
Abstract
The function and dynamics of proteins depend on their direct environment, and much evidence has pointed to a strong coupling between water and protein motions. Recently however, neutron scattering measurements on deuterated and natural-abundance purple membrane (PM), hydrated in H2O and D2O, respectively, revealed that membrane and water motions on the ns–ps time scale are not directly coupled below 260 K (Wood et al. in Proc Natl Acad Sci USA 104:18049–18054, 2007). In the initial study, samples with a high level of hydration were measured. Here, we have measured the dynamics of PM and water separately, at a low-hydration level corresponding to the first layer of hydration water only. As in the case of the higher hydration samples previously studied, the dynamics of PM and water display different temperature dependencies, with a transition in the hydration water at 200 K not triggering a transition in the membrane at the same temperature. Furthermore, neutron diffraction experiments were carried out to monitor the lamellar spacing of a flash-cooled deuterated PM stack hydrated in H2O as a function of temperature. At 200 K, a sudden decrease in lamellar spacing indicated the onset of long-range translational water diffusion in the second hydration layer as has already been observed on flash-cooled natural-abundance PM stacks hydrated in D2O (Weik et al. in J Mol Biol 275:632–634, 2005), excluding thus a notable isotope effect. Our results reinforce the notion that membrane-protein dynamics may be less strongly coupled to hydration water motions than the dynamics of soluble proteins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The purple membrane (PM) has provided biophysicists with an exceptional model of study. Natively forming a two-dimensional crystal, PM contains a unique protein, viz. the light-driven proton pump bacteriorhodopsin (BR), and various lipid species. From the external membrane of Halobacterium salinarum, PM can be purified relatively easily in large quantities necessary for neutron scattering measurements. PM dynamics has been well-characterised by neutron scattering at various time and space scales for different hydration levels (Ferrand et al. 1993; Fitter et al. 1996, 1997; Lehnert et al. 1998) and different lipid contents (Fitter et al. 1998). Isotope labelling of the membrane has been successfully performed (Engelman and Zaccai 1980; Patzelt et al. 1997), and combined with incoherent neutron scattering experiments, revealed the dynamical heterogeneity of PM on the ps–ns time scale, with the core of BR found to be stiffer than its surroundings (Reat et al. 1998).
Hydrated PM undergoes a so-called dynamical transition at around 230–260 K, evidenced in incoherent neutron scattering experiments as a sudden increase in the atomic mean square displacement (MSD) as a function of temperature (Ferrand et al. 1993; Fitter et al. 1997; Lehnert et al. 1998). Neutron scattering experiments on soluble proteins (Doster et al. 1989; Fitter 1999; Roh et al. 2006; Wood et al. 2008), on another membrane protein (Pieper et al. 2007) and on RNA (Caliskan et al. 2006) revealed the same phenomenon at a temperature between 180 and 250 K.
The dynamical transition only occurs in sufficiently hydrated systems, and changing the solvent composition shifts the transition temperature (Caliskan et al. 2004; Cordone et al. 1999; Cornicchi et al. 2005, 2006; Paciaroni et al. 2002; Tsai et al. 2000). The term “slaving” has been used to express that the solvent imposes its dynamics on the macromolecule (Fenimore et al. 2002, 2004; Iben et al. 1989). In particular, molecular dynamics (MD) simulations have shown that the onset of translational diffusion of hydration-water triggers the protein dynamical transition in soluble proteins (Tarek and Tobias 2002; Tournier et al. 2003). In the PM, however, the onset of translational motion in hydration water occurs at 200 K (Berntsen et al. 2005; Weik 2003; Weik et al. 2005; Wood et al. 2007), i.e. well below the dynamical transition of membrane motions. Membrane-protein motions might be controlled by lipid motions (Kamihira and Watts 2006) rather than by water motions (Wood et al. 2007). Nevertheless, water is required to allow the conformational changes necessary for BR to function (Buchsteiner et al. 2007; Dencher et al. 2000; Weik et al. 1998).
