
Abstract
A tryptophan-rich octapeptide, C8 (Ac-Trp-Glu-Asp-Trp-Val-Gly-Trp-Ile-NH2), modelled on the membrane-proximal external region of the feline immunodeficiency virus (FIV) gp36 glycoprotein ectodomain, exhibits potent antiviral activity against FIV. A mechanism has been proposed by which the peptide, being positioned on the surface of the cell membrane, inhibits its fusion with the virus. In the present work, peptide–lipid interactions of C8 with dimyristoyl phosphatidylcholine liposomes are investigated using electron spin resonance spectroscopy of spin-labelled lipids. Three other peptides, obtained from modifications of C8, have also been investigated, in an attempt to clarify the essential molecular features of the interactions involving the tryptophan residues. The results show that C8 adsorbs strongly on the bilayer surface. Membrane binding requires not only the presence of the Trp residues in the sequence, but also their common orientation on one side of the peptide that is engendered by the WX2 WX2 W motif. Membrane interaction correlates closely with peptide antiviral activity, indicating that the membrane is essential in stabilizing the peptide conformation that will be able to inhibit viral infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
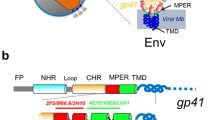
Both the human immunodeficiency virus (HIV) and the feline analogue (FIV) effect cell entry via a mechanism that involves a surface glycoprotein (Pancino et al. 1995; Serres 2000; Frey et al. 2001). These glycoproteins, named gp41 and gp36, respectively, exhibit a common structural architecture consisting of a transmembrane (TM) region and membrane-proximal segment (pre-TM) that is directly involved in the virus-target cell fusion. Furthermore, both proteins present an unusual clustering of tryptophan residues in the membrane-proximal external region (MPER) of the ectodomain of the TM region (Giannecchini et al. 2004; Suarez et al. 2000; Salzwedel et al. 1999). The presence of the MPER domain has been shown to be crucial, through a still unidentified mechanism, in the infection pathway (Barbato et al. 2003).
Peptides deriving from MPER of gp41 and gp36 are able to prevent HIV- and FIV-cell membrane fusion (Jiang et al. 1993; Jin et al. 2000; Wild et al. 1994; Kilby et al. 2002; Massi et al. 1998; Giannecchini et al. 2003). The antiviral activity is likely to derive from adsorption of the peptide on the external surface of the target-cell membrane, where it competes with the TM glycoprotein of the virus (Schibli et al. 2001). In this context, we have recently investigated the interactions between an antiviral peptide derived from the MPER domain of the FIV surface glycoprotein and phospholipid membranes. A 20-mer peptide named P59, corresponding to the 767L-G786 sequence of (TM) gp36, was synthesized and found to exert potent inhibitory activity on FIV replication (Esposito et al. 2006). Subsequently, we investigated interactions of P59 with phospholipid bilayers by electron spin resonance (ESR) experiments, using spin-labelled lipids (D’Errico et al. 2008). The results demonstrate that the P59 peptide associates with lipid bilayers by binding at the membrane surface or polar–apolar interface. This is consistent with high-resolution nuclear magnetic resonance (NMR) analysis of P59 in micellar environments, which revealed a region of secondary structural order in the sequence extending from Trp770 to Ile777 (Giannecchini et al. 2007). In particular, an amphipathic, turn-helical structure forms, in which the polar or charged residues are positioned on one side, with the opposite side occupied by hydrophobic residues including the three tryptophans, which have a propensity to localize to the polar–apolar interface of membranes (White and Wimley 1998; de Planque et al. 1998). This sequence seems to be fundamental in determining the peptide antiviral activity. Indeed, an octapeptide with the 770Trp-Ile777 sequence (named C8 or peptide 1; Table 1) was found to exert an antiviral activity comparable to that of the P59 20-mer (Giannecchini et al. 2003).
In the present work, we investigate the interaction between this C8 octapeptide and phospholipid bilayers, using spin-label ESR and fluorescence spectroscopy. Several analogues of the peptide were also tested, in order to find which structural features drive the interaction with the bilayer. Particularly, substitution of the Trp residues or change of the spacing between them was considered. Our aim is twofold: on one hand, we wish to test the connection between antiviral activity of the peptide and its interaction with the membrane; on the other hand, we wish to investigate more deeply the role played by the MPER domain in fusion of the virus and target-cell membranes.
Materials and methods
Materials
Dichloromethane and methanol, HPLC-grade solvents, were obtained from Merck (Darmstadt, Germany). The phospholipid dimyristoyl phosphatidylcholine (DMPC) was obtained from Avanti Polar Lipids (Birmingham, AL, USA). Spin-labelled phosphatidylcholines (n-PCSL) with the nitroxide group at different positions, n, in the sn-2 acyl chain were synthesized as described by Marsh and Watts (1982; Marsh 1997). The spin-labels were stored at −20°C in ethanol solutions at a concentration of 1 mg/ml.
