism maps to a splice site, which can result in a change in the overall domain composition of a protein (Blokhuis et al. 2009a). A recent example of co-evolution of MHC and KIR genes was documented for orangutans, and it was speculated that KIR with two extracellular domains evolved together with MHC-C orthologues (Guethlein et al. 2007).
Although certain individual Mamu-KIR sequences have been described, to date, information is limited with regard to alleles in the context of neighbouring ones. Studies on rhesus macaque KIR have resulted in the availability of some genotypes, but only one complete haplotype has been published (Sambrook et al. 2005). One reason for the lack of reports on haplotypes may be that it is necessary to have access to a pedigreed breeding colony to perform segregation analyses. Moreover, the same tools that are applied for fast and accurate KIR genotyping in humans do not yet exist for use with rhesus macaques. Ideally, to refine the use of rhesus macaques in biomedical research, it is required to characterise breeding colonies and to track the segregation of KIR haplotypes with speedy methods. In this manner, it will be possible to begin exploring the controlled effect of different KIR and MHC interactions in the context of, for instance, infectious diseases (Khakoo and Carrington 2006). To start this process, multiple families from a breeding colony of Indian rhesus macaques, which are genotyped for their MHC class I and II repertoire, were now analysed for KIR gene polymorphism and diversity.
Materials and methods
Animals and cells
For the purpose of this particular study, four families of pedigreed animals from the BPRC self-sustaining colony were analysed. Whole blood of 19 rhesus macaques was obtained during regular health checks, and peripheral blood mononuclear cells (PBMC) were isolated as described previously (Otting and Bontrop 1993). All animals had been haplotyped by different methods for their Mamu-A, -B and -DR allelic repertoire (Bontrop et al. 1995; Otting et al. 2005; Doxiadis et al. 2006).
DNA/RNA extraction, cDNA synthesis and amplification
Genomic DNA was extracted from heparinised blood samples or immortalised rhesus macaque B cell lines according to standardised protocols (Slierendregt et al. 1992). Total RNA was isolated from 5 × 106 PBMC with Trizol reagent (Invitrogen, Paisley, Scotland), according to the manufacturer’s instructions. First-strand cDNA synthesis was conducted based on an oligo(dT)18 primer and RevertAid H Minus M-MulV Reverse Transcriptase (Fermentas, St. Leon-Rot, Germany). Polymerase chain reaction (PCR) was performed with different primers specific for genomic KIR intron 4 and exon 5 and cDNA of Mamu-KIR2DL04, -KIR3DL and -KIR3DS (Table 1) synthesised by Invitrogen (Paisley, Scotland), using Phusion Hot Start High-Fidelity DNA Polymerase (Finnzymes, Espoo, Finland), according to the manufacturer’s instructions.
Cloning and sequencing
PCR products were subjected to gel electrophoresis, and bands of appropriate size were purified with a QIAquick PCR Purification Kit (Qiagen, Valencia, USA). Products were ligated in the cloning site of the Pjet1.2/blunt vector (Fermentas, St. Leon-Rot, Germany) and were used to transform XL1Blue Escherichia coli cells. A minimum of 48 clones were examined per PCR reaction. Plasmid was isolated and sequenced with forward and reverse primers supplied by the manufacturer, on a 3130XL ABI automatic sequencer (Applied Biosystems, Foster City, USA). Because KIR paralogues may be highly similar, recombination can occur frequently, potentially resulting in the formation of in vitro artefacts. Although a high-fidelity enzyme was used, polymerase-induced errors, represented as single nucleotide polymorphisms, were indeed observed. The reported sequences went through a stringent process of quality control; only sequences that were found to segregate or were obtained from more than two independent PCR reactions are reported, which might have resulted in an underestimation of their actual number.
Sequence alignments, in silico analysis and nomenclature
Sequences were aligned with the Seqman program (Dnastar, Inc., Madison, USA). Phylogenetic trees were drawn using the MEGA4.0 program (Tamura et al. 2007). The JTT algorithm (Jones et al. 1992) was used to construct a distance model for amino acid substitution. The neighbour-joining method (Saitou and Nei 1987) was used to build a phylogenetic tree, and a bootstrap reliability test was run with 1,000 iterations. Sequences were submitted to Genbank and given the unique accession numbers GU112255–GU12332 and GU134802. Formal names have been assigned, and annotated sequences will be available through the immunopolymorphism database (www.ebi.ac.uk/ipd; Robinson et al. 2009).