Here, we employed incoherent elastic neutron scattering and neutron diffraction to study hydration water in stacks of deuterated PM. The first part of this study extends previous neutron-diffraction measurements that showed inter-membrane heavy water (D2O) in the second hydration layers turns liquid-like at 200 K in flash-cooled natural-abundance PM (Weik et al. 2005). Here, the results have been reproduced for deuterated PM hydrated in light water (H2O). In the second part, atomic MSD of hydration-water and PM dynamics were measured separately by using deuterated PM in H2O and hydrogenated PM in D2O, respectively. In complement to an earlier study on samples with high hydration (three to four water layers between successive PM patches at room temperature) (Wood et al. 2007), we here present data on the same samples, yet at a lower hydration level corresponding to only two water layers between successive membrane fragments. Again, dynamical transitions of hydration-water and of membrane motions are observed at 200 and at 260 K, respectively, indicating that hydration-water and PM motions on the ns–ps time scale are uncoupled below 260 K.
Materials and methods
Sample preparation
Two PM samples were prepared, a fully deuterated one (denoted D-PM) and a natural-abundance one (denoted H-PM). Both were purified from Halobacterium salinarum (H. salinarum) as described previously (Oesterhelt and Stoeckenius 1974). To produce fully deuterated PM, H. salinarum were grown on deuterated algal medium (Patzelt et al. 1997). Two hundred milligram samples of both D-PM and H-PM were used, as deduced from the BR concentration determined by the absorbance at 565 nm on the PM suspensions obtained after purification. D2O in the deuterated PM sample (denoted D-PM-H2O) and H2O in the hydrogenated sample (denoted H-PM-D2O) were exchanged against H2O and D2O, respectively, by three successive centrifugation steps. The two concentrated membrane suspensions were then placed on 4 × 3 cm2 flat aluminium sample holders. The sample holders were placed in dessicators over silica gel for 12 h to achieve partial drying to approximately 0.5 g water/g membrane, as determined by weighing the holder regularly until it contained approximately 300 mg of sample. Vacuum was not applied to the dessicator at any stage of the sample preparation. The silica gel was exchanged for a saturated KNO3 solution [in H2O for the D-PM-H2O sample and in D2O for the H-PM-D2O sample, respectively; yielding a relative humidity (r.h.) of 93%] for 7 days until the weight was stable. The D-PM-H2O sample was then equilibrated over pure H2O (100% r.h.) for 2 days, called a “high-hydration” (Hh) level, and is hereafter denoted D-PM-H2O-Hh. The high-hydration level corresponds to about 0.3 g water/g membrane. The sample holder was sealed with indium and closed with an aluminium cover, resulting in a sample chamber of 0.3 mm thickness. After temperature-dependent neutron diffraction experiments (see below), the D-PM-H2O-Hh sample was opened and equilibrated for 3½ days over a saturated KCl solution (86% r.h.; in H2O), leading to a “low-hydration” (Lh) level of 0.1 g water/g membrane. The sample was then sealed, closed again (0.3 mm chamber thickness) and denoted D-PM-H2O-Lh. The H-PM-D2O sample was equilibrated for 2 days over pure D2O (100% r.h.) and then for 3½ days in a D2O atmosphere at 86 % r.h. (resulting in 0.1 g water/g membrane), before it was sealed and closed (0.3 mm chamber thickness): it is denoted H-PM-D2O-Lh. An estimate of the number of hydrogen and deuterium atoms in the different components of the H-PM-D2O-Lh and D-PM-H2O-Lh samples is given in Table 1.
Neutron diffraction as a function of temperature to determine lamellar spacing
As water evaporated from the membrane preparation, the PM patches oriented parallel to the surface of the sample holder, forming a stack of membranes with a characteristic repeat distance, called the lamellar spacing. The number of water layers intercalated between adjacent membrane fragments depends on the sample preparation protocol (r.h. and equilibration time) and can be determined by measuring the lamellar spacing using neutron diffraction. Lamellar spacings were determined on the diffractometer D16 at the Institut Laue-Langevin (ILL) by monitoring the first-order Bragg peak of the PM stack using a wavelength of 4.56 Å (Weik et al. 2005).
