
Abstract
Microbiomes play vital roles in insect fitness and health and can be influenced by interactions between insects and their parasites. Many studies investigate the microbiome of free-living insects, whereas microbiomes of endoparasitoids and their interactions with parasitised insects are less explored. Due to their development in the constrained environment within a host, endoparasitoids are expected to have less diverse yet distinct microbiomes. We used high-throughput 16S rRNA gene amplicon sequencing to characterise the bacterial communities of Dipterophagus daci (Strepsiptera) and seven of its tephritid fruit fly host species. Bacterial communities of D. daci were less diverse and contained fewer taxa relative to the bacterial communities of the tephritid hosts. The strepsipteran’s microbiome was dominated by Pseudomonadota (formerly Proteobacteria) (> 96%), mainly attributed to the presence of Wolbachia, with few other bacterial community members, indicative of an overall less diverse microbiome in D. daci. In contrast, a dominance of Wolbachia was not found in flies parasitised by early stages of D. daci nor unparasitised flies. Yet, early stages of D. daci parasitisation resulted in structural changes in the bacterial communities of parasitised flies. Furthermore, parasitisation with early stages of D. daci with Wolbachia was associated with a change in the relative abundance of some bacterial taxa relative to parasitisation with early stages of D. daci lacking Wolbachia. Our study is a first comprehensive characterisation of bacterial communities in a Strepsiptera species together with the more diverse bacterial communities of its hosts and reveals effects of concealed stages of parasitisation on host bacterial communities.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Insects have associations with diverse microbial communities that are important in host biology, host fitness and immunity and can provide protection against pathogens, parasitoids and toxins [1,2,3]. Symbiotic microbes can reside within the digestive tract, in particular the gut lumen [4, 5], on the surface of the insect host (ectosymbionts) and within host cells and tissues (endosymbionts) [6, 7]. The most common endosymbionts are maternally inherited Wolbachia (Alphaproteobacteria) that occur in over 50% of insects and other arthropod species [8, 9]. In many hosts, they manipulate host reproduction to enhance the production of infected females and thereby their prevalence in populations [9, 10]. For example, the induction of cytoplasmic incompatibility (CI) results in embryonic mortality when infected males mate with uninfected females or females infected with an incompatible Wolbachia strain; however, this CI is overcome in embryos of females infected with the same or a compatible Wolbachia strain, which can rescue the CI effect [9, 10]. Besides reproductive manipulation, Wolbachia strains may be beneficial to their hosts. Some strains confer protection to their hosts against parasites, viruses and other pathogens [11,12,13]. Other Wolbachia strains can synthesise vitamins deficient in host diets; for instance Wolbachia provides B vitamins to the bedbug, Cimex lectularius [14, 15]. Wolbachia can also influence the microbiome of hosts [16,17,18]. For instance, Wolbachia alters the relative abundance of bacterial taxa within microbial communities in the parasitoid wasp Nasonia vitripennis [18], the cabbage root fly Delia radicum [19] and adult mosquitoes [16]. Conversely, other symbiotic bacteria can influence Wolbachia prevalence and abundance, for example, Asaia can impede the establishment and stable transmission of Wolbachia in mosquitoes [20].
Many studies of host-microbe interactions have been performed on free-living insects, but less so on parasites [21] such as endoparasitoids that develop entirely within free-living insects. So far, the microbiomes of Strepsiptera, an endoparasitic insect order, have not yet been investigated [22], likely due to its extraordinary characteristic of extreme endoparasitism. Strepsiptera consists of 630 known species that parasitise hosts belonging to the seven insect orders Blattodea, Diptera, Hemiptera, Hymenoptera, Mantodea, Orthoptera and Zygentoma [23]. All strepsipteran species are obligate endoparasitoids and almost entirely complete their life cycle within their hosts [24, 25]. Adult strepsipterans display stark sexual dimorphism, with adult males that have external morphological features of a free-living adult insect, while the females are neotenic (lacking adult features) and fully endoparasitic within their hosts, except for adult females of one strepsipteran family, the Mengenillidae (suborder Mengenillidia) which are free-living [25, 26]. Parasitisation of the host occurs via the first instar larvae (planidia), which leave a parasitised host to then enter a new host where they undergo hypermetamorphosis to the fourth larval instar [26, 27]. In Mengenillidae, the fourth larval instars of both sexes leave and pupate on the outside of their hosts, while in all other families (all contained within the suborder Stylopidia), both male pupae and neotenic females extrude through the hosts’ cuticle [26, 27]. Males then emerge from the pupae within the host while females remain fully endoparasitic. Therefore, parasitisation by strepsipterans becomes visible as stylopisation in the later stages of strepsipteran development, while early stages of Strepsiptera may remain concealed in hosts and may only be detected by dissection or molecular tools [28]. Furthermore, strepsipterans are koinobionts which means that the hosts continue to live and feed while these endoparasitoids develop within their insect host [26].
Host-parasite interactions can be shaped by microbes associated with either the host or the parasite [21, 29]. Microbes can protect their hosts against parasites, for instance, a bacterial symbiont of aphids, Hamiltonella defensa, can protect its hosts against the parasitoid wasp Lysiphlebus fabarum [30, 31]. In contrast, the host microbiome can also aid in the establishment of parasites in their insect hosts, as seen in the interaction between the tapeworm Hymenolepis diminuta and its intermediate host, the grain beetle Tenebrio molitor [32]. Strepsiptera have an intimate relationship with their hosts and depend entirely on their hosts for nourishment [33]. Therefore, endoparasitoids may compete with the hosts’ microbiota for resources. Furthermore, host-associated microbes can also influence host immunity. For instance, altering the bacterial communities of the fruit fly Drosophila melanogaster by antibiotic treatment influenced its resistance to the parasitoid wasp Asobara tabida by moderating the encapsulation rate of the parasitoid eggs by the host [34]. Conversely, parasitisation can affect the host microbiome, such as seen in larvae of the two moth species Diatraea saccharalis and Spodoptera frugiperda parasitised by the parasitoid wasp Cotesia flavipes, which changed the bacterial community composition and structure of the moth larvae [35].
While some host species may have few or no microbial associations [36], parasites and parasitoids may have less diverse microbiomes than their hosts due to the relatively small size, their life cycle and exclusive dependence on their hosts for resources. For example, a parasitic plant, the obligate parasite Orobanche hederae, exhibits a reduced microbiome compared to its host plant Hedera [37]. Similarly, the bacterial alpha diversity is lower within the intestinal tapeworm Eubothrium than in its host, the Atlantic salmon [38], and the microbiomes associated with entomopathogenic nematodes used in the biological control of insect pests are of relatively low complexity [39]. Yet, an amplicon sequencing study of one of the smallest insects, the parasitoid wasp Megaphragma amalphitanum, has revealed that it still carries a diverse variety of bacteria, albeit different in composition from other larger parasitoid wasp species [40].
Strepsipteran neotenic females reproduce viviparously and obtain nutrients exclusively from the host hemolymph [33]. Strepsipteran larvae have a gut, and nutrient uptake from the host hemolymph occurs in the midgut; however, after extrusion of the females, the strepsipteran gut is degenerate and filled with hemolymph [41], and nutrient uptake from the host hemolymph occurs via a particular structure, the apron [42].Therefore, due to the complete dependence of Strepsiptera on their hosts, it could be predicted that they have a less diverse microbiome. Such microbiome simplicity in parasites may be a parallel feature to the reduced morphological and genomic characteristics observed in several parasitic, ectosymbiotic and endosymbiotic organisms [26, 43,44,45] including the reduced genomic characteristics exhibited by bacterial endosymbionts [46, 47].