Results and discussion
Mamu-KIR genotypes: correlation between transcripts and genomic sequences
Based on the cloning and sequencing of full-length KIR transcripts, genotype profiles were constructed (Fig. 1). As can be seen, genotypes are characterised by different combinations of activating and inhibitory Mamu-KIR. Moreover, transcript information correlates with the presence of partial genomic sequences as revealed by scanning for the presence of exon 5. However, exceptions emerged. In animal 1UE, five additional segregating exon 5 sequences were only observed by genomic analysis. These sequences match with an inhibitory Mamu-KIR3DL20, -KIR3DL02 and -KIR3DL07 gene, respectively. In animal 98016, two genomic exon 5 segments were observed that are homologous to an inhibitory Mamu-KIR3DL07 and an activating -KIR3DSW09 gene, respectively (Fig. 1). Nevertheless, the corresponding transcripts were not detected. Since KIR genes in humans are known to display quantitative transcription level differences, it could be that some transcripts in rhesus monkeys are present at a low frequency as well and fall under the threshold of detection. Alternatively, these genes could be pseudogenes, or epigenetic factors may have silenced their transcription.

Genotypic composition of a panel of Indian rhesus macaques. On the top, Mamu-KIR genes are displayed. Note that the order, in which genes are depicted for KIR3DL20, KIR1D and KIR2DL04, is based on the physical map. However, the copies of KIR3DL and KIR3DS are simply positioned numerically, since the order in which loci are positioned on a haplotype, is still unknown. Inhibitory KIR have been depicted in blue whereas activating ones are shown in red. KIR genes, for which only genomic exon 5 sequences are detected, are marked as gen and highlighted in red. KIR alleles, of which the observed transcript could be a splice variant, are indicated as sv, whereas a transcript that might be a recombinant is marked as rec
KIR genotypes: presence and absence of genes
Within the present panel of 19 animals, covering four families, a total of 70 different KIR sequences were observed, 49 of which have not been reported before in the scientific literature. The other 21 full-length KIR sequences have already been described, and thus, they verify the existence of a particular allele, although the relevant sequence information is often extended (Table 2).
The first KIR genotypes for rhesus macaques were deduced by a research team that studied five unrelated animals. In this case, the maximum number of KIR on one genotype was estimated to be five (Hershberger et al. 2001). Our genotyping data revealed that the number of KIR genes detected might vary substantially (Fig. 1). Overall, between 6 and 13 transcribed KIR genes were observed in any one animal, suggesting the presence of copy number variation.
Phylogenetic analysis and allelic variation of KIR genes
Phylogenetic analysis of full-length sequences of both activating KIR3DS and inhibitory 3DL alleles reveals extensive variation (Fig. 2). Based on the depth of branches and bootstrap values, the phylogenetic trees allow the definition of lineages for the KIR3DL and 3DS genes, and these clusters have been indicated. It is realised that the robustness of such clustering will become more definitive when more alleles become available. At this stage, the distinction between a locus/lineage cannot be made, and it was decided to introduce a “W” designation to define its workshop status, a tradition that is also used in the HLA field (Marsh et al. 2005). For instance, the Mamu-KIR3DLW09 lineage, which comprises *003 and *004 (bold, Fig. 2), for which at this stage it is unclear whether these represent alleles of one locus or define separate loci. Alternatively, the lineage that comprises GU112327 and GU112317 has been designated Mamu-KIR3DL20 because there is strong phylogenetic evidence and genomic data supporting the existence of a separate locus. Sequence analysis revealed that the sequence GU112302 (rec, Fig. 2a) is a recombinant between exons 1–7 of Mamu-KIR3DS05 and exons 8–9 of Mamu-KIR2DL04. It is for this reason that this sequence stands out within the tree of inhibitory KIR. A combined phylogenetic tree of all the Ig-like domain sequences revealed no new lineages. However, it did show that activating and inhibitory lineages cluster together instead of separate, which suggests that some of these lineages are closely related, for instance, Mamu-KIR3DLW03 and Mamu-KIR3DS05.