The D-PM-H2O-Hh sample was characterised by a lamellar spacing of 62 Å at room temperature, corresponding to an average inter-membrane water layer of 13 Å (the average thickness of a dry PM fragment is 49 Å). To determine its lamellar spacing after flash-cooling as a function of temperature upon heating (Weik et al. 2005), the sample holder was plunged into liquid nitrogen and then transferred into a cryostat pre-cooled at 100 K. The lamellar spacing was determined at 100 K and then on heating from 180 to 290 K. The time interval between successive data points was 28 min.
Subsequently, the D-PM-H2O-Hh sample was opened, re-equilibrated to a lower hydration level (Lh) (see preceding section) and re-sealed. The lamellar spacing of the D-PM-H2O-Lh and the H-PM-D2O-Lh samples were then measured, and the samples were opened and re-hydrated/dehydrated as necessary until a lamellar spacing of 54 Å at room temperature was reached, corresponding to a 5 Å thick inter-membrane water layer. Identical lamellar spacings of the two samples ensured identical hydration levels. Between neutron scattering experiments, the samples were stored at −20°C.
Elastic incoherent neutron scattering (EINS) experiments
Owing to their particular wavelengths and energies, thermal and cold neutrons are an important probe for macromolecular structure and thermal dynamics (Gabel et al. 2002). The incoherent cross section of hydrogen atoms is about 40 times larger than that of deuterium, and much larger than those of all other atoms present in biological macromolecules. Using deuterium labelling, it is therefore possible to focus on different components of a complex system. In the present work, studying fully deuterated PM in H2O allowed us to predominantly measure hydration water dynamics. Natural-abundance PM hydrated in D2O, on the other hand, provided information about membrane motions. Indeed, 60% of the total incoherent scattering cross section of D-PM-H2O-Lh is due to the contribution of inter-membrane water. For H-PM-D2O-Lh, the contribution of the D2O is negligible, and protein (BR) and lipid make up 77 and 23%, respectively. In the estimations, we consider that 60% of the exchangeable hydrogen/deuterium atoms in bacteriorhodopsin exchange, and that all of the exchangeable hydrogen/deuterium atoms in the lipid head groups do so (Zaccai and Gilmore 1979).
So-called “elastic” scans (the elastic intensity only is recorded) were performed as a function of temperature on the IN16 backscattering spectrometer at the ILL, with an energy resolution of 0.9 μeV (full width at half maximum of the elastic peak) (Frick and Gonzalez 2001) and a wavelength of 6.275 Å. The Q-range accessible is 0.02–1.9 Å−1, corresponding to movements on the angstrom (Å) scale. At the given instrumental resolution, hydrogen movements with characteristic times faster than 1 ns are monitored, which reflect the dynamics of the larger groups to which they are attached. A sudden drop in the elastic signal is indicative of an increase in movements faster than 1 ns on the angstrom (Å) length scale.
D-PM-H2O-Lh and H-PM-D2O-Lh samples were placed in a standard “orange” cryostat at room temperature at an angle of 135° with respect to the incident beam and then cooled to 50 K in about 2 h. Subsequently, the elastic energy was recorded on heating from 50 to 300 K at constant rates of 0.16 and 0.17 K/min for the D-PM-H2O-Lh and the H-PM-D2O-Lh samples, respectively. The acquisition was continuous on heating and the data were subsequently binned into points corresponding to a temperature step of about 5 K. The signal from the empty sample holder was subtracted, and the data were normalised to the intensity at 50 K.