Our study focused on Dipterophagus daci, a strepsipteran endoparasitoid of tephritid fruit flies [48] belonging to the family of Halictophagidae [28]. To date, D. daci is the only described strepsipteran endoparasitoid of Diptera (besides other undescribed strepsipteran endoparasitoids of platystomatid flies from Papua New Guinea) and has been reported from 22 species of the tephritid subfamily of Dacini in Australia and the Solomon Islands [28, 48, 49]. A recent study revealed that the presence of two Wolbachia strains previously detected in flies of seven Australian tephritid species [50, 51] was due to concealed parasitisation of these flies with early developmental stages of D. daci [28]. This recent study also concluded that D. daci is the actual host of the two Wolbachia strains wDdac1 and wDdac2, which occur at high prevalence in this host, and D. daci without Wolbachia is found in only about 10% of parasitised flies [28]. The two strains wDdac1 and wDdac2 belong to the Wolbachia supergroup A and have previously been characterised using the Wolbachia surface protein (wsp) gene and five multi locus sequence typing (MLST) loci [50].
Tephritid fruit fly species are diverse and can infest diverse host plants but also different plant parts [52, 53]. Furthermore, tephritids have bacterial communities that can vary in diversity and structure depending on life stage, fly species and phylogeny, host plant species, diet and rearing environment [54,55,56]. For example, the bacterial communities of the island fly, Dirioxa pornia, differs from those of Bactrocera species, and this could be due to their different life histories [54]. Furthermore, bacterial communities with diverse compositions were observed among different Bactrocera species suggesting that several factors such as host plant specialisation and domestication play a role in shaping the microbiome of tephritid fruit flies [54].
Our study aimed to explore the diversity and composition of bacterial communities of the strepsipteran D. daci. We hypothesised that due to its endoparasitic life cycle, the bacterial communities of D. daci consist of only few taxa and are distinct from the bacterial communities of its fruit fly hosts. Furthermore, we expected that Wolbachia would dominate the microbiome of D. daci but not of the parasitised fruit fly species. We also tested whether early stages of D. daci parasitisation influenced the fruit fly microbiome, and whether this was influenced by the presence of Wolbachia in D. daci. To address these questions, we performed high-throughput next generation amplicon sequencing analyses of the commonly used and conserved bacterial marker gene, the 16S rRNA gene, of (i) D. daci male pupae, (ii) fruit flies parasitised by early stages of Wolbachia-positive D. daci, (iii) fruit flies parasitised by early stages of Wolbachia-negative D. daci and (iv) unparasitised fruit flies.
Materials and Methods
Fruit Fly Collection and DNA Extraction
We sequenced and analysed the bacterial 16S rRNA gene diversity of total genomic DNA extracts of 84 adult male fruit flies and 17 D. daci male pupae (Table S1) [28, 50, 51]. The 84 adult male fruit flies comprised individuals of seven species including Bactrocera bryoniae (n = 4), Bactrocera decurtans (n = 2), Bactrocera frauenfeldi (n = 11), Bactrocera neohumeralis (n = 22), Bactrocera tryoni (n = 32), Dacus axanus (n = 2) and Zeugodacus strigifinis (n = 11), collected from Queensland in 1998, 2001, 2012, 2013 and 2019 using male lure traps with malathion as part of fruit fly monitoring programs (Table S1) [57]. After emptying of traps, the trapped fruit fly specimens were kept dry and at room temperature for identification and then stored in ethanol at − 20 °C until DNA extraction. The D. daci male pupae were dissected from visibly parasitised (stylopised) male fruit flies of six species (Bactrocera breviaculeus, B. frauenfeldi, Bactrocera mayi, Bactrocera pallida, B. neohumeralis and B. tryoni; Table S1) collected from Queensland in 2019 [28]. The pupae were removed from the cephalotheca extruding from the abdomen of the parasitised fruit flies (Fig. 1a and b). In contrast to these stylopised individuals, the 84 adult male flies were either parasitised by concealed stages of D. daci (D. daci-positive by PCR) or unparasitised (D. daci-negative by PCR) (Fig. 1c) [28]. Prior to DNA extraction, the male fruit fly specimens and D. daci male pupae were surface-treated with 4% sodium hypochlorite to remove any external microorganisms and then washed with 0.2% Triton-X and rinsed thoroughly using Milli-Q water [51].

Field-caught male tephritid fruit flies collected using male lure traps. A Stylopised male fruit fly (Bactrocera neohumeralis); B Dipterophagus daci male pupa dissected from a stylopised male fruit fly (green circle shows the cephalotheca containing a male pupa); C non-stylopised male fruit fly (Bactrocera bryoniae)
Total genomic DNA was extracted from individual fruit fly male abdomens and individual whole D. daci pupae using GenElute DNA Miniprep Kit (Sigma-Aldrich) as per the manufacturer’s instructions. The DNA quality was determined using NanoDrop and gel electrophoresis, and the extract was then stored at − 20 °C for subsequent experiments. The fruit fly and D. daci DNA extracts were screened by PCR using specific primers for the wsp and 16S rRNA genes [50, 51] and the D. daci cytochrome c oxidase I (cox1) gene [28]. Based on the PCR results, the samples were categorised into four groups: (i) D. daci male pupae (Dd) which were all positive for Wolbachia (n = 17), (ii) fruit flies parasitised by early stages of Wolbachia-positive D. daci (FliesDdW) (n = 30), (iii) fruit flies parasitised by early stages of D. daci without detectable Wolbachia (FliesDd) (n = 19) and (iv) unparasitised fruit flies (Flies) (n = 35) (Table S1). It is noted that Wolbachia-negative D. daci are relatively rare [28], and, therefore, flies parasitised by early stages of D. daci without detectable Wolbachia were preferentially included in our amplicon sequencing study to obtain a fair representation when compared to flies parasitised by early stages of Wolbachia-positive D. daci.
Bacterial 16S rRNA Gene Amplification and Sequencing
The DNA extracts were submitted for 16S rRNA gene amplicon sequencing on an Illumina MiSeq platform at the Western Sydney University Next Generation Sequencing Facility. Primers 341F (5′ CCTACGGGNGGCWGCAG) and 805R (5′ GACTACHVGGGTATCTAATCC) were used to amplify the V3–V4 region of the 16S rRNA gene with a total read length of 2 × 301 bp. A bacterial mock community (Microbial Community DNA Standard, ZymoBiomics) provided by the sequencing facility was included.