Peptide design and synthesis
Four peptides were designed, synthesized and tested for membrane interaction. Their amino-acid sequences are shown in Table 1. Peptide 1, also named C8, corresponds to the 770Trp-Ile777 sequence of FIV gp36 with three Trp residues separated by two dipeptide units. The other three peptides have been strategically chosen among a series of C8 mutants whose antiviral activity was tested as reported in a previous study (D’Ursi et al. 2006). The changes in their molecular structure allow the analysis of the peptide molecular features driving the peptide/membrane interaction. Particularly, we have focused our attention on the spacing between the Trp residues and on their replacements with other non-natural aromatic residue. In peptide 2, the two dipeptides have been replaced by a shorter spacer: 3-aminopropionic acid (or β-alanine; βAla), which reduces the distance along the peptide backbone by two atoms for each dipeptide replaced by βAla. In order to compensate for the increased hydrophobicity of peptide 2, an additional Lys residue has been added at the N-terminus. Lys is the amino acid occurring naturally at the corresponding position of the gp36 sequence. In peptide 3, the Trp residues have been replaced by a non-natural aromatic residue: 3-biphenyl-4-yl-l-alanine (Bip, see Scheme 1 for the molecular structure). In peptide 4, the Trp residues have been replaced with a naphthylalanine isomer: 3-naphthalen-2-yl-l-alanine (Nal2).

Structures of non-natural aromatic amino-acid residues. Bip 3-biphenyl-4-yl-l-alanine, Nal2 3-naphthalen-2-yl-l-alanine
Peptides were synthesized manually on a solid phase, using standard Fmoc/tBu chemistry, as described elsewhere (D’Ursi et al. 2006). Analytical RP-HPLC indicated a >97% purity level. The peptides were characterized by mass spectrometry on a Finningan LCQ-Deca ion trap instrument equipped with an electrospray source (LCQ Deca Finnigan, San José, CA, USA); samples were injected directly in the ESI source using a syringe pump at a flow rate of 5 μl/min, and spectral data were analysed using Xcalibur software.
Sample preparation
For ESR experiments, appropriate amounts of DMPC in dichloromethane/methanol (2/1 v/v) and spin-labelled phosphatidylcholines in ethanol were mixed in small test tubes. A thin film of the lipid was produced by evaporating the solvent with dry nitrogen gas. Final traces of solvent were removed by subjecting the sample to vacuum desiccation for at least 3 h. The samples were then hydrated with 20–50 μl of 10 mM HEPES, 5 mM EDTA, 150 mM NaCl, pH 7.4 buffer (HBS) and vortexed. The lipid suspension thus obtained was transferred to a 100-μl glass capillary and pelleted in a tabletop centrifuge. The supernatant was removed and the capillary was flame sealed. Samples containing peptide were prepared in a similar manner, except that the lipid film was hydrated directly with the peptide solution in HBS (at a concentration of 10 mg/ml). In some cases, as discussed below, it was found that the samples containing peptide 1 (C8) became optically clear and could no longer be pelleted; therefore, the sample was used as such for ESR study. In view of this limitation, the volume of the peptide solution added was minimized using a higher concentration of the peptide (20 mg/ml, obtained by gentle warming and sonication), such that the amount of sample in the ESR cavity was maximized.
ESR spectroscopy
Electron spin resonance spectra of lipid and lipid/peptide samples were recorded on a 9-GHz Bruker EMX EPR spectrometer with a model ER 041 XK-D microwave bridge. Capillaries containing the samples were placed in a standard 4-mm quartz sample tube containing light silicone oil for thermal stability. The temperature of the sample was regulated and maintained constant during the measurement by blowing thermostated nitrogen gas through a quartz dewar. The instrumental settings were as follows: sweep width 120 G, resolution 1,024 points, modulation frequency 100 kHz, modulation amplitude 1.0 G, time constant 20.5 ms, incident power 5.0 mW. Several scans, typically 16, were accumulated to improve the signal-to-noise ratio. Values of the outer hyperfine splitting, 2A max, were determined by measuring the difference between the low-field maximum and the high-field minimum (Marsh and Horváth 1998; Schorn and Marsh 1997). The main source of error on the 2A max value is the uncertainty in composition of samples prepared by mixing few microlitres of mother solutions. For this reason, reproducibility of 2A max determination was estimated by evaluating its value for selected independently prepared samples with the same nominal composition. It was found to be ± 0.2–0.3 G.
Circular dichroism spectroscopy
Circular dichroism (CD) experiments were performed at 30°C on a 810-Jasco spectropolarimeter using a quartz cuvette with a path length of 1 mm. CD spectra of peptides 1 and 2 in water were measured in HBS and in the presence of DMPC unilamellar vesicles. Samples prepared for CD measurements contained the peptide (1.5 × 10−4 M) and the phospholipid at a tenfold higher concentration (1.5 × 10−3 M). Unilamellar vesicles were obtained from multilamellar vesicles by sonication for 3–4 h and successive extrusion through a polycarbonate membrane of 100-nm pore size. During all the measurements, the trace of the high tension voltage was checked to be <700 V, which should ensure reliability of the data obtained (Kelly et al. 2005). Base lines of either solvent or vesicular suspensions without peptide were subtracted from each respective sample to yield the peptide contribution.
Fluorescence spectroscopy
In the case of peptide 1 (C8), peptide–liposome interactions were studied by monitoring the changes in the Trp fluorescence emission spectra in the presence of unilamellar vesicles formed from DMPC. Samples prepared for fluorescence measurements contained the peptide (3.5 × 10−5 M) and the phospholipid at a tenfold higher concentration (3.5 × 10−4 M).
Fluorescence measurements were performed at 30°C using a Fluorolog-3 spectrofluorimeter (Jobin Yvon). The excitation wavelength was 280 nm and emission spectra were recorded between 310 and 450 nm with slit widths of 2 nm.