From the wave-vector (Q) dependence of the elastic intensity (Q = 4πsin θ/λ, in which 2θ is the angle of the scattered neutron and λ is the neutron wavelength), the mean square displacement (MSD) \( {\left\langle {u^{2} } \right\rangle } \) of the investigated movements can be calculated according to the Gaussian approximation, which is valid for \( Q^{2} {\left\langle {u^{2} } \right\rangle } < 2 \):
\( I(Q,\omega = 0) \) is the normalised elastically scattered intensity and I 0 is the value of the scattering at Q = 0.
MSD were calculated in the Q-ranges 0.2 < Q 2 < 1.5 Å−2 for H-PM-D2O-Lh and 0.2 < Q 2 < 0.9 Å−2 for D-PM-H2O-Lh. At high Q, the range used for the deuterated PM sample is limited by a coherent peak centered at approximately Q = 1.4 Å−1 arising from the spacing of deuterated lipids at 4.5 Å (Henderson 1975), which is not constant with temperature.
The elastic intensity summed over all Q-values used for MSD determination, which was first used as a guide for interpreting MSD in neutron biological dynamics studies by Reat et al. (2000), is also presented as a function of temperature. Limited to the first term, the expansion of the exponential of Eq. 1 gives
where b is a constant depending only on the Q values over which the sum is performed. In a first-order approximation, the summed elastic intensity is therefore proportional to the MSD \( {\left\langle {u^{2} } \right\rangle } \). The quantity was normalised to unity at low temperature by dividing the sum by the number of detectors over which it was performed.
Results and discussion
Lamellar spacing of flash-cooled D-PM-H2O-Hh as a function of temperature
The samples described here are formed of stacked PM fragments, intercalated by a number of water layers at room temperature that can be adjusted according to different equilibration protocols (see “Materials and methods”). If the lamellar spacing at room temperature is larger than 54 Å, excess inter-membrane water leaves the membrane stack upon slow-cooling at temperatures around 260 K until a minimal spacing of 54 Å is reached and forms crystalline ice outside the stack (Lechner et al. 1998). Only two layers of water (corresponding to 54–49 Å = 5 Å) remain thus in the inter-membrane space below 260 K. Upon flash-cooling, however, 9 Å of water (three to four layers) can be trapped in natural-abundance PM hydrated in D2O (Weik et al. 2005). On heating, two of the four water layers are observed to leave the stack at 200 K to form crystalline ice, and the lamellar spacing decreases to 54 Å. The decrease in lamellar spacing at 200 K has been attributed to an onset of translational mobility of inter-membrane water in the second hydration shells. Above 260 K, swelling of the membrane stacks is observed as crystalline ice melts and returns in the inter-membrane space. Samples with a lamellar spacing of 54 Å at room temperature do not exhibit major changes, and the spacing remains constant (to within 0.3 Å) as a function of temperature (Lechner et al. 1998). The different scenarios are schematised in Fig. 1.

Schematic representation of the lamellar spacing d of a hydrated purple membrane stack as a function of temperature (after Lechner et al. 1998; Weik et al. 2005). Purple membranes are sketched as open rectangles intercalated by hydration water layers depicted as waved lines (A–E). Low-hydration (Lh) samples (dashed line) have a constant lamellar spacing of 54 Å in the temperature range from 100 K (A) to 300 K (B). High-hydration (Hh) samples (full and dotted lines) are characterised by a lamellar spacing of 62 Å at 300 K (C). Upon heating the Hh sample after slow-cooling (full line; C and D), the lamellar spacing increases at 260 K. Part of the hydration water had left the inter-membrane space during slow-cooling and is present as crystalline ice (depicted by stars in D) outside the membrane stacks at 100 K (D). Upon heating the Hh sample after flash-cooling (dotted line; C, E), water leaves the inter-membrane space at 200 K, which causes a decrease in the lamellar spacing, and forms ice. At 260 K, the lamellar spacing increases again. A fraction of inter-membrane water had been trapped during flash-cooling, and less crystalline ice has formed in E than in D
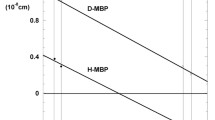
The earlier studies measured D2O in natural-abundance PM (Weik et al. 2005). Here, we examined the temperature-dependent lamellar spacing of the D-PM-H2O-Hh sample to address possible isotope effects. Figure 2 shows the lamellar spacing of flash-cooled D-PM-H2O-Hh upon heating, which exhibits a similar temperature dependence to the one of H-PM-D2O-Hh measured earlier (Weik et al. 2005). The lamellar spacing of about 57.5 Å remains constant up to 200 K, then decreases to reach a minimal value of 54 Å at 250 K (only first hydration layers remain) and increases again above 260 K to reach the initial value of 62 Å at room temperature. The lamellar spacing of H-PM-D2O-Hh (Weik et al. 2005) and D-PM-H2O-Hh samples thus display an identical temperature-dependence within the experimental temperature error estimated to be about 5 K. Second hydration-layer water, H2O or D2O, thus turns liquid-like at 200 K.