Sequence Analysis
After sequencing, the sequence reads were pre-processed, quality filtered and analysed using Quantitative Insight into Microbial Ecology (QIIME 2, v. 2019.7). Raw demultiplexed Illumina fastq sequence (Phred33 applied for quality control) and mapping files were imported into QIIME 2 for downstream processes. The manifest file was created by concatenating the forward and the reverse sequences. The DADA2 pipeline was used for denoising, quality filtering, dereplication and chimera removal [58]. Quality analysis was performed by trimming the primers and truncating the reads using the commands –p-trim-left-f 17, –p-trim-left-r 21, –p-trunc-len-f 290 and –p-trunc-len-r 210. A naive Bayes classifier was trained using the Greengenes 99% sequence similarity threshold for calling operational taxonomic units (OTUs) at the V3–V4 region of the 16S rRNA gene. Amplicon sequence variants (ASVs) from DADA2 were used for taxonomic classification at a 99% similarity threshold using QIIME 2 q2-feature-classifier plugin [59] and sample taxonomic composition, and structure was visualised using QIIME 2 bar plot and plotted in R version 3.6.3 (R core Team, 2020, https://www.R-project.org/). The core-metrics-phylogenetic pipeline was used to construct the phylogenetic tree. A rarefaction curve was used to assess adequate sampling of the microbial communities. Based on the rarefaction curve, the overall alpha and beta diversity analyses were performed at a sampling depth of 6120, and at 1000 upon filtering out the Wolbachia reads, to avoid biases using the q2-diversity plugin. We estimated the alpha diversity among the four groups of samples using Shannon diversity index and Pielou’s evenness. Beta diversity was assessed using weighted UniFrac distance (phylogenetic relationships and relative abundance) and Bray–Curtis distance (relative abundance) to determine the microbial community variation in the four sample groups (Dd, FliesDdW, FliesDd and Flies) with pairwise comparisons (PERMANOVA) using qiime diversity beta-group-significance in QIIME 2 (v. 2019.7). Beta diversity results were also visualised using principal coordinates analysis (PCoA) plots in R. To confirm that the Wolbachia ASV of our study corresponded to the Wolbachia previously characterised from D. daci, we compared it in a multiple sequence alignment using CLUSTALW together with 16S rRNA gene sequences of wDdac1 and wDdac2 extracted from genome reads obtained from the Wolbachia-positive sample B. frauenfeldi 485 as part of a whole genome sequencing project [28, 43] and with a cloned Wolbachia 16S rRNA gene (GenBank accession KC775794) sequence obtained from B. neohumeralis [51].
Differential Relative Abundance Analysis
To determine whether early stages of D. daci parasitisation had an impact on the microbiome of the host fruit fly, we compared the relative abundance of bacterial taxa in the fruit flies parasitised by early stages of D. daci without detectable Wolbachia (FliesDd) to the unparasitised fruit flies (Flies) (Table S1). Similarly, we assessed whether parasitisation by early stages of Wolbachia-positive D. daci had an impact on the host fruit fly microbiome by comparing the FliesDd samples to the FliesDdW samples (Table S1). For these comparisons, we used the original taxonomic assignments of ASVs (at 99% identity) with the Wolbachia reads excluded. OTU datasets generated in QIIME and summarised at genus level were imported into Phyloseq for downstream analysis. The differential relative abundance was then performed in edgeR [60].
Results
Sequence Read Analysis
The 101 sequenced 16S rRNA gene amplicon libraries (Table S1) included 17 D. daci male pupae (Dd), 30 fruit flies parasitised by Wolbachia-positive D. daci (FliesDdW), 19 fruit flies parasitised by D. daci without detectable Wolbachia (FliesDd) and 35 unparasitised fruit flies (Flies) across seven tephritid species (B. bryoniae, B. decurtans, B. frauenfeldi, B. neohumeralis, B. tryoni, D. axanus and Z. strigifinis). After quality control and filtering, we obtained a total of 2,274,402 sequence reads, with a mean sequence read number of 22,519 per sample (between 42 and 120,845 sequence reads per sample). After normalising the sequence read number at a sampling depth of 6120 to minimise biases, we excluded one fruit fly specimen that contained fewer than 6120 sequences (one D. axanus Flies sample with 42 sequence reads) from the subsequent analysis (Table S1). A total of 1808 ASVs were identified in this study (Table S2).
Bacterial Community of D. daci
The bacterial community of D. daci pupae was dominated by the class Alphaproteobacteria, accounting for 79.2% of the total sequence reads (Fig. 2a, Table S3). Other classes include Gammaproteobacteria (16.2%), Bacilli (2.1%), Deltaproteobacteria (0.6%), Flavobacteria (0.66%), Bacteroidia and various other classes with a combined relative abundance of < 1% (Fig. 2a, Table S3). The ASV with the highest relative abundance was one Wolbachia 16S rRNA gene sequence accounting for 78.7% of all sequence reads and was present in all 17 D. daci pupae (Fig. 2b, Table S4). The 16S rRNA gene sequences from wDdac1 and wDdac2 obtained from a previous whole genome sequencing project did not vary in the V3–V4 region (402 bp) and were identical to the dominant Wolbachia ASV obtained in this study (Fig. S1). An additional 15 Wolbachia ASVs, all singletons, consisted of sequences with up to 2 mismatches to the dominant ASV. The Wolbachia 16S rRNA gene sequence previously obtained in a molecular cloning experiment from the Wolbachia-positive B. neohumeralis was also identical to the wDdac1 and wDdac2 16S rRNA gene sequences albeit in another region (349 bp) (Fig. S1) further confirming that the two Wolbachia strains cannot be differentiated in the V3–V4 region. Other genera that were relatively abundant included Serratia (5.6%), Trabulsiella (2.4%), Enterobacter (1.6%), one unknown Pasteurellales ASV (2.4%), one unknown Enterobacteriaceae ASV (1.4%) and Lactococcus (1.09%) (Fig. 2b, Table S4).

Relative abundance of bacterial taxa in Dipterophagus daci. A Barplot of the relatively abundant bacterial classes in the four categories of samples: D. daci male pupae (Dd), fruit flies parasitised by early stages of Wolbachia-positive D. daci (FliesDdW), fruit flies parasitised by early stages of D. daci without detectable Wolbachia (FliesDd) and unparasitised fruit flies (Flies); B barplot of the relatively abundant bacterial genera in the 17 D. daci pupae samples. The highest available classification was used for taxa with no assigned genus
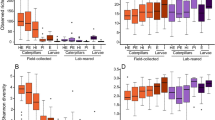
Alpha diversity analysis revealed low Shannon diversity and Pielou’s evenness indices in D. daci, whereas both indices were higher for flies (Kruskal–Wallis, p < 0.05, Fig. 3a and b, Table S5). Beta diversity analysis of bacterial communities using weighted UniFrac and Bray–Curtis PCoAs showed that D. daci bacterial communities clustered separately from those of the host fruit flies (Fig. 3c and d, Table S6 and S7). PERMANOVA analyses based on Bray–Curtis results also revealed the distinct clustering of D. daci bacterial communities from those of host fruit flies (Table 1).