Results
ESR spectra
Association of the four peptides with lipid membranes can be detected from the perturbation of the chain mobility of spin-labelled lipids using ESR spectroscopy (Ramakrishnan et al. 2003; Swamy and Marsh 2001; Sankaram and Marsh 1993). The samples investigated were spin-labelled phosphatidylcholine (5-PCSL) in DMPC membranes, in the presence and absence of peptides. As an example, ESR spectra from DMPC membranes in the presence and absence of peptide 1 (C8), recorded at temperatures above and below the chain-melting transition of DMPC (24°C), are shown in Fig. 1. Significant perturbations due to the peptide are detectable (compare solid and dashed lines). A quantitative analysis of this effect can be done by plotting the temperature dependences of the outer hyperfine splittings (2A max) of 5-PCSL, as shown in Fig. 2. In the absence of any peptide, a sharp decrease in 2A max is evident at the chain-melting temperature, corresponding to the increase in the spin-label rotational mobility on transition from the gel to the fluid phase (Ramakrishnan et al. 2003). Addition of 0.7:1 wt/wt peptide to DMPC membranes causes changes in 2A max, the extent of which depends on the particular peptide. This peptide/lipid ratio has been chosen because, in the case of interacting peptides, it ensures the bilayer to be completely perturbed, see the following subsection. Peptide 1 (C8) causes the largest effects of the peptides studied. The chain-melting transition is broadened almost beyond detection by the interaction with C8: 2A max decreases in the gel phase and increases strongly in the fluid phase. Peptide 2 shows negligible interaction, hardly changing the transition and values of 2A max. Peptide 3 also produces only small perturbation of 2A max, i.e. little interaction with lipid membranes. In the presence of peptide 4, the co-operativity of the DMPC chain-melting transition is strongly reduced, even if its position remains approximately the same. Above the transition temperature, peptide 4 causes a very significant increase in 2A max, although less than that observed for peptide 1.

Electron spin resonance spectra of 5-PCSL in dimyristoyl phosphatidylcholine bilayer membranes, in the presence (solid line) and in the absence (dashed line) of 0.7:1 wt/wt peptide 1 (C8). The total scan range is 100 G

Temperature dependence of the outer hyperfine splitting, 2A max, of 5-PCSL in DMPC membranes in the absence (bold line) and in the presence of 0.7:1 wt/wt peptide: peptide 1 (solid circles), peptide 2 (solid squares), peptide 3 (open circles), peptide 4 (open squares). For comparison, the temperature dependence in the case of peptide P59 is given by the thin line (adapted from D’Errico et al. 2008)
As a comparison, data for the 20-mer peptide P59, whose sequence includes that of C8, are also shown in Fig. 2 (D’Errico et al. 2008). It is clear that C8 perturbs the mobility of the membrane to an equal, or even greater, extent than does the longer, parent P59 peptide.
Peptide–lipid titration
Because peptides 1 and 4 showed appreciable effects on lipid mobility, we investigated their interaction with DMPC bilayers further. Figure 3 shows the increase in outer hyperfine splitting, Δ2A max, of 5-PCSL in DMPC membranes with increasing peptide concentration. With both peptides, a typical saturation trend is registered (Ramakrishnan et al. 2003): the decrease in lipid chain mobility saturates with a value of Δ2A max ≈ 3.3 G for peptide 1 and Δ2A max ≈ 1.7 G for peptide 4. The stoichiometry of the interaction can be estimated by extrapolation of the increase in Δ2A max, on initial tight binding, to the saturation value of Δ2A max. In both cases, this yields a stoichiometry of one peptide molecule bound per six lipids. On a per-residue basis (0.75 lipids), this is the same stoichiometry as obtained for the 20-mer P59 peptide (D’Errico et al. 2008).

Dependence of the increase, Δ2A max, in outer hyperfine splitting of 5-PCSL at 30°C on peptide/DMPC ratio for peptide 1 (solid circles) and peptide 4 (open squares). Data for 20-mer peptide P59 (continuous line) is also shown, adapted from D’Errico et al. 2008. Dashed lines refer to the procedure used to evaluate the stoichiometry of the peptide–lipid interaction, see text
It should be noted that the addition of peptide 1 to multi-lamellar dispersions of DMPC solubilizes the bilayer structures, so that systems with a peptide to lipid ratio higher than 0.8 wt/wt appear transparent, a phenomenon already observed for the P59 peptide (D’Errico et al. 2008). However, the ESR spectra of the spin-labelled lipid are insensitive to this change, showing that the lipid chains continue to be substantially restricted in their motion. No evidence of liposome solubilization was found for the other peptides investigated in this work.
Lipid chain flexibility profiles
Perturbation of the ESR spectra of lipids spin-labelled at different positions, n, in the sn-2 chain that is induced by binding peptide 1 or peptide 4 was also investigated. Figure 4 gives the ESR spectra of the n-PCSL phosphatidylcholine spin-label positional isomers in fluid DMPC bilayer membranes (T = 30°C), in the presence and absence of peptide 1 at a peptide to lipid ratio of 0.7:1 wt/wt. In the absence of peptide, the outer hyperfine splitting decreases progressively with increasing n, as the spin-label position is stepped down the chain towards the centre of the membrane. This flexibility gradient in segmental chain mobility is a characteristic hallmark of the liquid-crystalline state of fluid phospholipid bilayers (Ramakrishnan et al. 2003).