Lamellar spacing of D-PM stacks, equilibrated in H2O, after flash cooling in liquid nitrogen, upon heating from 100 to 290 K
Comparison of PM dynamics (H-PM-D2O-Lh sample) and dynamics of the first layer of hydration water (D-PM-H2O-Lh sample)
A direct comparison of PM and hydration-water dynamics has been made recently by EINS using deuterated membranes hydrated in H2O (the same D-PM-H2O-Hh sample as the one examined in the previous section) and natural-abundance membranes in D2O (Wood et al. 2007). A transition in the mean square displacements of hydration water at 200 K, attributed by MD simulations to the onset of water translational mobility, did not trigger a dynamical transition in PM that, in fact, took place at 260 K. The samples were characterised by a high-hydration level corresponding to a lamellar spacing of 62 Å at room temperature (i.e. 13 Å of inter-membrane water) that is not constant as a function of (cryo-) temperature (see Fig. 1). Here, we present data on samples with a lower hydration level corresponding to a lamellar spacing of 54 Å (i.e. 5 Å of inter-membrane water), which varies by less than 0.3 Å between 300 and 50 K. Furthermore, upon heating and cooling, no crystalline ice is formed (Lechner et al. 1998).
Figure 3 shows the elastic intensity summed over the Q-range examined (see “Materials and methods”) and the extracted MSD of D-PM-H2O-Lh and HPM-H2O-Lh. The elastic intensities (Fig. 3a) are very different for both samples, with inflections seen at approximately the same temperatures for which they were seen in the “high-hydration” samples (Wood et al. 2007): 120 and 260 K for the H-PM-D2O-Lh sample, 200 and 260 K for the D-PM-H2O-Lh sample.

Comparison of purple membrane dynamics (H-PM-D2O-Lh; open diamonds) and hydration water dynamics (D-PM-H2O-Lh; filled diamonds). a Sum of the scattered normalised elastic intensity as a function of temperature. Inflections can be seen at approximately 120 and 260 K in the purple membrane data and at 200 and 260 K in the hydration water data. b Mean square displacements as a function of temperature
Using the summed elastic intensities (where the error on each point is of the size of the points in Fig. 3a) as a guide, the atomic MSD of water and PM motions can be divided into regions with different characteristic temperatures. Below 120 K, MSD of both membrane (H-PM-D2O-Lh sample) and water (D-PM-H2O-Lh) are similar. Following an inflection at 120 K, the membrane’s MSD are clearly above the water’s MSD up to 260 K—the membrane has larger amplitude motions than its hydration water. The hydration water MSD evolves linearly with temperature to a higher temperature than PM, and undergo a broad transition at 200 K. At 260 K, the water MSD catches up with the membrane MSD and are almost identical to them up until 288 K. Above 288 K, it becomes impossible to calculate MSD for the water molecules, since their motions become too large to be resolved, and the Gaussian approximation is no longer valid.