Phylogenetic trees of KIR3DL (a) and -3DS (b) genes depicting evolutionary distances. The scale bar represents amino acid substitutions per site. The JTT matrix method was used to calculate evolutionary distances. The neighbour-joining method was used to construct the phylogenetic tree. Bootstraps were calculated with 1,000 iterations, and confidence values above 50% are depicted. Genbank accession numbers are depicted of most reported macaque KIR sequences. The sequences in bold with an allele designation represent those observed in this study. Based on the clustering of sequences, KIR lineages have been indicated next to the alleles they comprise
The number of transcribed KIR3DL and 3DS copies can vary substantially in one animal; however, some lineages appear to be more common. For instance, Mamu-KIR3DL01 alleles were observed in 16 out of 19 animals, while members of the lineages Mamu-KIR3DL08 and -3DS02 were reported in 10 and 11 animals, respectively. The current sample size might well be too small to give these numbers any significance, but the observed trend may be a result of recent selective pressures, ensuring the presence of these lineages in at least part of the population. In combination with allelic polymorphism, this will result in a degree of genotypic complexity that may rival the situation observed in humans (Yawata et al. 2002).
In the four macaque families, 12 alleles were observed that cluster together within the KIR3DL20 lineage (Fig. 2). On the one haplotype that has been published (Sambrook et al. 2005), this locus is positioned most centromeric. This locus appears to be an orthologue of human KIR3DL3, which is considered a framework gene of the KIR gene cluster (Wilson et al. 2000). The gene transcript was observed in all animals except for 1UE, which would suggest that haplotypes exist where Mamu-KIR3DL20 is not expressed or that lack this locus. No genotypes were observed with more than two copies of this gene (Fig. 1), although copy number variations of the locus cannot be excluded. However, this locus displays allelic variation (Fig. 2). This report brings the total number of 3DL20 alleles to 15. This amount represents “the tip of the iceberg” as only a limited number of samples has been analysed.
A locus that was also observed on the genomic haplotype that has been sequenced (Sambrook et al. 2005) is that of Mamu-KIR1D. In ten genotypes, transcripts of this gene were observed, and a total of four alleles are reported, three of which are novel, thus doubling the number of reported alleles (Fig. 1).
One KIR gene that is observed in many primates is named KIR2DL4 in humans, of which the orthologue in macaques has been named Mamu-KIR2DL04. A previous study reported extensive Mamu-KIR2DL04 polymorphism in a large cohort of Indian rhesus macaques (Blokhuis et al. 2009b), and in the present set of animals, no unreported 2DL04 alleles were detected. However, genotyping revealed two animals (1UE, R02091) that lack the KIR2DL04 gene, the absence of which was established by combined genomic and transcription analysis. Although KIR2DL04 was thought to be a framework gene in rhesus macaques, the recent analysis of a cohort of Mauritian cynomolgus macaques has also demonstrated the absence of the orthologue in animals of this species (Bimber et al. 2008). Moreover, retrospective analysis demonstrated that 1UE has given successful birth to at least three offspring, suggesting that the presence of KIR2DL04 is not essential for a successful pregnancy. This is similar to humans, with respect to women who have been reported to lack a functional KIR2DL4 gene but have given birth successfully (Gomez-Lozano et al. 2003). One animal (95041) was found to possess three KIR2DL04 sequences, suggesting that the locus has been duplicated on one haplotype. This is a novel observation in macaques but has been described for humans as well (Williams et al. 2003). It was also noted that identical 2DL04 alleles are shared by different genotypes, and as such, most likely are present on different haplotypes. This phenomenon was not observed, in the present panel, for any other gene besides KIR2DL04 and -1D.
Mamu-KIR genes are subject to considerable splicing
Complex splicing events were observed frequently for all KIR gene transcripts except for Mamu-KIR2DL04, where frequency was much lower. Alternatively spliced transcripts usually comprise a complete or partial deletion of one of exons 3, 4, 5, 6 and 7. These splice variants were observed to segregate, for instance, if a parental animal had a deletion of exon 5 in one transcript; a similar transcript would also be observed in the offspring, if the gene were inherited. It is yet unclear what determines the induction of splice variants. One possible explanation is the prevalence of GU-AG donor–acceptor splice motifs: These are highly frequent in the exons of many KIR genes, which could lead to a general proneness to alternative splicing in KIR transcripts. Because these products are shorter than the corresponding full-length transcript, they can be amplified more efficiently, which may result in an increase in the frequency of observed splice products in final cloned products.