Alpha and beta diversity analysis of Dipterophagus daci male pupae (Dd), unparasitised fruit flies (Flies), fruit flies parasitised by early stages of D. daci without detectable Wolbachia (FliesDd) and fruit flies parasitised by early stages of Wolbachia-positive D. daci (FliesDdW). A Shannon diversity, B Pielou’s evenness, C weighted UniFrac and D Bray–Curtis principle coordinate analysis (PCoA) plots. Different letters indicate significant differences in Kruskal–Wallis comparisons (p < 0.05)
Comparison of Bacterial Communities Among Fruit Fly Species
The weighted UniFrac analysis showed no distinct clustering pattern in the fruit fly species (Fig. 4a, Table S8). However, Bray–Curtis PCoA revealed a distinct separation in the bacterial communities of Z. strigifinis and the remaining six fruit fly species (Fig. 4b, Table S9). Therefore, the fruit fly bacterial communities of Z. strigifinis, B. bryoniae, B. frauenfeldi, B. neohumeralis and B. tryoni were investigated to determine the differences in relative abundances (B. decurtans and D. axanus were not included due to low sample numbers). Prior to this, the alpha diversity analysis of the five fruit fly species was performed. Both Shannon diversity and Pielou’s evenness indices revealed a significant difference in bacterial communities of B. frauenfeldi and Z. strigifinis compared to other fruit fly species (Kruskal–Wallis, p < 0.05, Fig. S2, Table S10).

Analyses of fruit fly samples groups. A Weighted UniFrac and B Bray–Curtis beta diversity principle coordinate analysis (PCoA) plots to visualise the clustering and similarity of the fruit fly sample groups. The ellipses drawn based on the standard deviation show the clustering of the Bactrocera and Zeugodacus samples. C Bar plot of the most common bacterial genera in the host fruit flies Bactrocera bryoniae, Bactrocera frauenfeldi, Bactrocera neohumeralis, Bactrocera tryoni and Zeugodacus strigifinis. Analysis was performed on fruit fly specimens from all sample groups including unparasitised fruit flies (Flies), fruit flies parasitised by early stages of Dipterophagus daci without detectable Wolbachia (FliesDd) and fruit flies parasitised by early stages of Wolbachia-positive D. daci (FliesDdW)
Bacterial genera with the highest abundance in all fruit flies were Vagoccoccus (27.8%), one unknown Pasteurellales ASV (19.0%), one unknown Enterobacteriacea ASV (7.7%), Acinetobacter (6.4%), Providencia (6.2%), Enterobacter (4.6%), one unknown Desulfovibrionaceae ASV (3.9%), Dysgonomonas (3.6%), Klebsiella (2.5%), Citrobacter (2.1%), Serratia (1.7%), Trabulsiella (1.6%), Lactococcus (0.63%) and others (all remaining bacteria combined, 12.4%) (Fig. 4c, Table S11). Bar plots representing the diversity of the fruit fly bacterial communities revealed variability in the relative abundance of bacteria (Fig. 4c). The most striking difference was the low relative abundance of the one unknown Pasturellales ASV in Z. strigifinis (1%) compared to the other fruit fly species which contained this bacterium at relative abundances ranging from 13 to 35% (Fig. 4c, Table S11). Interestingly, Acinetobacter bacteria were relatively more abundant in Z. strigifinis (21%) compared to B. bryoniae (0.2%), B. frauenfeldi (8.9%), B. neohumeralis (0.01%) and B. tryoni (1.4%) (Fig. 4c, Table S11). PERMANOVA pairwise analyses based on both weighted UniFrac and Bray–Curtis results showed significant differences (p < 0.05, PERMANOVA) in bacterial communities of B. frauenfeldi compared to the other fruit fly species (Table 2). This could be attributed to the one unknown Enterobacteriaceae ASV that had a relative abundance of 15.1% in B. frauenfeldi, while the other fruit fly species had relative abundances of this unknown Enterobacteriaceae ASV ranging from 2.05 to 9.7% (Fig. 4c, Table S11). No significant differences were observed in comparisons among the bacterial communities of B. bryoniae, B. neohumeralis and B. tryoni (Table 2).
Influence of Early Stages of D. daci Parasitisation on Bacterial Communities in Fruit Fly Hosts
A comparison of the fruit flies parasitised by early stages of D. daci without detectable Wolbachia (FliesDd) and unparasitised flies (Flies) was performed to determine the impact of early D. daci parasitisation on the fruit fly bacterial diversity. The OTU datasets used were retrieved from QIIME and summarised to genus level. This comparison revealed an impact of early parasitisation by D. daci on the relative abundance of Pseudomonadota (formerly Proteobacteria) and Bacillota (formerly Firmicutes) in fruit flies (Fig. 5a, Table S12). The relative abundance of nine bacterial genera including Proteus, one unknown Enterobacteriaceae ASV, Klebsiella, one unknown Acetobacteriacea ASV, Ochrobactrum, Morganella, Providencia, three unknown Pasteurellales ASVs and Enterococcus were increased in FliesDd, while three bacterial genera (Enterobacter, Citrobacter and one unknown Halomonadacea ASV) decreased in FliesDd (Fig. 5a, Table S12).

Scatter plot of the bacterial taxa with differential relative abundance in A fruit flies parasitised by early stages of Dipterophagus daci without detectable Wolbachia (FliesDd) compared to unparasitised fruit (Flies) and in B fruit flies parasitised by early stages of Wolbachia-positive Dipterophagus daci (FliesDdW) compared to fruit flies parasitised by early stages of D. daci without detectable Wolbachia (FliesDd). A log fold change of logFC > 0 indicates that the abundance of the genera increased, whereas logFC < 0 indicates that the abundance of the genera decreased. The taxa with significantly different relative abundances are coloured by phylum
Similarly, we compared the relative abundance of bacterial taxa between flies parasitised by D. daci with and without detectable Wolbachia. Sequences used in this analysis were corrected for Wolbachia and normalised to a sequencing depth of 1000, based on the minimum number of reads after excluding Wolbachia. We found that the relative abundance of 11 genera comprising Proteus, Providencia, Dysgonomonas, Morganella, one unknown Acetobacteriacea ASV, two unknown Pasteurellales ASVs, Vagococcus, Serratia, one unknown Enterobacteriaceae ASV, Staphylococcus and Enterobacter were decreased in FliesDd, while the relative abundances of Klebsiella, Trabulsiella, Myroides and Citrobacter were increased (Fig. 5b, Table S13).
Discussion
We used 16S rRNA gene amplicon sequencing to characterise the bacterial communities involved in the interactions between tephritid fruit flies and the strepsipteran endoparasitoid D. daci. With this we have performed, according to our knowledge, the first comprehensive characterisation of bacterial communities in a species of the endoparasitic insect order Strepsiptera [23]. The bacterial communities of D. daci were dominated by Wolbachia; however, this dominance was not observed in fruit flies parasitised by early stages of Wolbachia-positive D. daci (and Wolbachia was completely absent in flies not parasitised by D. daci), supporting previous findings that D. daci is the host of Wolbachia in this host-parasitoid interaction [28]. We found that the bacterial communities of D. daci are not as diverse but distinct when compared to the more diverse bacterial communities of the fruit fly hosts. Furthermore, early stages of D. daci parasitisation and presence of Wolbachia in D. daci altered the microbiome of parasitised fruit flies. We also found that the bacterial communities of Z. strigifinis were distinct from the bacterial communities of the Bactrocera species and this may be linked to their different ecologies, with Z. strigifinis developing in cucurbit flowers, whereas the analysed Bactrocera species develop in fruit [53, 61].