Although the same transition temperatures are observed for the low-hydration samples studied here and the high-hydration samples measured earlier (Wood et al. 2007), small differences in the dynamics of water in the two hydration states can be seen. Water MSDs for the high-hydration and low-hydration samples are plotted for comparison in Fig. 4, where two temperature regions with significant differences in dynamics are evident. The first is between 150 and approximately 200 K, where the low-hydration sample displays larger MSD than the high-hydration sample. This indicates that the two inter-membrane water layers, present in both high- and low-hydration samples, are more mobile than ice in the same temperature region. The second temperature region where a large difference is observed is above 260 K, temperature above which in the case of high-hydration samples the ice present in the sample melts and flows back into the membrane stack and 13 Å of water is in the inter-lamellar space. Above 260 K, water in the high-hydration sample has much larger MSD than water in the low-hydration sample, implying that four layers of water confined in PM stacks show on average higher mobility on the ps–ns time scale than two water layers.

Comparison of water dynamics in high-hydration (Wood et al. 2007) and low-hydration samples (from Fig. 3b). Atomic mean square displacements of D-PM-H2O-Lh (filled diamonds) and D-PM-H2O-Hh (open circles) are shown as a function of temperature. For the low-hydration sample, all of the water is inter-membrane in the whole temperature region and contributes to 60% of the scattering cross section (40% is due to PM atoms). For the high-hydration samples, below 250 K, the water is in two forms: inter-membrane water and ice, which contribute, respectively, 31 and 49% to the scattering. Above 270 K, 80% of the scattering comes from inter-membrane water in the high-hydration sample (20% is due to PM atoms). In this estimation, it is assumed that all the exchangeable protons/deuterons in the lipid head-groups exchange and that 60% of those in the protein do so
The main conclusion by Wood et al. (2007) is confirmed by the present experiments at a lower hydration level: the dynamical transition in the hydration water at 200 K does not trigger a transition in the membrane at the same temperature. Since BR contributes most to the scattering cross section of PM, it can be supposed that the absence of a transition at 200 K in the whole PM indicates that no transition occurs in the protein. The result suggests that, in contrast to soluble proteins, the dynamics of water and membrane proteins on the ns–ps time scale are decoupled below 260 K, possibly suggesting an important role of lipids in the dynamical interdependency of BR, lipids and water.
Abbreviations
- BR:
-
Bacteriorhodopsin
- PM:
-
Purple membrane
- EINS:
-
Elastic incoherent neutron scattering
- MD:
-
Molecular dynamics
- MSD:
-
Mean square displacements
References
Berntsen P, Bergman R, Jansson H, Weik M, Swenson J (2005) Dielectric and calorimetric studies of hydrated purple membrane. Biophys J 89:3120–3128
Buchsteiner A, Lechner RE, Hauss T, Dencher NA (2007) Relationship between structure, dynamics and function of hydrated purple membrane investigated by neutron scattering and dielectric spectroscopy. J Mol Biol 371:914–923
Caliskan G, Briber RM, Thirumalai D, Garcia-Sakai V, Woodson SA, Sokolov AP (2006) Dynamic transition in tRNA is solvent induced. J Am Chem Soc 128:32–33
Caliskan G, Mechtani D, Roh JH, Kisliuk A, Sokolov AP, Azzam S, Cicerone MT, Lin-Gibson S, Peral I (2004) Protein and solvent dynamics: how strongly are they coupled? J Chem Phys 121:1978–1983