Members of the Mamu-KIR3DL20 gene produce splice variants in which exon 4 is missing (Hershberger et al. 2001). The consequence is an expressed protein that would have the characteristic D0-D2 domain structure reminiscent of human KIR2DL4 and -2DL5. Indeed, Mamu-KIR3DL20 genes contain an exon 3 that seems to be orthologous to KIR2DL5, so it is possible that by alternative splicing, a similar gene product is formed in macaques. This would be an example of convergent evolution, as KIR2DL5-like structures are observed in different primate species (Khakoo et al. 2000). At this stage, no ligand or function is known; however, one might speculate from its conservation that the ligand, too, has maintained certain structural characteristics that would enable co-evolution.
The Mamu-KIR1D gene product is also generated by alternative splicing. On a genomic level, KIR1D has three exons that potentially can encode for Ig-like domains. However, much as in human KIR2DL1, 2 and 3, exon 3 is a pseudo exon, so the 1D transcript comprises exons 1–2 and 4–9. Since this gene contains a deletion in exon 5, resulting in a premature stop codon, the corresponding gene product might be soluble. In human, a common allele of KIR2DS4, which has a 22-bp deletion in exon 5, is thought to also encode a soluble product (Maxwell et al. 2002). In animals 95041 and 96077, only an alternatively spliced transcript of the KIR1D gene is observed, resulting in a 1DL conformation which has retained its intracellular tail. No function or ligand is known for these short KIR.
The mosaic of KIR haplotypes: extensive gene copy number variation in rhesus macaques
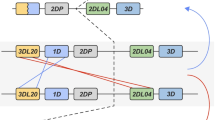
Genotyping in concert with segregation analyses revealed that different combinations of KIR genes segregate in rhesus monkey families according to Mendelian rules (Fig. 3). This approach allowed the definition in detail of 14 of 18 haplotypes that are present in the parental animals. For four haplotypes, marked H-02, -07, -13 and -18, informative segregation profiles were absent (Fig. 3). The minimal gene content could be deduced, since genes that are not present on the haplotype, for which segregation data is available, should be present on the opposite one. However, the gene content of the haplotype could represent an underestimation because homozygosity issues are not resolved by segregation. One should also keep in mind that low-frequency transcripts observed in these animals, which could not be confirmed by segregation analysis, were not reported.

Segregation of KIR haplotypes in a pedigree of four families of Indian rhesus macaques. Depicted are the sires and dames with their offspring, and H-01 to H-18 represent haplotypes. Four haplotypes, H-02, -07, -13 and -18, were not inherited by the offspring
For the other haplotypes, the discrimination power is high, as segregation of genes that differ by single nucleotide polymorphisms could be observed. As such, highly detailed haplotypes could be defined in 14 cases (Fig. 4). On these given haplotypes, the highest number of transcribed genes appears to be eight (H-17) while the average seems to be approximately six for most haplotypes (Fig. 4).

Definition of Mamu-KIR haplotypes H-01 to H-18 based on segregation analysis. On the top axis, the different Mamu-KIR genes are shown, and allele designation are provided in the boxes. Although haplotype gene content is reported, the order of the loci on the chromosome, however, is not known. For haplotypes H-02, -07, -13 and -18 the gene content could not be determined by segregation analysis, and a minimal haplotype is represented. If for one animal it is undeterminable whether an allele is present on both haplotypes or a single one, the allele is represented covering both haplotypes with an interrupted outline. On haplotype H-09, H-10 and H-15, genes are depicted half full because only an exon 5 sequence has been detected on a genomic level. This exon 5 sequence is similar to that of the particular lineage to which it has been appointed
For instance, it was possible to distinguish highly similar Mamu-KIR2DL04 alleles. Nevertheless, interpretation of segregation profiles may be hampered if both parents share the same genes/alleles. For example, this was observed for 2DL04 in animal 95041; since both offspring inherited an identical allele from the father, it was impossible to determine which haplotype comprises that of the mother. To complicate matters further, all family members were genotyped positively for the same alleles of the lineages Mamu-KIR3DSW08 and -3DS05, which means that these genes are present on at least one haplotype in both father and mother. A similar situation occurred in the family of 96077 and 98024, where the same allele of KIR3DS01 was observed in all animals. Finally, an allele of KIR3DS03 was observed in all members of the family with parents 1UE and 8719. Although it is a coincidence that these alleles are shared between parents, many alleles are not shared.