Electron spin resonance spectra of n-PCSL positional isomers of spin-labelled phosphatidylcholine in fluid-phase dimyristoyl phosphatidylcholine bilayer membranes, in the presence (solid line) and in the absence (dashed line) of 0.7:1 wt/wt peptide 1 (C8) at 30°C
In the presence of bound peptides 1 or 4, the outer hyperfine splitting is increased at all spin-label chain positions. Figure 5 shows the dependence of the outer hyperfine splitting, 2A max, on chain position, n, for the n-PCSL spin labels in fluid DMPC membranes, with and without a saturating amount of peptide. For both peptides, the characteristic flexibility gradient with chain position of the fluid lipid bilayer membranes is preserved. In the presence of bound peptides 1 or 4, the outer hyperfine splitting is increased at all spin-label chain positions. This indicates that the increase in lipid packing density, which is induced by surface association of the peptide (Ramakrishnan et al. 2003; Swamy and Marsh 2001), propagates throughout the chain region. For either peptide, there is no appearance of a second, more motionally restricted component in the spectra of the spin labels positioned towards the terminal methyl ends of the chains. This is evidence that the peptides bind solely at the membrane surface and do not penetrate appreciably into the membrane interior, as does, for instance, the myelin basic protein or apocytochrome c (Sankaram and Marsh 1993). A similar behaviour was observed in the case of the P59 peptide (D’Errico et al. 2008).

Dependence on spin-label position, n, of the outer hyperfine splitting, 2A max (left-hand ordinate), of n-PCSL in fluid-phase membranes of DMPC, in the absence (bold line) and in the presence of 0.7:1 wt/wt peptide 1 (solid circles) or peptide 4 (open squares). T = 30°C. Right-hand ordinate: increase in outer hyperfine splitting, Δ2A max, on adding peptide 1 (solid circles) or peptide 4 (open squares). Corresponding data for peptide P59, adapted from D’Errico et al. 2008, are also shown (thin lines)
To verify the effect of the peptide/lipid ratio on the perturbations of the lipid chain flexibility profile, a set of measurements was also performed at a lower C8/DMPC weight ratio (0.12:1 wt/wt). The results, not shown, are qualitatively similar to those discussed above, but the changes in Δ2A max are almost one-half those reported in Fig. 5 at all spin-label chain positions.
Circular dichroism spectra
An investigation on the conformational preferences of peptides 1 and 2 in HBS and in the presence of DMPC liposomes was performed by means of CD spectroscopy. Both peptides in buffer present a spectrum with the typical shape of a random coil structure, including a negative band at 202 nm. The presence of DMPC liposomes does not significantly affect the CD spectrum of peptide 2. In contrast, the spectrum of peptide 1 assumes the double-well shape typical of a turn-helical structure, including negative bands at 205 and 217 nm (Fig. 6).

Circular dichroism spectra (range 190–260 nm) of peptide 1 in aqueous phosphate buffer (dashed line) and in DMPC unilamellar liposomes (solid line)
Fluorescence spectra
The fluorescence intensities of some fine vibronic structures in the tryptophan fluorescence spectrum show strong environmental dependence (Konev 1967; Ambrosone et al. 1997). In particular, the emission maximum shifts from 354 to 329 nm when going from water to an apolar medium. The quantum yield can also undergo large changes, the direction and extent of which depend on the system under consideration (Callis and Liu 2004). Consequently, fluorescence experiments allow evaluation of the polarity experienced by the tryptophans of peptide 1 in DMPC liposomes.
C8 in water (Fig. 7) gives a Trp emission spectrum typical of an aqueous environment (λ max ≈ 352 nm), indicating that the Trp side chains are exposed to the aqueous medium. The presence of DMPC liposomes causes a weak shift of the emission maximum to shorter wavelength (λ max = 347 nm) and a reduction of the fluorescence quantum yield. These results are similar to those observed for P59 (D’Errico et al. 2008) and indicate that, despite the evident interaction between C8 and DMPC liposomes, the Trp residues of this peptide are not significantly inserted the apolar inner core of the bilayer.

Tryptophan emission spectra (range 310–450 nm) of peptide 1 in aqueous phosphate buffer (dashed line) and in DMPC unilamellar liposomes (solid line)
Discussion
The C8 octapeptide, derived from the MPER of the FIV envelope glycoprotein gp36 has been found to exert potent antiviral activity (Giannecchini et al. 2003; D’Ursi et al. 2006 and see Table 1), a remarkable feature for an oligopeptide of such small size. It has been hypothesized that the inhibitory action is due to adsorption of the peptide on the plasma membrane of the target cell, where it can inhibit interaction with the virus fusion glycoprotein. To check this hypothesis, we studied the interactions of C8 and analogues with lipid membranes using ESR spectroscopy (Marsh and Watts 1982). When combined with site-specifically spin-labelled lipids, this technique has proved to be a powerful tool to monitor peptide–membrane interactions. However, it is not suitable to discriminate the different kinetic steps in the interaction process (Shai 1999), so that in the following discussion we shall assume a simple two-state model, in which only interacting and non-interacting peptides are distinguished.