Dipterophagus daci Has a Less Diverse Microbiome
The most abundant bacterial phylum in D. daci was Pseudomonadota comprising 96.2% of the total bacterial sequence reads, followed by Bacillota and Bacteroidota at a substantially lower relative abundance. A high relative abundance of Pseudomonadota and Bacillota has previously been detected in fruit flies [54, 62] and other insect species [55, 63]; however, the relative abundance of Pseudomonadota in D. daci pupae found in our study was generally higher and mostly just consisted of Wolbachia. This indicates that bacterial communities in D. daci are not very diverse, which is perhaps due to its parasitic life cycle. The strepsipteran D. daci and all other Strepsiptera are almost fully endoparasitic in their host and depend exclusively on the host for nourishment [24, 26, 33]. The high presentation of Pseudomonadota in D. daci was due to Wolbachia, a member of the Alphaproteobacteria, in combination with Gammaproteobacteria, Bacilli, Deltaproteobacteria, Bacteroidia and Flavobacteriia at substantially lower relative abundance. It needs to be noted that we were only able to characterise the bacterial communities of D. daci in isolation from its host by carefully dissecting pupae out of the cephalotheca extruding from the abdomen of parasitised fruit flies, followed by surface treatment to minimise contamination. We do not know how bacterial communities in D. daci change throughout its development. Given the endoparasitic life cycle of D. daci, it is likely that exposure to environmental bacteria is limited, which could impact the observed low levels of bacterial diversity in D. daci. Therefore, bacterial symbionts detected in D. daci pupae are either maternally inherited or horizontally acquired from the host fly or from the environment during the short period that planidial larvae search for new hosts. It is perhaps less likely that bacteria acquired by adult D. daci males are then paternally transmitted. We did not obtain free living males of D. daci as they would require different sampling techniques such as light trapping or collection of adult males emerging from parasitised flies and are, therefore, more difficult to collect than parasitised flies.
The Microbiome of D. daci is Dominated by Wolbachia and Distinct from the Fruit Fly Hosts' Bacterial Communities
Wolbachia is a common maternally inherited endosymbiont of insects and other arthropods that can manipulate host reproduction to increase its prevalence in host populations [8, 64, 65]. In several host species, Wolbachia provides fitness benefits which can also maintain this endosymbiont in host populations [66]. For several insect species, it has been found that, when present, Wolbachia can dominate bacterial communities within hosts [67, 68]. Our findings of the dominance of Wolbachia in bacterial communities within D. daci (but not in the bacterial communities within fruit flies) further confirms that the two Wolbachia strains first detected in fruit flies [50, 51] are actually associated with D. daci and had previously been detected in these fruit flies because of parasitisation by concealed early stages of Wolbachia-positive D. daci [28]. Additionally, alpha diversity analysis revealed low Shannon diversity and Pielou’s evenness values in D. daci bacterial communities (a consequence of the Wolbachia dominance) while this was not observed in fruit flies parasitised by early stages of Wolbachia-positive D. daci.
Previous analyses have found that Wolbachia occurs at high prevalence in D. daci [28], and D. daci is depauperate in mitogenome diversity across large parts of its geographic distribution [28, 43]. Because of maternal co-inheritance with mitochondria, Wolbachia may have caused a selective sweep of mitochondria due to either reproductive manipulation or beneficial host fitness effects. Previous analyses of whole genome sequenced specimens have not detected the presence of Wolbachia genes involved in reproductive manipulations, and, therefore, it is likely that Wolbachia confers a fitness benefit to D. daci [28], for example, by providing a key nutrient and/or supporting immunity; however, this will need further investigation. Most strepsipteran life stages are fully endoparasitic except for the free-living first instar larvae (planidia) and adult males and are therefore fully dependent on the host for nourishment [42]. The host may not always provide all the essential nutrition, and therefore endoparasitoids may form beneficial interactions with maternally inherited endosymbionts like Wolbachia. In the bedbug, Cimex lectularius, a Wolbachia supergroup F strain, provides B vitamins which are deficient in the bedbug’s diet [14]. Similarly, a Wolbachia supergroup A strain provides D. melanogaster with metabolic support in periods of nutritional stress [69], and Wolbachia supergroup B strains have been associated with synthesis of biotin and riboflavin to increase host fitness in the small brown planthopper Laodelphax striatellus and the brown planthopper Nilaparvata lugens [15]. Furthermore, Wolbachia supergroup A strains provide protection against pathogens such as RNA viruses in several insect species such as Drosophila [11, 13, 70] and mosquitoes [71, 72]. Throughout its entire development, D. daci is exposed to the fruit flies’ immune system and the host’s viruses. It has recently been found that the tephritid host species of D. daci have a very high incidence and prevalence of insect-specific RNA viruses [73] with vertical and horizontal transmission modes [74]. Future research should investigate how these viruses interact with fruit fly hosts and D. daci.
Furthermore, the weighted UniFrac and Bray–Curtis beta diversity analyses revealed that the bacterial community of D. daci was distinct from the bacterial communities of its fruit fly host species. This may be due to the phylogenetic distance between the strepsipteran and its fruit fly hosts, or the differences in host life cycle and diet. It also indicates that D. daci and the fruit fly hosts do not share microbiome components.
Variable Bacterial Communities in Fruit Fly Host Species
Our analyses of the bacterial communities in fruit flies with the presence of several Enterobacteriaceae taxa (one unknown Enterobacteriaceae ASV, Acinetobacter, Providencia, Enterobacter, Klebsiella, Citrobacter and Serratia) confirmed their importance in tephritid fruit fly microbiomes as found in previous studies [54, 56, 75]. However, we also found an abundance of bacterial taxa such as Vagoccoccus, one unknown Pasteurellales ASV, Trabulsiella, one unknown Desulfovibrionaceae ASV and Dysgonomonas, which were different bacterial community members when compared to previous studies on tephritid fruit flies. This difference could be attributed to our sample collection and handling procedures (samples were collected in male lure traps with an insecticide and kept dry and at room temperature until identification). Additionally, for our study, we specifically selected individuals that were parasitised by early stages of D. daci and this could also have resulted in a sampling bias.
Tephritid fruit fly species exhibit diverse life histories and host plant preferences [52, 53, 61], and these can affect their microbiomes [53, 54, 76]. The Shannon diversity and Pielou’s evenness showed significant difference in B. frauenfeldi and Z. strigifinis bacterial communities compared to the other fruit flies. Additionally, the Bray–Curtis PCoA revealed that bacterial communities associated with Z. strigifinis were distinct from those of Bactrocera species, possibly suggesting a fly genus effect, albeit we only included one Zeugodacus species in our study. Furthermore, there could be a host plant effect as Z. strigifinis is a pest of Cucurbitaceae flowers while B. tryoni, B. neohumeralis, B. frauenfeldi and B. bryoniae infest fruits [53, 61]. The weighted UniFrac analysis, however, did not show any distinct clustering, suggesting that the variation between the bacterial communities of Z. strigifinis and the Bactrocera species may only be in the relative abundance of the bacterial taxa that may have similar function. This difference may be due to the unknown ASVs of Pasteurellales and Acinetobacter. In addition to the different bacterial communities observed in Z. strigifinis, PERMANOVA revealed that B. frauenfeldi were also different in bacterial community structure when compared to B. bryoniae, B. tryoni and B. neohumeralis. Bactrocera tryoni and B. neohumeralis are closely related sibling species [77]; hence, this may explain the similarity of their bacterial communities [54], while it is unclear why B. bryoniae grouped with these two species.