The earlier observation that KIR2DL04 may be absent on a genotype was confirmed at the haplotype level for animals 1UE and R02091. Segregation analyses confirmed that there are also parental animals that have KIR2DL04 present on only one of their haplotypes (96077, 98024, 98016, 94056).
The exact mechanisms are still unclear as to how the expansion and contraction of haplotypes is generated, but it is thought that meiotic recombination events are involved (Norman et al. 2009). Unequal crossing-over in particular is a prominent mechanism that may influence gene copy number variation. This would account for the plasticity observed in the region, and as a consequence, it could very well be that activating and inhibitory KIR loci may be located juxtaposed on a haplotype.
One should realise that the haplotypes described here reflect gene content and not the localisation on the chromosome. Therefore, the terms downstream and upstream are based on the manner in which genes have been represented on the reported haplotypes. If, for instance, the region upstream of KIR2DL04 would recombine with a similar more telomeric one, this could account for a haplotype without KIR2DL04, but with the genes telomeric of 2DL04, like haplotype H-16. As a consequence, another recombinant haplotype would be formed that would contain a duplicated 2DL04 locus as one of haplotypes H-03 or H-04 might possibly posses. In a similar manner, if a recombination occurs upstream of 3DL20 with a region downstream of 2DL04, this would result in the formation of a haplotype that lacks its most telomeric genes: for instance, as observed in haplotypes H-09 and H-10. As can be seen (Fig. 4), some haplotypes share particular segments, for instance, in H-06, H-10, H-11 and H-16, the 3DL01 and 3DL08 genes segregate together. Likewise, 3DS02 and 3DS06 are both present on haplotypes H-04, H-09 and H-17.
In conclusion, it seems likely that unequal crossing-over promotes the generation of mosaic haplotypes and rearrangement of gene content on a regular basis. The reported haplotypes display extensive gene copy number variation and gene polymorphism (Fig. 4). The present approach to obtain detailed alleles and discriminative haplotypes is highly accurate but also time consuming and could be complemented by an approach to track segregation of identified alleles. In the current issue of Immunogenetics, Kruse and co-workers report a rapid typing method to identify the presence or absence of KIR lineage specific markers (Kruse et al. 2010). In this manner, informative genotypes and haplotypes could be detected. Based on expansion of detailed knowledge of allelic variation, additional refinement can be added to sequence-specific typing to make distinctions between highly similar sequences, for instance, alleles *009 and *010 of Mamu-KIR3DS02.
Activating and inhibitory haplotypes in rhesus macaques
In human, a distinction can be made between group A and B haplotypes, the former comprising on average six KIR genes and being more inhibitory than the latter (Uhrberg et al. 1997). In human, nine common B haplotypes contain between seven and eleven KIR genes (Uhrberg et al. 2002). In rhesus macaque, such clear distinction cannot be made. However, some haplotypes seem to comprise at least one inhibitory KIR, except for haplotype H-04 but appears to lack activating KIR (e.g. see haplotypes H-11, H-12 and possibly H-06 (Fig. 4)). In this panel, possibly only H-06 and one or two of H-10, -11 and -12 comprises solely inhibitory KIR genes. Overall, it appears that rhesus macaque haplotypes comprise a combination of activating and inhibitory KIR genes, and these can originate from several lineages. This mosaic pattern is also observed by another research group (Kruse et al. 2010). Possibly because recombination events have taken place, there is no definite trend for a group A or B haplotype. Although it is true that haplotypes without activating KIR are reported, in humans the frequency of group A and B haplotypes is about 50%. It would take a much larger population study to estimate the ratio of A- and B-like haplotypes in rhesus macaques.