Phospholipid bilayers are a simple model system which allows a detailed analysis of the molecular features influencing the peptide–membrane interaction. The presence of two acidic residues in the peptide sequence led us to choose liposomes formed by the ubiquitous zwitterionic phospholipid phosphatidylcholine rather than by anionic lipids. Indeed, in previous work, we found that the electrostatic contribution to the interaction between the 20-mer peptide P59, whose sequence includes C8, and phospholipid bilayers is negligible (D’Errico et al. 2008). In order to allow a straightforward interpretation of the data and a direct comparison with the results already reported for P59, we also chose not to include other lipids in the bilayer composition. Indeed, the presence of cholesterol and sphingomyelin is reported to enhance the interaction between membranes and peptides derived from the MPER domain of HIV (Sáez-Cirión et al. 2002; Veiga and Castanho 2007).
Inspection of the sequence of C8 shows that it is mainly characterized by the presence of three equally distant tryptophans in a WX2WX2W motif (Table 1). The presence of these tryptophan residues and their spatial arrangement has been found to be essential for the antiviral activity (D’Ursi et al. 2006). In particular, the substitution of the indole ring of tryptophan, and/or a change of the ring orientation, led to a dramatic decrease of the peptide activity. Since tryptophan residues are usually strongly involved in the interaction with biological membranes, it is possible that the variation in the peptide antiviral activity is due to their different ability to interact with the membranes. In order to investigate this aspect, we also studied the behaviour of three analogues, each of them differing from C8 for a single molecular feature. Peptide 2 has a shorter spacing between the Trp residues. In peptides 3 and 4 the Trp residues have been replaced by a non-natural aromatic amino acid, Bip or Nal2, with different side chain topology (Scheme 1). Specifically, Bip is characterized by a non-planar relative orientation of the two phenyl rings, while Nal2 contains a planar naphthyl group, very similar to the native indole group of Trp.
The ESR results show that C8 perturbs the lipid chain motion of DMPC membranes. Particularly, it increases the chain mobility in the gel phase and reduces it in the fluid phase so that the chain-melting transition becomes almost undetectable. Peptide 4 produces a similar, although weaker effect, whereas peptides 2 and 3 do not affect the lipid chain-melting transition, indicating that they do not interact with the membrane. For peptides 1 and 4, the perturbation of lipid motion extends along the whole lipid chain. However, the lack of any specific motionally restricted spin-labelled lipid component in the spectra from 14-PCSL suggests this perturbation to be an indirect effect from interfacial association rather than penetration of the peptide into the membrane.
Taken together, the experimental evidence indicates that the tryptophan residues play a major role in driving the interaction between C8 and lipid membranes. The presence of two conjugated aromatic rings seems important because, whereas an attenuated interaction is preserved if the indole group is replaced by a naphthyl one, the interaction is lost completely if a biphenyl group is substituted. However, simply the presence of three Trp residues (as in peptide 2) is insufficient; they must be spaced such that they are located on the same side of the putative helix. Conformational studies by CD and NMR show that all four peptides, which adopt a prevalent random coil conformation in water, assume a turn-folded conformation in DMSO/water mixtures (Giannecchini et al. 2003; D’Ursi et al. 2006). In the case of C8 an amphiphilic helix forms, in which the polar or charged residues are positioned on one side, the opposite being occupied by hydrophobic residues and by the three Trp residues, which are known to have a propensity to locate to the polar–apolar interface. A comparative conformational analysis shows that while the structures of peptides 1 and 4 are almost superimposable, i.e. the Trp and Nal2 side chains are iso-sterically oriented, this is not true for peptide 2, because the shorter spacer induces a backbone arrangement that minimizes interactions between the Trp rings, resulting in oppositely oriented side chains.
Our CD data show that C8 assumes a turn-folded conformation in the presence of liposomes, while peptide 2 still retains a random coil conformation. Thus, the ability of C8 to assume a helical conformation with the Trp residues exposed on the same side of the helix promotes its interaction with bilayers, which, in turn, stabilizes the peptide conformation. In contrast, because the Trp residues are on different sides of peptide 2 in the helical conformation, this peptide is not able to interact with the membrane, preferring to remain in aqueous solution in the random coil configuration.
Table 1 includes results from FIV inhibition assays, expressed as 50% inhibitory peptide concentrations, IC50 (highest antiviral activity corresponds to lowest IC50). There is a strong correlation between inhibitory potency and membrane interaction of the four peptides. Peptides 1 and 4 have significant and comparable antiviral activities, which is completely absent for the non-binding peptides 2 and 3. This correlation supports the proposal that the antiviral activity is exerted through a mechanism of peptide adsorption onto the membrane of the target cell. Adsorbed peptide protects the cell from the fusion glycoprotein of the virus. The role played by the membrane in this process is fundamental in stabilizing the peptide conformation. In turn, once the peptide has assumed the correct conformation, it can exert antiviral activity by interacting with the glycoproteins present on the surface of the approaching viruses (Lorizate et al. 2008).
Mutagenesis studies and crystal structure determinations identify a cavity in the TM glycoprotein of immunodeficiency viruses, to which peptide inhibitors could bind. Particularly, in the case of HIV gp41, the ectodomain core is a trimer of helical hairpins in which three C helices pack against a central coiled coil. Each C helix is preferentially in contact with one of three symmetry-related hydrophobic cavities on the surface of the coiled coil. In principle, compounds that are able to insert stably in these cavities should actively block the protein function, thus inhibiting viral infection. However, small peptides designed to bind in such a cavity exhibit no antiviral activity, and this has been attributed to conformational instability (Chan et al. 1998; Eckert et al. 1999). Indeed, satisfactory antiviral activity has been shown only by peptides composed of 30 or more residues (Jin et al. 2000; Yu et al. 2008).