Cordone L, Ferrand M, Vitrano E, Zaccai G (1999) Harmonic behavior of trehalose-coated carbon-monoxy-myoglobin at high temperature. Biophys J 76:1043–1047
Cornicchi E, Marconi M, Onori G, Paciaroni A (2006) Controlling the protein dynamical transition with sugar-based bioprotectant matrices: a neutron scattering study. Biophys J 91:289–297
Cornicchi E, Onori G, Paciaroni A (2005) Picosecond-time-scale fluctuations of proteins in glassy matrices: the role of viscosity. Phys Rev Lett 95:158104
Dencher NA, Sass HJ, Buldt G (2000) Water and bacteriorhodopsin: structure, dynamics, and function. Biochim Biophys Acta 1460:192–203
Doster W, Cusack S, Petry W (1989) Dynamical transition of myoglobin revealed by inelastic neutron scattering. Nature 337:754–756
Engelman DM, Zaccai G (1980) Bacteriorhodopsin is an inside-out protein. Proc Natl Acad Sci USA 77:5894–5898
Fenimore PW, Frauenfelder H, McMahon BH, Parak FG (2002) Slaving: solvent fluctuations dominate protein dynamics and functions. Proc Natl Acad Sci USA 99:16047–16051
Fenimore PW, Frauenfelder H, McMahon BH, Young RD (2004) Bulk-solvent and hydration-shell fluctuations, similar to alpha- and beta-fluctuations in glasses, control protein motions and functions. Proc Natl Acad Sci USA 101:14408–14413
Ferrand M, Dianoux AJ, Petry W, Zaccai G (1993) Thermal motions and function of bacteriorhodopsin in purple membranes: effects of temperature and hydration studied by neutron scattering. Proc Natl Acad Sci USA 90:9668–9672
Fitter J (1999) The temperature dependence of internal molecular motions in hydrated and dry alpha-amylase: the role of hydration water in the dynamical transition of proteins. Biophys J 76:1034–1042
Fitter J, Lechner RE, Buldt G, Dencher NA (1996) Internal molecular motions of bacteriorhodopsin: hydration-induced flexibility studied by quasielastic incoherent neutron scattering using oriented purple membranes. Proc Natl Acad Sci USA 93:7600–7605
Fitter J, Lechner RE, Dencher NA (1997) Picosecond molecular motions in bacteriorhodopsin from neutron scattering. Biophys J 73:2126–2137
Fitter J, Verclas SA, Lechner RE, Seelert H, Dencher NA (1998) Function and picosecond dynamics of bacteriorhodopsin in purple membrane at different lipidation and hydration. FEBS Lett 433:321–325
Frick B, Gonzalez M (2001) Five years operation of the second generation backscattering spectrometer IN16—a retrospective, recent developments and plans. Phys B Condens Matter 301:8–19
Gabel F, Bicout D, Lehnert U, Tehei M, Weik M, Zaccai G (2002) Protein dynamics studied by neutron scattering. Q Rev Biophys 35:327–367
Henderson R (1975) The structure of the purple membrane from Halobacterium hallobium: analysis of the X-ray diffraction pattern. J Mol Biol 93:123–138
Iben IE, Braunstein D, Doster W, Frauenfelder H, Hong MK, Johnson JB, Luck S, Ormos P, Schulte A, Steinbach PJ, Xie AH, Young RD (1989) Glassy behavior of a protein. Phys Rev Lett 62:1916–1919
Kamihira M, Watts A (2006) Functionally relevant coupled dynamic profile of bacteriorhodopsin and lipids in purple membranes. Biochemistry 45:4304–4313
Lechner RE, Fitter J, Dencher NA, Hauss T (1998) Dehydration of biological membranes by cooling: an investigation on the purple membrane. J Mol Biol 277:593–603
Lehnert U, Reat V, Weik M, Zaccai G, Pfister C (1998) Thermal motions in bacteriorhodopsin at different hydration levels studied by neutron scattering: correlation with kinetics and light-induced conformational changes. Biophys J 75:1945–1952
Oesterhelt D, Stoeckenius W (1974) Isolation of the cell membrane of Halobacterium halobium and its fractionation into red and purple membrane. Methods Enzymol 31:667–678
Paciaroni A, Cinelli S, Onori G (2002) Effect of the environment on the protein dynamical transition: a neutron scattering study. Biophys J 83:1157–1164