KIR3D expansion, restricted haplotype distribution and their impact on MHC repertoire
The present data suggest that the KIR3DS and 3DL families have experienced several rounds of duplications (Fig. 2). Based on segregation studies, the gene content of some KIR haplotypes has been established. As can be seen, these haplotypes comprise different numbers of KIR3DL and 3DS genes, ranging in the order of 0–3 and 0–4, respectively. The observed limit to the amount of genes on one haplotype implicates that there is a restricted haplotype distribution of KIR3DS and 3DL genes with regard to copy number. How can one explain the expansion of KIR3D lineages and the observed mosaic of KIR haplotypes from an evolutionary perspective? At this stage, it is understood that products of the KIR locus may exert different types of functions. On one hand, KIR on NK cells scan changes in the MHC class I expression status of cells. On the other hand, it is thought that some KIR recognise epitopes of pathogens or set apart pathogen derived peptides in the context of MHC class I. Such a complex task is difficult to achieve with one gene. Moreover, copy number variation of KIR3D minimises the chance that particular pathogens may escape immune recognition. However, if all individuals in a population share the same KIR repertoire, they could again become vulnerable to the adaptation of pathogens. As such, haplotypes with differential gene content would provide a selective advantage. The next issue is how haplotypes with differential gene content maintained in the population. Selection by pathogens may enrich or even deplete particular haplotypes, altering gene frequencies in a population. NK cells play also a pivotal role during reproductive biology. Pregnancy can be regarded as a complex model for transplantation as the foetus consists from an immunological point of view 50% of nonself (paternal) material. NK cell activation plays an important role in successful gestation (Trowsdale and Moffett 2008). If this is true, then incompatibility between the KIR gene repertoire of the mother and inherited MHC repertoire of the father is favoured. As a consequence, an overrepresentation of MHC heterozygous individuals in the population can be expected. One can envision that this would in turn affect co-evolution of the KIR gene cluster.
In conclusion, to cope with a highly polymorphic gene system that is variable in both quality and quantity - such as the rhesus macaque MHC class I - a complex class of receptors is required that can detect such differences. This prerequisite is fulfilled by the highly plastic KIR gene system in rhesus macaques which displays copy number variation as well as extensive levels of polymorphism.
References
Bimber BN, Moreland AJ, Wiseman RW, Hughes AL, O’Connor DH (2008) Complete characterization of killer Ig-like receptor (KIR) haplotypes in Mauritian cynomolgus macaques: novel insights into nonhuman primate KIR gene content and organization. J Immunol 181:6301–6308
Blokhuis JH, Doxiadis GG, Bontrop RE (2009a) A splice site mutation converts an inhibitory killer cell Ig-like receptor into an activating one. Mol Immunol 46:640–648
Blokhuis JH, van der Wiel MK, Doxiadis GG, Bontrop RE (2009b) Evidence for balancing selection acting on KIR2DL4 genotypes in rhesus macaques of Indian origin. Immunogenetics 61:503–512
Bontrop RE, Watkins DI (2005) MHC polymorphism: AIDS susceptibility in non-human primates. Trends Immunol 26:227–233
Bontrop RE, Otting N, Slierendregt BL, Lanchbury JS (1995) Evolution of major histocompatibility complex polymorphisms and T-cell receptor diversity in primates. Immunol Rev 143:33–62
Cadavid LF, Lun CM (2009) Lineage-specific diversification of killer cell Ig-like receptors in the owl monkey, a new world primate. Immunogenetics 61:27–41