Our data indicate that the unusually high antiviral activity shown by a short peptide such as C8 is strictly related to its ability to interact with membranes. Thus, it is likely that C8 or other derived peptides are able to exert antiviral activity only if bound to the target cell membrane, which stabilizes its conformation enabling it to interact with the gp36 protein of the approaching FIV.
Our results might also give some indication of the role played by the Trp-rich MPER domain of the viral glycoprotein in the process of fusion between FIV and the target-cell membrane. This is an energy-costing event that involves destabilization of the bilayer and dehydration of the interfacial regions. Our results show that C8 (and also P59) destabilizes phospholipid bilayer membranes, producing smaller micelle-like aggregates at peptide to lipid weight ratios >0.8. This effect is due to the synergistic action of the three tryptophan residues. In fact, a similar behaviour has been recently reported for Trp-Trp and Trp-Trp-Trp oligomers (Stano et al. 2005). This suggests the existence of a specific interaction between membranes formed by phosphatidylcholine lipids and Trp-rich peptides. However, the mechanism and microstructural details of this interaction are not satisfactorily understood. Our spin-label results exclude a deep insertion of the peptide in the bilayer, rather indicating adsorption of the peptide at the membrane interface. This is further supported by fluorescence measurements, showing that, even in the presence of excess DMPC, the tryptophan residues of C8 are not embedded in the hydrophobic bilayer inner core, continuing to be located in a polar environment, such as that defined by the fully hydrated lipid headgroups. These results are in agreement with those in the literature. In fact, Yau et al. (1998) showed that the Trp residues of peptides interacting with biomembranes tend to be positioned at the interface. This is related to the aromatic nature of indole (comprehensive of dipole and quadrupole, H-bonding, cation-π and steric interactions), which confer an amphipathic behaviour on the Trp residue, so that contact with water is disfavoured and, at the same time, deep penetration into the bilayer hydrophobic core is also precluded. Molecular dynamic calculations have also shown that indoles are preferably positioned in the region defined by the headgroups and upper acyl chain sections of phosphatidylcholine lipids (Grossfield and Woolf 2002), and linear dichroism measurements have indicated that the indole plane is at an oblique angle with respect to the membrane normal (Esbjörner et al. 2007).
Thus, a possible mechanism through which C8 can destabilize the bilayer is its intercalation between the lipid headgroups. This disrupts the hydration structure of phosphocholine groups (Grossfield and Woolf 2002) and, also because of steric reasons, increases the distance between them. Consequently, the result of the peptide solubilization is an altered lipid packing, so that the bilayer evolves towards smaller aggregates with higher surface curvature. This perturbation is more effective because of the Trp residues closeness. It is likely that, similar to what is observed by small-angle neutron scattering measurements with P59 (D’Errico et al. 2008), phospholipid liposomes are converted by binding of C8 into smaller rod-like micelles. This destabilizing effect is not observed for the other peptides used here, because of their diminished or non-existent capacity to interact with the bilayer. Consequently, this effect also should be ascribed to the presence of the Trp residues in their correct orientation.
The destabilizing effect exerted by the MPER domain, and specifically by the Trp cluster, on the membrane could be fundamental to the fusion process (Garg et al. 2004). Indeed, the presence of the MPER domain has been shown to be crucial in the infection pathway (Barbato et al. 2003). In the surface glycoproteins of various viruses, including not only FIV and HIV, but also that responsible for the severe acute respiratory syndrome (Guillén et al. 2007) and Ebola infection (Sáez-Cirión et al. 2003), this Trp-rich domain is located very close to the viral membrane, but definitely protruding from it. Once the fusion peptide has entered the target-cell membrane and subsequent conformational changes in the glycoprotein have brought the two membranes into close proximity, the MPER domain could then destabilize the bilayer of the target cell, thus promoting membrane fusion.