Dipterophagus daci Parasitisation Alters Structure of Bacterial Communities
We observed a significant decrease in the relative abundance of nine bacterial genera in fruit flies parasitised by D. daci without Wolbachia, while three bacterial genera increased in their relative abundance, suggesting that D. daci parasitisation affects the relative abundance of bacterial taxa in host bacterial communities. Furthermore, despite the low relative abundance of Wolbachia in fruit flies parasitised by Wolbachia-positive D. daci, we found a decrease in the relative abundance of 11 bacterial genera in flies parasitised by Wolbachia-positive D. daci, while the relative abundance of four bacterial genera increased. This suggests that parasitisation by D. daci and presence of Wolbachia in D. daci affect bacterial communities in flies. This is in line with other research that has shown that microbes can influence host-parasite interactions [19, 29, 32, 35, 78]
Conclusions
According to our knowledge, our study is the first comprehensive characterisation of the bacterial communities of a strepsipteran using a next generation amplicon sequencing approach. We demonstrated that bacterial communities of D. daci are not very diverse, dominated by Wolbachia and distinct from those of its host fruit fly species, and this could be attributed to the differences in host life cycles, life histories and phylogeny. Further studies should investigate the role of the two Wolbachia strains in D. daci, in particular as it is clear from previous genome analyses that they lack the capacity to manipulate host reproduction yet have an overall high prevalence in D. daci [28]. Furthermore, we observed variability in the relative abundance of bacterial taxa across fruit fly species, irrespective of parasitisation by D. daci, suggesting that phylogeny, host plant preference and host plant use play a role in shaping bacterial communities in fruit flies [54]. In addition, early stages of D. daci parasitisation affected the relative abundance of bacteria in microbial communities of host fruit flies. Hence, parasitisation can also shape the microbiome of insects and should therefore be considered in host-microbiome studies.
Data Availability
The raw bacterial 16S rRNA gene sequence reads have been deposited in GenBank Sequence Read Archive (GenBank accession SAMN26586984- SAMN26587084).
References
Akman Gündüz E, Douglas AE (2009) Symbiotic bacteria enable insect to use a nutritionally inadequate diet. Proc R Soc B 276:987–991. https://doi.org/10.1098/rspb.2008.1476
Cheng D, Guo Z, Riegler M et al (2017) Gut symbiont enhances insecticide resistance in a significant pest, the oriental fruit fly Bactrocera dorsalis (Hendel). Microbiome 5:13. https://doi.org/10.1186/s40168-017-0236-z
Eleftherianos I, Atri J, Accetta J, Castillo JC (2013) Endosymbiotic bacteria in insects: guardians of the immune system? Front Physiol 4:46. https://doi.org/10.3389/fphys.2013.00046
Behar A, Yuval B, Jurkevitch E (2008) Gut bacterial communities in the Mediterranean fruit fly (Ceratitis capitata) and their impact on host longevity. J Insect Physiol 54:1377–1383. https://doi.org/10.1016/j.jinsphys.2008.07.011
Ben-Yosef M, Jurkevitch E, Yuval B (2008) Effect of bacteria on nutritional status and reproductive success of the Mediterranean fruit fly Ceratitis capitata. Physiol Entomol 33:145–154. https://doi.org/10.1111/j.1365-3032.2008.00617.x
Aharon Y, Pasternak Z, Ben Yosef M et al (2013) Phylogenetic, metabolic, and taxonomic diversities shape Mediterranean fruit fly microbiotas during ontogeny. Appl Environ Microbiol 79:303–313. https://doi.org/10.1128/AEM.02761-12
Saridaki A, Bourtzis K (2010) Wolbachia: more than just a bug in insects genitals. Curr Opin Microbiol 13:67–72. https://doi.org/10.1016/j.mib.2009.11.005
Hilgenboecker K, Hammerstein P, Schlattmann P et al (2008) How many species are infected with Wolbachia? A statistical analysis of current data. FEMS Microbiol Lett 281:215–220. https://doi.org/10.1111/j.1574-6968.2008.01110.x
Kaur R, Shropshire JD, Cross KL et al (2021) Living in the endosymbiotic world of Wolbachia: A centennial review. Cell Host Microbe 29:879–893. https://doi.org/10.20944/preprints202103.0338.v1
Werren JH (1997) Biology of Wolbachia. Annu Rev Entomol 42:587–609. https://doi.org/10.1146/annurev.ento.42.1.587
Hedges LM, Brownlie JC, O’Neill SL, Johnson KN (2008) Wolbachia and virus protection in insects. Science 322:702. https://doi.org/10.1126/science.1162418
Hughes GL, Koga R, Xue P et al (2011) Wolbachia infections are virulent and inhibit the human malaria parasite Plasmodium falciparum in Anopheles gambiae. PLoS Pathog 7:e1002043. https://doi.org/10.1371/journal.ppat.1002043
Teixeira L, Ferreira Á, Ashburner M (2008) The bacterial symbiont Wolbachia induces resistance to RNA viral infections in Drosophila melanogaster. PLoS Biol 6:e1000002. https://doi.org/10.1371/journal.pbio.1000002
Hosokawa T, Koga R, Kikuchi Y et al (2010) Wolbachia as a bacteriocyte-associated nutritional mutualist. Proc Natl Acad Sci U S A 107:769–774. https://doi.org/10.1073/pnas.0911476107
Ju JF, Bing XL, Zhao DS et al (2020) Wolbachia supplement biotin and riboflavin to enhance reproduction in planthoppers. ISME J 14:676–687. https://doi.org/10.1038/s41396-019-0559-9
Audsley MD, Seleznev A, Joubert DA et al (2018) Wolbachia infection alters the relative abundance of resident bacteria in adult Aedes aegypti mosquitoes, but not larvae. Mol Ecol 27:297–309. https://doi.org/10.1111/mec.14436
Dittmer J, Bouchon D (2018) Feminizing Wolbachia influence microbiota composition in the terrestrial isopod Armadillidium vulgare. Sci Rep 8:6998. https://doi.org/10.1038/s41598-018-25450-4
Duan XZ, Sun JT, Wang LT et al (2020) Recent infection by Wolbachia alters microbial communities in wild Laodelphax striatellus populations. Microbiome 8:104. https://doi.org/10.1186/s40168-020-00878-x
Ourry M, Crosland A, Lopez V et al (2021) Influential insider: Wolbachia, an intracellular symbiont, manipulates bacterial diversity in its insect host. Microorganisms 9:1313. https://doi.org/10.3390/microorganisms9061313
Hughes GL, Dodson BL, Johnson RM et al (2014) Native microbiome impedes vertical transmission of Wolbachia in Anopheles mosquitoes. Proc Natl Acad Sci 111:12498–12503. https://doi.org/10.1073/pnas.1408888111
Dheilly NM, Martínez Martínez J, Rosario K et al (2019) Parasite microbiome project: Grand challenges. PLOS Pathog 15:e1008028. https://doi.org/10.1371/journal.ppat.1008028