Doxiadis GG, Rouweler AJ, de Groot NG, Louwerse A, Otting N, Verschoor EJ, Bontrop RE (2006) Extensive sharing of MHC class II alleles between rhesus and cynomolgus macaques. Immunogenetics 58:259–268
Gomez-Lozano N, de Pablo R, Puente S, Vilches C (2003) Recognition of HLA-G by the NK cell receptor KIR2DL4 is not essential for human reproduction. Eur J Immunol 33:639–644
Guethlein LA, Older Aguilar AM, Abi-Rached L, Parham P (2007) Evolution of killer cell Ig-like receptor (KIR) genes: definition of an orangutan KIR haplotype reveals expansion of lineage III KIR associated with the emergence of MHC-C. J Immunol 179:491–504
Hershberger KL, Shyam R, Miura A, Letvin NL (2001) Diversity of the killer cell Ig-like receptors of rhesus monkeys. J Immunol 166:4380–4390
Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8:275–282
Karl JA, Wiseman RW, Campbell KJ, Blasky AJ, Hughes AL, Ferguson B, Read DS, O’Connor DH (2008) Identification of MHC class I sequences in Chinese-origin rhesus macaques. Immunogenetics 60:37–46
Kelley J, Walter L, Trowsdale J (2005) Comparative genomics of natural killer cell receptor gene clusters. PLoS Genet 1:129–139
Khakoo SI, Carrington M (2006) KIR and disease: a model system or system of models? Immunol Rev 214:186–201
Khakoo SI, Rajalingam R, Shum BP, Weidenbach K, Flodin L, Muir DG, Canavez F, Cooper SL, Valiante NM, Lanier LL, Parham P (2000) Rapid evolution of NK cell receptor systems demonstrated by comparison of chimpanzees and humans. Immunity 12:687–698
Knechtle SJ, Burlingham WJ (2004) Metastable tolerance in nonhuman primates and humans. Transplantation 77:936–939
Kruse PH, Rosner C, Walter L (2010) Characterization of rhesus macaque KIR genotypes and haplotypes. Immunogenetics doi:10.1007/s00251-010-0433-4
Lanier LL (1998) NK cell receptors. Annu Rev Immunol 16:359–393
Marsh SG, Parham P, Dupont B, Geraghty DE, Trowsdale J, Middleton D, Vilches C, Carrington M, Witt C, Guethlein LA, Shilling H, Garcia CA, Hsu KC, Wain H (2003) Killer-cell immunoglobulin-like receptor (KIR) nomenclature report, 2002. Immunogenetics 55:220–226
Marsh SG, Albert ED, Bodmer WF, Bontrop RE, Dupont B, Erlich HA, Geraghty DE, Hansen JA, Hurley CK, Mach B, Mayr WR, Parham P, Petersdorf EW, Sasazuki T, Schreuder GM, Strominger JL, Svejgaard A, Terasaki PI, Trowsdale J (2005) Nomenclature for factors of the HLA system, 2004. Hum Immunol 66:571–636
Martin MP, Single RM, Wilson MJ, Trowsdale J, Carrington M (2008) KIR haplotypes defined by segregation analysis in 59 Centre d’Etude Polymorphisme Humain (CEPH) families. Immunogenetics 60:767–774
Maxwell LD, Wallace A, Middleton D, Curran MD (2002) A common KIR2DS4 deletion variant in the human that predicts a soluble KIR molecule analogous to the KIR1D molecule observed in the rhesus monkey. Tissue Antigens 60:254–258
Moretta A, Bottino C, Mingari MC, Biassoni R, Moretta L (2002) What is a natural killer cell? Nat Immunol 3:6–8
Norman PJ, Abi-Rached L, Gendzekhadze K, Hammond JA, Moesta AK, Sharma D, Graef T, McQueen KL, Guethlein LA, Carrington CV, Chandanayingyong D, Chang YH, Crespi C, Saruhan-Direskeneli G, Hameed K, Kamkamidze G, Koram KA, Layrisse Z, Matamoros N, Mila J et al (2009) Meiotic recombination generates rich diversity in NK cell receptor genes, alleles, and haplotypes. Genome Res 19:757–769
Otting N, Bontrop RE (1993) Characterization of the rhesus macaque (Macaca mulatta) equivalent of HLA-F. Immunogenetics 38:141–145
Otting N, Heijmans CM, Noort RC, de Groot NG, Doxiadis GG, van Rood JJ, Watkins DI, Bontrop RE (2005) Unparalleled complexity of the MHC class I region in rhesus macaques. Proc Natl Acad Sci USA 102:1626–1631
Otting N, de Vos-Rouweler AJ, Heijmans CM, de Groot NG, Doxiadis GG, Bontrop RE (2007) MHC class I A region diversity and polymorphism in macaque species. Immunogenetics 59:367–375