References
Ambrosone L, D’Errico G, Ragone R (1997) Interaction of tryptophan and N-acetyltryptophanamide with dodecylpentaoxyethyleneglycol ether micelles. Spectrochim Acta 53:1615–1620. PII:S 1386-1425(97)00096-6
Barbato G, Bianchi E, Ingallinella P, Hurni WH, Miller MD, Ciliberto G, Cortese R, Bazzo R, Shiver JW, Pessi A (2003) Structural analysis of the epitope of the anti-HIV antibody 2F5 sheds light into its mechanism of neutralization and HIV fusion. J Mol Biol 330:1101–1115. doi:10.1016/S0022-2836(03)00611-9
Callis PR, Liu T (2004) Quantitative prediction of fluorescence quantum yields for tryptophan in proteins. J Phys Chem B 108:4248–4259. doi:10.1021/jp0310551
Chan DC, Chutkowski CT, Kim PS (1998) Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc Natl Acad Sci USA 95:15613–15617
de Planque MRR, Greathouse DV, Koeppe REII, Schäfer H, Marsh D, Killian JA (1998) Influence of lipid/peptide hydrophobic mismatch on the thickness of diacylphosphatidylcholine bilayers. A 2H NMR and ESR study using designed transmembrane R-helical peptides and gramicidin A. Biochemistry 37:9333–9345. doi:10.1021/bi980233r
D’Errico G, D’Ursi AM, Marsh D (2008) Interaction of a peptide derived from glycoprotein gp36 of feline immunodeficiency virus and its lipoylated analogue with phospholipid membranes. Biochemistry 47:5317–5327. doi:10.1021/bi7025062
D’Ursi AM, Giannecchini S, Esposito C, Alcaro MC, Sichi O, Armenante MR, Carotenuto A, Papini AM, Bendinelli M, Rovero P (2006) Development of antiviral fusion inhibitors: short modified peptides derived from the transmembrane glycoprotein of feline immunodeficiency virus. Chembiochem 7:774–779. doi:10.1002/cbic.200500390
Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS (1999) Inhibiting HIV-1 entry: discovery of d-peptide inhibitors that target the gp41 coiled-coil pocket. Cell 99:103–115. doi:10.1016/S0092-8674(00)80066-5
Esbjörner EK, Caesar CEB, Albinsson N, Lincoln P, Nordén B (2007) Tryptophan orientation in model lipid membranes. Biochem Biophys Res Commun 361:645–650. doi:10.1016/j.bbrc.2007.07.135
Esposito C, D’Errico G, Armenante MR, Giannecchini S, Bendinelli M, Rovero P, D’Ursi AM (2006) Physicochemical characterization of a peptide deriving from the glycoprotein gp36 of the feline immunodeficiency virus and its lipoylated analogue in micellar systems. Biochim Biophys Acta 1758:1653–1661. doi:10.1016/j.bbamem.2006.06.017
Frey SC, Hoover EA, Mullins JI (2001) Feline immunodeficiency virus cell entry. J Virol 75:5433–5440. doi:10.1128/JVI.75.11.5433-5440.2001
Garg H, Fuller FJ, Tompkins WAF (2004) Mechanism of feline immunodeficiency virus envelope glycoprotein-mediated fusion. Virology 321:274–286. doi:10.1016/j.virol.2004.01.006
Giannecchini S, Di Fenza A, D’Ursi AM, Matteucci D, Rovero P, Bendinelli M (2003) Antiviral activity and conformational features of an octapeptide derived from the membrane-proximal ectodomain of the feline immunodeficiency virus transmembrane glycoprotein. J Virol 77:3724–3733. doi:10.1128/JVI.77.6.3724-3733.2003
Giannecchini S, Bonci F, Pistello M, Matteucci D, Sichi O, Rovero P, Bendinelli M (2004) The membrane-proximal tryptophan-rich region in the transmembrane glycoprotein ectodomain of feline immunodeficiency virus is important for cell entry. Virology 320:156–166. doi:10.1016/j.virol.2003.12.001
Giannecchini S, D’Ursi AM, Esposito C, Scrima M, Zabogli E, Freer G, Rovero P, Bendinelli M (2007) Antibodies generated in cats by a lipopeptide reproducing the membrane proximal external region of the feline immunodeficiency virus transmembrane enhance virus infectivity. Clin Vaccine Immunol 14:944–951. doi:10.1128/CVI.00140-07
Grossfield A, Woolf TB (2002) Interaction of tryptophan analogs with POPC lipid bilayers investigated by molecular dynamic calculations. Langmuir 18:198–210. doi:10.1021/la0106485
Guillén J, Moreno MR, Pérez-Berná AJ, Bernabeu A, Villalain J (2007) Interaction of a peptide from the pre-transmembrane domain of the severe acute respiratory syndrome coronavirus spike protein with phospholipid membranes. J Phys Chem 111:13714–13725. doi:10.1021/jp073675y
Jiang S, Lin K, Strick N, Neurath AR (1993) HIV-1 inhibition by a peptide. Nature 365:113. doi:10.1038/365113a0
Jin BS, Ryu JR, Ahn K, Yu YG (2000) Design of a peptide inhibitor that blocks the cell fusion mediated by glycoprotein 41 of human immunodeficiency virus type 1. AIDS Res Hum Retroviruses 16:1797–1804. doi:10.1089/08892220050195757
Kelly SM, Jess TJ, Price NC (2005) How to study proteins by circular dichroism. Biochim Biophys Acta 1751:119–139
Kilby JM, Lalezari JP, Eron JJ, Carlson M, Cohen C, Arduino RC, Goodgame JC, Gallant JE, Volberding P, Murphy RL, Valentine F, Saag MS, Nelson EL, Sista PR, Dusek A (2002) The safety, plasma pharmacokinetics, and antiviral activity of subcutaneous enfuvirtide (T-20), a peptide inhibitor of gp41-mediated virus fusion, in HIV-infected adults. AIDS Res Hum Retroviruses 18:685–693. doi:10.1089/088922202760072294
Konev SV (1967) Fluorescence and phosphorescence of proteins and nucleic acids. Plenum Press, New York