Hammer TJ, Moran NA (2019) Links between metamorphosis and symbiosis in holometabolous insects. Philos Trans R Soc B Biol Sci 374:20190068. https://doi.org/10.1098/rstb.2019.0068
Kathirithamby J (1998) Host-parasitoid associations of Strepsiptera: Anatomical and developmental consequences. Int J Insect Morphol Embryol 27:39–51. https://doi.org/10.1016/S0020-7322(97)00031-7
Kathirithamby J (1991) Strepsiptera. In: The Insects of Australia. Volume II. Second edition. CSIRO, Melbourne University Press, Carlton, Victoria, Australia, pp 685–695
Kathirithamby J (1989) Review of the order Strepsiptera. Syst Entomol 14:41–92. https://doi.org/10.1111/j.1365-3113.1989.tb00265.x
Kathirithamby J (2009) Host-parasitoid associations in Strepsiptera. Annu Rev Entomol 54:227–249. https://doi.org/10.1146/annurev.ento.54.110807.090525
Kathirithamby J (2018) Biodiversity of Strepsiptera. In: Foottit RG, Adler PH, editors. Insect Biodiversity: Science and Society. Volume II, first edition. John Wiley & Sons, Chichester, UK, pp 673–703
Towett-Kirui S, Morrow JL, Close S et al (2021) Host-endoparasitoid-endosymbiont relationships: Concealed Strepsiptera provide new twist to Wolbachia in Australian tephritid fruit flies. Environ Microbiol 23:5587–5604. https://doi.org/10.1111/1462-2920.15715
Dheilly NM, Poulin R, Thomas F (2015) Biological warfare: Microorganisms as drivers of host-parasite interactions. Infect Genet Evol 34:251–259. https://doi.org/10.1016/j.meegid.2015.05.027
Cayetano L, Vorburger C (2015) Symbiont-conferred protection against hymenopteran parasitoids in aphids: How general is it? Ecol Entomol 40:85–93. https://doi.org/10.1111/een.12161
Oliver KM, Russell JA, Morant NA, Hunter MS (2003) Facultative bacterial symbionts in aphids confer resistance to parasitic wasps. Proc Natl Acad Sci U S A 100:1803–1807. https://doi.org/10.1073/pnas.0335320100
Fredensborg BL, Fossdal Í, Kálvalíð I, Johannesen TB et al (2020) Parasites modulate the gut-microbiome in insects: A proof-of-concept study. PLoS One 15:e0227561
Kathirithamby J, Ross LD, Johnston JS (2003) Masquerading as self? Endoparasitic Strepsiptera (Insecta) enclose themselves in host-derived epidermal bag. Proc Natl Acad Sci U S A 100:7655–7659. https://doi.org/10.1073/pnas.1131999100
Chaplinska M, Gerritsma S, Dini-Andreote F et al (2016) Bacterial communities differ among Drosophila melanogaster populations and affect host resistance against parasitoids. PLoS ONE 11:1–21. https://doi.org/10.1371/journal.pone.0167726
Cavichiolli De Oliveira N, Cônsoli FL (2020) Beyond host regulation: Changes in gut microbiome of permissive and non-permissive hosts following parasitization by the wasp Cotesia flavipes. FEMS Microbiol Ecol 96:fiz206. https://doi.org/10.1093/FEMSEC/FIZ206
Hammer TJ, Sanders JG, Fierer N (2019) Not all animals need a microbiome. FEMS Microbiol Lett 366:fnz117. https://doi.org/10.1093/femsle/fnz117
Fitzpatrick CR, Schneider AC (2020) Unique bacterial assembly, composition, and interactions in a parasitic plant and its host. J Exp Bot 71:2198–2209. https://doi.org/10.1093/jxb/erz572
Brealey JC, Lecaudey LA, Kodama M et al (2022) Microbiome “inception”: an intestinal cestode shapes a hierarchy of microbial communities nested within the host. MBio 13:e00679-e722. https://doi.org/10.1128/mbio.00679-22
Ogier JC, Pagès S, Frayssinet M, Gaudriault S (2020) Entomopathogenic nematode-associated microbiota: From monoxenic paradigm to pathobiome. Microbiome 8:25. https://doi.org/10.1186/s40168-020-00800-5
Nedoluzhko AV, Sharko FS, Tsygankova SV et al (2017) Metagenomic analysis of microbial community of a parasitoid wasp Megaphragma amalphitanum. Genomics Data 11:87–88. https://doi.org/10.1016/j.gdata.2016.12.007
Giusti F, Dallai L, Beani L et al (2007) The midgut ultrastructure of the endoparasite Xenos vesparum (Rossi) (Insecta, Strepsiptera) during post-embryonic development and stable carbon isotopic analyses of the nutrient uptake. Arthropod Struct Dev 36:183–197. https://doi.org/10.1016/j.asd.2007.01.001
Kathirithamby J (2000) Morphology of the female Myrmecolacidae (Strepsiptera) including the apron, and an associated structure analogous to the peritrophic matrix. Zool J Linn Soc 128:269–287. https://doi.org/10.1111/j.1096-3642.2000.tb00164.x
Towett-Kirui S, Morrow JL, Riegler M (2022) Substantial rearrangements, single nucleotide frameshift deletion and low diversity in mitogenome of Wolbachia-infected strepsipteran endoparasitoid in comparison to its tephritid hosts. Sci Reports 12:477. https://doi.org/10.1038/s41598-021-04398-y
Johnston JS, Ross LD, Beani L et al (2004) Tiny genomes and endoreduplication in Strepsiptera. Insect Mol Biol 13:581–585. https://doi.org/10.1111/j.0962-1075.2004.00514.x
Sundberg LR, Pulkkinen K (2015) Genome size evolution in macroparasites. Int J Parasitol 45:285–288. https://doi.org/10.1016/j.ijpara.2014.12.007
Moran NA, Baumann P (2000) Bacterial endosymbionts in animals. Curr Opin Microbiol 3:270–275. https://doi.org/10.1016/S1369-5274(00)00088-6
Wernegreen JJ (2002) Genome evolution in bacterial endosymbionts of insects. Nat Rev Genet 3:850–861
Drew RAI, Allwood AJ (1985) A new family of Strepsiptera parasitizing fruit flies (Tephritidae) in Australia. Syst Entomol 10:129–134. https://doi.org/10.1111/j.1365-3113.1985.tb00523.x
Allwood AJ, Drew RAI (1996) Seasonal abundance, distribution, hosts and taxonomic placement of Dipterophagus daci (Strepsiptera: Dipterophagidae). Aust Entomol 23:61–71
Morrow JL, Frommer M, Shearman DCA, Riegler M (2014) Tropical tephritid fruit fly community with high incidence of shared Wolbachia strains as platform for horizontal transmission of endosymbionts. Environ Microbiol 16:3622–3637. https://doi.org/10.1111/1462-2920.12382
Morrow JL, Frommer M, Royer JE et al (2015) Wolbachia pseudogenes and low prevalence infections in tropical but not temperate Australian tephritid fruit flies: manifestations of lateral gene transfer and endosymbiont spillover? BMC Evol Biol 15:202. https://doi.org/10.1186/s12862-015-0474-2
Bragard C, Dehnen-Schmutz K, Di Serio F et al (2020) Pest categorisation of non-EU Tephritidae. EFSA J 18:e05931. https://doi.org/10.2903/j.efsa.2020.5931
Vargas RI, Piñero JC, Leblanc L (2015) An overview of pest species of Bactrocera fruit flies (Diptera: Tephritidae) and the integration of biopesticides with other biological approaches for their management with a focus on the Pacific region. Insects 6:297–318. https://doi.org/10.3390/insects6020297