Otting N, Heijmans CM, van der Wiel M, de Groot NG, Doxiadis GG, Bontrop RE (2008) A snapshot of the Mamu-B genes and their allelic repertoire in rhesus macaques of Chinese origin. Immunogenetics 60:507–514
Parham P (2005) MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol 5:201–214
Rajalingam R, Parham P, Abi-Rached L (2004) Domain shuffling has been the main mechanism forming new hominoid killer cell Ig-like receptors. J Immunol 172:356–369
Rajalingam R, Du Z, Meenagh A, Luo L, Kavitha VJ, Pavithra-Arulvani R, Vidhyalakshmi A, Sharma SK, Balazs I, Reed EF, Pitchappan RM, Middleton D (2008) Distinct diversity of KIR genes in three southern Indian populations: comparison with world populations revealed a link between KIR gene content and pre-historic human migrations. Immunogenetics 60:207–217
Robinson J, Mistry K, McWilliam H, Lopez R, Marsh SG (2009) IPD—the immuno polymorphism database. Nucleic Acids Res 38:D863–D869
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sambrook JG, Bashirova A, Palmer S, Sims S, Trowsdale J, Abi-Rached L, Parham P, Carrington M, Beck S (2005) Single haplotype analysis demonstrates rapid evolution of the killer immunoglobulin-like receptor (KIR) loci in primates. Genome Res 15:25–35
Single RM, Martin MP, Gao X, Meyer D, Yeager M, Kidd JR, Kidd KK, Carrington M (2007) Global diversity and evidence for coevolution of KIR and HLA. Nat Genet 39:1114–1119
Slierendregt BL, van Noort JT, Bakas RM, Otting N, Jonker M, Bontrop RE (1992) Evolutionary stability of transspecies major histocompatibility complex class II DRB lineages in humans and rhesus monkeys. Hum Immunol 35:29–39
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Trowsdale J, Moffett A (2008) NK receptor interactions with MHC class I molecules in pregnancy. Semin Immunol 20:317–320
Uhrberg M, Valiante NM, Shum BP, Shilling HG, Lienert-Weidenbach K, Corliss B, Tyan D, Lanier LL, Parham P (1997) Human diversity in killer cell inhibitory receptor genes. Immunity 7:753–763
Uhrberg M, Parham P, Wernet P (2002) Definition of gene content for nine common group B haplotypes of the Caucasoid population: KIR haplotypes contain between seven and eleven KIR genes. Immunogenetics 54:221–229
Vierboom MP, Jonker M, Bontrop RE, Hart B (2005) Modeling human arthritic diseases in nonhuman primates. Arthritis Res Ther 7:145–154
Vilches C, Parham P (2002) KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol 20:217–251
Vossen MT, Westerhout EM, Soderberg-Naucler C, Wiertz EJ (2002) Viral immune evasion: a masterpiece of evolution. Immunogenetics 54:527–542
Williams F, Maxwell LD, Halfpenny IA, Meenagh A, Sleator C, Curran MD, Middleton D (2003) Multiple copies of KIR 3DL/S1 and KIR 2DL4 genes identified in a number of individuals. Hum Immunol 64:729–732
Wilson MJ, Torkar M, Haude A, Milne S, Jones T, Sheer D, Beck S, Trowsdale J (2000) Plasticity in the organization and sequences of human KIR/ILT gene families. Proc Natl Acad Sci USA 97:4778–4783
Witt CS, Dewing C, Sayer DC, Uhrberg M, Parham P, Christiansen FT (1999) Population frequencies and putative haplotypes of the killer cell immunoglobulin-like receptor sequences and evidence for recombination. Transplantation 68:1784–1789
Yawata M, Yawata N, Abi-Rached L, Parham P (2002) Variation within the human killer cell immunoglobulin-like receptor (KIR) gene family. Crit Rev Immunol 22:463–482
Acknowledgements
The authors wish to thank Donna Devine for editing the manuscript, and Henk van Westbroek for preparing the figures.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Blokhuis, J.H., van der Wiel, M.K., Doxiadis, G.G.M. et al. The mosaic of KIR haplotypes in rhesus macaques. Immunogenetics 62, 295–306 (2010). https://doi.org/10.1007/s00251-010-0434-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-010-0434-3