Lorizate M, Huarte N, Sáez-Cirión A, Nieva JL (2008) Interfacial pre-transmembrane domains in viral proteins promoting membrane fusion and fission. Biochim Biophys Acta 1778:1624–1639. doi:10.1016/j.bbamem.2007.12.018
Marsh D (1997) Magnetic resonance of lipids and proteins in membranes. Curr Opin Colloid Interface Sci 2:4–14
Marsh D, Horváth LI (1998) Structure, dynamics and composition of the lipid–protein interface. Perspectives form spin-labelling. Biochim Biophys Acta 1376:267–296. doi:10.1016/S0304-4157(98)00009-4
Marsh D, Watts A (1982) Spin-labelling and lipid-protein interactions in membranes. In: Jost PC, Griffith OH (eds) Lipid–protein interactions, vol 2. Wiley Interscience, New York, pp 53–126
Massi C, Indino E, Lami C, Fissi A, Pieroni O, La Rosa C, Esposito F, Galoppini C, Rovero P, Bandecchi P, Bendinelli M, Garzelli C (1998) The antiviral activity of a synthetic peptide derived from the envelope SU glycoprotein of feline immunodeficiency virus maps in correspondence of an amphipathic helical segment. Biochem Biophys Res Commun 246:160–165. doi:10.1006/bbrc.1998.8580
Pancino G, Camoin L, Sonigo P (1995) Structural analysis of the principal immunodominant domain of the feline immunodeficiency virus transmembrane glycoprotein. J Virol 69:2110–2118
Ramakrishnan M, Jensen PH, Marsh D (2003) α-Synuclein association with phosphatidylglycerol probed by lipid spin labels. Biochemistry 42:12919–12926. doi:10.1021/bi035048e
Sáez-Cirión A, Nir S, Lorizate M, Agirre A, Cruz A, Pérez-Gil J, Nieva JL (2002) Sphingomyelin and cholesterol promote HIV-1 gp41 pretransmembrane sequence surface aggregation and membrane restructuring. J Biol Chem 277:21776–21785. doi:10.1074/jbc.M202255200
Sáez-Cirión A, Gómara MJ, Agirre A, Nieva JL (2003) Pretransmembrane sequence of Ebola glycoprotein: interfacial hydrophobicity distribution and interaction with membranes. FEBS Lett 533:47–53. PII:S0014-5793(02)03747-X
Salzwedel K, West JT, Hunter E (1999) A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J Virol 73:2469–2480
Sankaram MB, Marsh D (1993) Protein–lipid interactions with peripheral membrane proteins. In: Watts A (ed) New comprehensive biochemistry: protein–lipid interactions, vol 25. Elsevier, Amsterdam, pp 127–162. doi:10.1016/S0167-7306(08)60235-5
Schibli DJ, Montelaro RC, Vogel HJ (2001) The membrane-proximal tryptophan-rich region of the HIV glycoprotein, gp41, forms a well-defined helix in dodecylphosphocholine micelles. Biochemistry 40:9570–9578. doi:10.1021/bi010640u
Schorn K, Marsh D (1997) Extracting order parameters from powder EPR lineshapes for spin-labelled lipids in membranes. Spectrochim Acta A 53:2235–2240. PII:S1386-1425(97)00136-4
Serres PF (2000) Molecular mimicry between the trimeric ectodomain of the transmembrane protein of immunosuppressive lentiviruses (HIV-SIV-FIV) and interleukin 2. C R Acad Sci III 323:1019–1029. doi:10.1016/S0764-4469(00)01277-4
Shai Y (1999) Mechanism of binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim Biophys Acta 1462:55–70. PII:S0005-2736(99)00200-X
Stano P, Bufali S, Domazou AS, Luisi PL (2005) Effect of tryptophan oligopeptides on the size distribution of POPC liposomes: a dynamic light scattering and turbidimetric study. J Liposome Res 15:29–47. doi:10.1081/LPR-200064956
Suarez T, Nir S, Goni FM, Saez-Cirion A, Nieva JL (2000) The pre-transmembrane region of the human immunodeficiency virus type-1 glycoprotein: a novel fusogenic sequence. FEBS Lett 477:145–149. doi:10.1016/S0014-5793(00)01785-3
Swamy MJ, Marsh D (2001) Spin-label electron paramagnetic resonance studies on the interaction of avidin with dimyristoyl-phosphatidylglycerol membranes. Biochim Biophys Acta 1513:122–130. PII:S0005-2736(01)00316-9
Veiga AS, Castanho MARB (2007) The influence of cholesterol on the interaction of HIV gp41 membrane proximal region-derived peptides with lipid bilayers. FEBS J 274:5096–5104. doi:10.1111/j1742-4758.2007.06029.x
White SH, Wimley WC (1998) Hydrophobic interactions of peptides with membrane interfaces. Biochim Biophys Acta 1376:339–352. PII:S0304-4157(98)00021-5
Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ (1994) Peptides corresponding to a predictive α-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc Natl Acad Sci USA 91:9770–9774
Yau WC, Wimley WC, Gawrisch K, White SH (1998) The preference of tryptophan for membrane interfaces. Biochemistry 37:14713–14718. doi:10.1021/bi980809c
Yu H, Tudor D, Alfsen A, Labrosse B, Clavel F, Bomsel M (2008) Peptide P5 (residues 628–683), comprising the entire membrane proximal region of HIV-1 gp41 and its calcium-binding site, is a potent inhibitor of HIV-1 infection. Retrovirology 5:93. doi:10.1186/1742-4690-5-93
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
D’Errico, G., Vitiello, G., D’Ursi, A.M. et al. Interaction of short modified peptides deriving from glycoprotein gp36 of feline immunodeficiency virus with phospholipid membranes. Eur Biophys J 38, 873–882 (2009). https://doi.org/10.1007/s00249-009-0454-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00249-009-0454-9