Patzelt H, Ulrich AS, Egbringhoff H, Düx P, Ashurst J, Simon B, Oschkinat H, Oesterhelt D (1997) Structural investigation on isotope labelled native bacteriorhodopsin in detergent micelles by NMR spectroscopy. J Biomol NMR 10:95–106
Pieper J, Hauss T, Buchsteiner A, Baczynski K, Adamiak K, Lechner RE, Renger G (2007) Temperature- and hydration-dependent protein dynamics in photosystem II of green plants studied by quasielastic neutron scattering. Biochemistry 46:11398–11409
Reat V, Dunn R, Ferrand M, Finney JL, Daniel RM, Smith JC (2000) Solvent dependence of dynamic transitions in protein solutions. Proc Natl Acad Sci USA 97:9961–9966
Reat V, Patzelt H, Ferrand M, Pfister C, Oesterhelt D, Zaccai G (1998) Dynamics of different functional parts of bacteriorhodopsin: H-2H labeling and neutron scattering. Proc Natl Acad Sci USA 95:4970–4975
Roh JH, Curtis JE, Azzam S, Novikov VN, Peral I, Chowdhuri Z, Gregory RB, Sokolov AP (2006) Influence of hydration on the dynamics of lysozyme. Biophys J 91:2573–2588
Tarek M, Tobias DJ (2002) Role of protein-water hydrogen bond dynamics in the protein dynamical transition. Phys Rev Lett 88:138101
Tournier AL, Xu J, Smith JC (2003) Translational hydration water dynamics drives the protein glass transition. Biophys J 85:1871–1875
Tsai AM, Neumann DA, Bell LN (2000) Molecular dynamics of solid-state lysozyme as affected by glycerol and water: a neutron scattering study. Biophys J 79:2728–2732
Weik M (2003) Low-temperature behavior of water confined by biological macromolecules and its relation to protein dynamics. Eur Phys J E Soft Matter 12:153–158
Weik M, Lehnert U, Zaccai G (2005) Liquid-like water confined in stacks of biological membranes at 200 k and its relation to protein dynamics. Biophys J 89:3639–3646
Weik M, Zaccai G, Dencher NA, Oesterhelt D, Hauss T (1998) Structure and hydration of the M-state of the bacteriorhodopsin mutant D96N studied by neutron diffraction. J Mol Biol 275:625–634
Wood K, Caronna C, Fouquet P, Hauessler W, Natali F, Ollivier J, Orecchini A, Plazanet M, Zaccai G (2008) A benchmark for protein dynamics: ribonuclease A measured by neutron scattering in a large wavevector-energy transfer range. Chem Phys. doi:10.1016/j.chemphys.2007.09.012
Wood K, Plazanet M, Gabel F, Kessler B, Oesterhelt D, Tobias DJ, Zaccai G, Weik M (2007) Coupling of protein and hydration-water dynamics in biological membranes. Proc Natl Acad Sci USA 104:18049–18054
Zaccai G, Gilmore DJ (1979) Areas of hydration in the purple membrane of Halobacterium halobium: a neutron diffraction study. J Mol Biol 132:181–191
Acknowledgments
We are grateful to Heloisa Nunes Bordallo, Bruno Demé and Bernhard Frick for their help during data collection at the ILL. We also thank Marek Koza and Douglas Tobias for fruitful discussions. Financial support by the CEA, the CNRS and the UJF is acknowledged, as well as a grant to MW from the Agence Nationale de la Recherche (project number JC05_45685). The study was supported by the EU DLAB program under contract numbers HPRI-CT-2001-50035 and RII3-CT-2003-505925.
Author information
Authors and Affiliations
Corresponding author
Additional information
Advanced neutron scattering and complementary techniques to study biological systems. Contributions from the meetings, “Neutrons in Biology”, STFC Rutherford Appleton Laboratory, Didcot, UK, 11–13 July and “Proteins At Work 2007”, Perugia, Italy, 28–30 May 2007.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wood, K., Plazanet, M., Gabel, F. et al. Dynamics of hydration water in deuterated purple membranes explored by neutron scattering. Eur Biophys J 37, 619–626 (2008). https://doi.org/10.1007/s00249-008-0285-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00249-008-0285-0