Morrow JL, Frommer M, Shearman DCA, Riegler M (2015) The microbiome of field-caught and laboratory-adapted Australian tephritid fruit fly species with different host plant use and specialisation. Microb Ecol 70:498–508. https://doi.org/10.1007/s00248-015-0571-1
Colman DR, Toolson EC, Takacs-Vesbach CD (2012) Do diet and taxonomy influence insect gut bacterial communities? Mol Ecol 21:5124–5137. https://doi.org/10.1111/j.1365-294X.2012.05752.x
Woruba DN, Morrow JL, Reynolds OL et al (2019) Diet and irradiation effects on the bacterial community composition and structure in the gut of domesticated teneral and mature Queensland fruit fly, Bactrocera tryoni (Diptera: Tephritidae). BMC Microbiol 19(Suppl 1):281. https://doi.org/10.1186/s12866-019-1649-6
Royer JE, Hancock DL (2012) New distribution and lure records of Dacinae (Diptera: Tephritidae) from Queensland, Australia, and description of a new species of Dacus fabricius. Aust J Entomol 51:239–247. https://doi.org/10.1111/j.1440-6055.2012.00864.x
Callahan BJ, McMurdie PJ, Rosen MJ et al (2016) DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. https://doi.org/10.1038/nmeth.3869
Bokulich NA, Kaehler BD, Rideout JR et al (2018) Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90. https://doi.org/10.1186/s40168-018-0470-z
Robinson MD, McCarthy DJ, Smyth GK (2009) edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. https://doi.org/10.1093/bioinformatics/btp616
Doorenweerd C, Leblanc L, Norrbom AL et al (2018) A global checklist of the 932 fruit fly species in the tribe Dacini (Diptera, Tephritidae). Zookeys 730:19–56. https://doi.org/10.3897/zookeys.730.21786
Deutscher AT, Chapman TA, Shuttleworth LA et al (2019) Tephritid-microbial interactions to enhance fruit fly performance in sterile insect technique programs. BMC Microbiol 19(Suppl 1):287. https://doi.org/10.1186/s12866-019-1650-0
Yun JH, Roh SW, Whon TW et al (2014) Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl Environ Microbiol 80:5254–5264. https://doi.org/10.1128/AEM.01226-14
Stouthamer R, Breeuwer JA, Hurst GD (1999) Wolbachia pipientis: microbial manipulator of arthropod reproduction. Annu Rev Microbiol 53:71–102. https://doi.org/10.1146/annurev.micro.53.1.71
Werren JH, Baldo L, Clark ME (2008) Wolbachia: Master manipulators of invertebrate biology. Nat Rev Microbiol 6:741–751. https://doi.org/10.1038/nrmicro1969
Fry AJ, Palmer MR, Rand DM (2004) Variable fitness effects of Wolbachia infection in Drosophila melanogaster. Heredity 93:379–389. https://doi.org/10.1038/sj.hdy.6800514
Diouf M, Miambi E, Mora P et al (2018) Variations in the relative abundance of Wolbachia in the gut of Nasutitermes arborum across life stages and castes. FEMS Microbiol Lett 365:fny046. https://doi.org/10.1093/femsle/fny046
Gottlieb Y, Ghanim M, Gueguen G et al (2008) Inherited intracellular ecosystem: symbiotic bacteria share bacteriocytes in whiteflies. FASEB J 22:2591–2599. https://doi.org/10.1096/fj.07-101162
Brownlie JC, Cass BN, Riegler M et al (2009) Evidence for metabolic provisioning by a common invertebrate endosymbiont, Wolbachia pipientis, during periods of nutritional stress. PLoS Pathog 5:e1000368. https://doi.org/10.1371/journal.ppat.1000368
Stevanovic AL, Arnold PA, Johnson KN (2015) Wolbachia-mediated antiviral protection in Drosophila larvae and adults following oral infection. Appl Environ Microbiol 81:8215–8223. https://doi.org/10.1128/AEM.02841-15
Bian G, Xu Y, Lu P et al (2010) The endosymbiotic bacterium Wolbachia induces resistance to dengue virus in Aedes aegypti. PLoS Pathog 6:e1000833. https://doi.org/10.1371/journal.ppat.1000833
Pimentel AC, Cesar CS, Martins M, Cogni R (2021) The antiviral effects of the symbiont bacteria Wolbachia in insects. Front Immunol 11:626329. https://doi.org/10.3389/fimmu.2020.626329
Sharpe SR, Morrow JL, Brettell LE et al (2021) Tephritid fruit flies have a large diversity of co-occurring RNA viruses. J Invertebr Pathol 186:107569. https://doi.org/10.1016/j.jip.2021.107569
Morrow JL, Sharpe SR, Tilden G et al (2023) Transmission modes and efficiency of iflavirus and cripavirus in Queensland fruit fly Bactrocera tryoni. J Invertebr Pathol 197:107874. https://doi.org/10.1016/J.JIP.2022.107874
Majumder R, Taylor PW, Chapman TA (2022) Dynamics of the Queensland fruit fly microbiome through the transition from nature to an established laboratory colony. Microorganisms 10:291. https://doi.org/10.3390/microorganisms10020291
Ben-Yosef M, Pasternak Z, Jurkevitch E, Yuval B (2014) Symbiotic bacteria enable olive flies (Bactrocera oleae) to exploit intractable sources of nitrogen. J Evol Biol 27:2695–2705. https://doi.org/10.1111/jeb.12527
Yeap HL, Lee SF, Robinson F et al (2020) Separating two tightly linked species-defining phenotypes in Bactrocera with hybrid recombinant analysis. BMC Genet 21:132. https://doi.org/10.1186/s12863-020-00936-1
Koch H, Schmid-Hempel P (2011) Socially transmitted gut microbiota protect bumble bees against an intestinal parasite. Proc Natl Acad Sci U S A 108:19288–19292. https://doi.org/10.1073/pnas.1110474108
Acknowledgements
The authors would like to thank Caroline Janitz from the Western Sydney University Next-Generation Sequencing Facility for the Miseq sequencing. We also thank Melissa Starkie and Caterina Torrisi from the Queensland Department of Agriculture and Fisheries for their help with fruit fly collection and identification and two reviewers for their constructive feedback on our manuscript.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions This research was supported by the Australian Research Council (ARC) Industrial Transformation Training Centre (ITTC) Fruit Fly Biosecurity Innovation (IC150100026). STK was supported by an ARC ITTC scholarship and the 2018 E.A. Southee Award of the Hawkesbury Foundation.
Author information
Authors and Affiliations
Contributions
STK, MR and JLM conceived the study. The research was designed by STK, MR and JLM; STK performed the experiments and data analysis with input from MR and JLM; JR and SC provided the insect specimens. STK and MR wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Towett-Kirui, S., Morrow, J.L., Close, S. et al. Bacterial Communities Are Less Diverse in a Strepsipteran Endoparasitoid than in Its Fruit Fly Hosts and Dominated by Wolbachia. Microb Ecol 86, 2120–2132 (2023). https://doi.org/10.1007/s00248-023-02218-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-023-02218-6